
Abstract
Background:
This phase 2 study evaluated trebananib (AMG 386), an investigational peptide-Fc fusion protein that neutralises the interaction between angiopoietins-1/2 and the Tie2 receptor, plus FOLFIRI as second-line treatment for patients with metastatic colorectal cancer.
Methods:
Patients had adenocarcinoma of the colon or rectum with progression within 6 months of receiving only one prior fluoropyrimidine/oxaliplatin-based chemotherapy regimen for metastatic disease. All patients received FOLFIRI and were randomised 2 : 1 to also receive intravenous trebananib 10 mg kg−1 once weekly (QW) (Arm A) or placebo QW (Arm B). The primary end point was investigator-assessed progression-free survival (PFS).
Results:
One hundred and forty-four patients were randomised (Arms A/B, n=95/49). Median PFS in Arms A and B was 3.5 and 5.2 months (hazard ratio (HR) 1.23; 95% CI, 0.81–1.86; P=0.33) and median overall survival (OS) was 11.9 and 8.8 months, respectively (HR 0.90; 95% CI; 0.53–1.54; P=0.70). Objective response rate (ORR) was 14% and 0% in Arms A and B, respectively. Incidence of grade ⩾3 adverse events was similar between treatment arms (Arm A, 61%; Arm B, 65%) and included pulmonary embolism (1%/4%), deep vein thrombosis (5%/2%), and hypertension (1%/0%).
Conclusion:
Administration of trebananib plus FOLFIRI did not prolong PFS compared with placebo plus FOLFIRI. Toxicities were manageable and consistent with those known for FOLFIRI and trebananib.
Similar content being viewed by others


Main
Current first- and second-line therapies for metastatic colorectal cancer (mCRC) include a variety of different oxaliplatin- and irinotecan-based chemotherapy regimens (Van Cutsem et al, 2010). Improved outcomes have been demonstrated with chemotherapy combined with therapies targeting the epidermal growth factor receptor (EGFR) or vascular endothelial growth factor (VEGF) (Van Cutsem et al, 2010). However, overall survival (OS) times remain relatively short and investigation of alternative treatment strategies is warranted.
Angiogenesis is a complex process that has an important role in tumour development, growth, and metastasis (Carmeliet and Jain, 2011). The VEGF pathway and the angiopoietin-Tie2 receptor axis have distinct roles in the regulation of pathologic angiogenesis (Huang et al, 2010). Evidence suggests that the angiopoietins may be implicated in colorectal cancer. Elevated serum angiopoietin-2 has been reported in patients with colorectal cancer compared with healthy controls (Goede et al, 2010), and increased serum angiopoietin-2 has been associated with poorer survival outcomes (Volkova et al, 2011). Furthermore, higher tumour angiopoietin-2 expression has been associated with lymph node metastasis, venous invasion, and microvascular density (Chung et al, 2006). Preclinical evidence suggests there may be interactions between the angiopoietin axis and other signalling pathways, including the EGFR pathway (Fujiyama et al, 2001) that could contribute to tumour angiogenesis. Potentially, inhibiting angiogenesis via blockade of the angiopoietin axis may represent a novel treatment approach for colorectal cancer.
Trebananib (formerly known as AMG 386) is an investigational, intravenously administered peptide-Fc fusion protein that neutralises the interaction between angiopoietin-1 and angiopoietin-2 and the Tie2 receptor. In a Colo205 colorectal cancer tumour xenograft model, blocking the angiopoietin-2/Tie2 interaction inhibited tumour growth (Oliner et al, 2004). Importantly, administration of peptibodies targeting angiopoietin-1 or angiopoietin-2 was less effective in inhibiting Colo205 xenograft growth than dual inhibition of angiopoietin-1 and angiopoietin-2 (either by combined administration of anti-angiopoietin-1– and anti-angiopoietin-2–peptibodies or by administration of trebananib) (Coxon et al, 2010). Trebananib has shown encouraging antitumour activity and exhibited a specific toxicity profile when administered as monotherapy (Herbst et al, 2009) or in combination with various chemotherapy regimens (Mita et al, 2010), including weekly paclitaxel in patients with recurrent ovarian cancer (Karlan et al, 2012). The primary objective of our study was to estimate the treatment effect (as assessed by progression-free survival (PFS)) of second-line trebananib plus FOLFIRI vs placebo plus FOLFIRI in patients with mCRC.
Materials and Methods
Patients
Eligible patients (⩾18 years) had histologically confirmed, metastatic adenocarcinoma of the colon or rectum, had received only one prior fluoropyrimidine- and oxaliplatin-based chemotherapy regimen for metastatic disease, had measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 (Therasse et al, 2000), and had radiographically documented disease progression per RECIST during or within 6 months of their last chemotherapy dose. Other eligibility criteria included Eastern Cooperative Oncology Group (ECOG) performance status ⩽1; life expectancy ⩾3 months; and adequate haematologic, renal, hepatic, and haemostatic function. Key exclusion criteria were arterial or deep venous thromboembolism within 12 months of randomisation; clinically significant bleeding within 6 months; clinically significant cardiovascular disease within 12 months; and nonhealing wound, ulcer, or fracture; radiotherapy within 14 days (patients must have recovered from all radiotherapy-related toxicities); prior therapy with angiopoietin-Tie2 axis inhibitors; and prior irinotecan therapy. Prior treatment with anticancer agents other than irinotecan was allowed with a sufficient washout period before randomisation (30 days for proteins/antibodies (including bevacizumab) and 21 days for other agents) and prior adjuvant chemotherapy (in addition to first-line therapy) was allowed if it preceded the onset of metastatic disease. All patients provided written informed consent; study procedures were approved by independent ethics committees/institutional review boards.
Study design and treatment
This randomised, double-blind, placebo-controlled phase 2 study was conducted at 38 international sites. Using an interactive voice response system, patients were randomly assigned 2 : 1 to receive (Arm A) intravenous (IV) trebananib 10 mg kg−1 once weekly (QW) plus FOLFIRI (irinotecan 180 mg m−2 IV plus leucovorin 400 mg m−2 IV plus 5-FU 400 mg m−2 IV bolus followed by 2400 mg m−2 continuous IV infusion) once every 2 weeks (Q2W), or (Arm B) placebo QW plus FOLFIRI Q2W. Randomisation was stratified by ECOG status (0 vs 1). Patients, investigators, and study staff were blinded to treatment assignments. Treatment continued until disease progression, unacceptable toxicity, or withdrawal of consent. Dose modifications were not permitted for trebananib or placebo. Treatment was permanently discontinued if withheld for >28 days or >2 times because of treatment-related toxicity or in the event of the following toxicities: central nervous system haemorrhage (any grade), haemorrhage (grade ⩾3), grade 4 symptomatic venous thromboembolic event, or arterial thrombosis (any grade).
The primary end point was PFS. Secondary end points included objective response rate (ORR) per RECIST (Therasse et al, 2000), duration of response, time to response, OS, incidence of adverse events (AEs), incidence of anti-trebananib antibodies, and assessment of trebananib pharmacokinetics. Exploratory end points included assessment of pharmacodynamic biomarkers. Furthermore, PFS was also evaluated by KRAS mutational status.
Efficacy assessments
Radiologic tumour measurement (computed tomography/magnetic resonance imaging) was performed at baseline and every 8±1 weeks thereafter. Responses were assessed according to RECIST version 1.0 (Therasse et al, 2000) by investigators and confirmed ⩾28 days after the initial criteria for response were met. Patients who discontinued treatment without progressive disease or withdrew consent continued scheduled response assessments until disease progression or initiation of new therapy. For patients discontinuing treatment because of progression or unacceptable toxicity, long-term follow-up is being conducted every 3 months through 30 months from the date the last patient was randomised.
Toxicity assessments
Adverse events were recorded and graded for severity according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. A safety follow-up visit occurred 30–37 days after a patient discontinued the study for any reason. Serum samples for measurement of human anti-trebananib binding and neutralising antibodies (evaluated as described previously) (Herbst et al, 2009) were collected predose on day 1 of weeks 1, 5, and 9; every 16 weeks thereafter; and at the safety follow-up visit.
Pharmacokinetics and biomarkers
Methods for pharmacokinetic analysis of trebananib, 5-FU, SN-38, and irinotecan and analysis of the biomarkers angiopoietin-1, angiopoietin-2, placental growth factor (PLGF), soluble vascular cell adhesion molecule-1 (sVCAM-1), VEGF-A, soluble VEGF receptors 1 and 2, and soluble Kit are described in the Supplementary Material.
Statistical analysis
In this phase 2 study, the planned enrolment of 138 patients (Arm A, n=92; Arm B, n=46) was intended to generate treatment effect estimates of trebananib plus FOLFIRI vs placebo plus FOLFIRI. The primary analysis was planned for when 100 PFS events had occurred. The primary statistical analysis was estimation of the hazard ratio (HR) of PFS. With a hypothesised HR of 0.69, this allowed estimation of the HR for PFS with a two-sided 80% CI with a maximum half-width of 0.22. Efficacy end points were analysed for the intent-to-treat population (all randomised patients). The safety analysis set included all patients who received ⩾1 dose of study treatment.
A Cox regression model stratified by ECOG performance status was used to estimate HRs and two-sided 80% CIs and 95% CIs for PFS (time from randomisation to disease progression per RECIST or death) and 95% CIs for OS (time from randomisation to death). Kaplan–Meier estimates of median (95% CI) PFS, OS, time to response, and duration of response were also derived (Brookmeyer and Crowley, 1982). Exact binomial two-sided 95% CIs were generated for ORR for both treatment arms. The 95% CI for the difference in ORR between treatment arms was calculated using Wilson’s score method with continuity correction (Newcombe, 1998). Analyses of PFS, OS, and ORR by KRAS mutation status were performed for each KRAS subgroup using similar methods.
Results
Patients
Between December 2008 and May 2010, 144 patients were randomised (Arm A, n=95; Arm B, n=49). All but one patient in Arm A received ⩾1 dose of trebananib/placebo. Baseline demographics and clinical characteristics were generally balanced in both treatment arms (Table 1). Twenty-one patients in Arm A and 9 in Arm B had received antiangiogenic agents before the study, including 20 and 8 patients, respectively, who had received bevacizumab. The proportion of patients with wild-type, mutant, or unknown KRAS status was 49%, 36%, and 15%, respectively, in Arm A, and 59%, 29%, and 12%, respectively, in Arm B. At the time of this primary analysis, 15 patients in Arm A and 12 in Arm B continued to receive study treatment; reasons for discontinuation are shown in Figure 1. Patients in Arm A received a median (range) of 9 (1–57) trebananib infusions; patients in Arm B received 16 (2–55) placebo infusions. A median of 6 and 8 cycles of FOLFIRI were administered in Arms A and B, respectively. Median follow-up time was 27.3 weeks for Arm A and 24.4 weeks for Arm B. Forty-six per cent of patients in Arm A and 38% of patients in Arm B received anticancer treatment after progression. More patients in Arm A than Arm B received chemotherapy plus anti-EGFR (12% vs 3%) or anti-VEGF (5% vs 0%) therapy post-progression.

Disposition of study patients. Noncompliance includes patients who did not comply with study drug administration, visit schedule, or other protocol requirement(s). QW=once weekly.
PFS and OS
At the time of this analysis, 72 (76%) and 35 (71%) patients in Arms A and B, respectively, had had disease progression or died. The HR for PFS was 1.23 (95% CI, 0.81–1.86; P=0.33) and median PFS was 3.5 months in Arm A vs 5.2 months in Arm B (Table 2; Figure 2). Overall survival data were not mature at the time of this primary analysis: 40% of patients in Arm A and 43% of patients in Arm B had died. Median estimated OS in Arms A and B was 11.9 and 8.8 months, respectively (HR, 0.90; 95% CI, 0.53–1.54; P=0.70; Table 2).

Progression-free survival among patients randomised to trebananib 10 mg kg−1 QW plus FOLFIRI or placebo plus FOLFIRI. QW=once weekly.
ORR
The confirmed ORR was 14% in Arm A (including two complete responses) and 0% in Arm B (Table 2). The median duration of response for patients in Arm A was 27.1 weeks, and the mean time to response was 12.9 weeks. The proportion of patients with reductions in tumour size from baseline was 64% and 59% in Arms A and B, respectively.
Outcomes by KRAS status
Among patients with wild-type KRAS tumours (n=76), median PFS was 5.2 months in Arm A and 4.5 months in Arm B (HR, 0.96; 95% CI, 0.56–1.67; P=0.89). For those with mutant KRAS tumours (n=48), median PFS was 2.8 months in Arm A and 5.5 months in Arm B (HR, 2.10; 95% CI, 0.84–5.25; P=0.12). Corresponding median OS times were 11.9 months in Arm A and 12.1 months in Arm B for patients with wild-type KRAS (HR, 0.86; 95% CI, 0.40–1.85; P=0.70) and 9.6 and 8.8 months, respectively, for those with mutant KRAS (HR, 1.04; 95% CI, 0.39–2.77; P=0.94). The ORR for Arm A patients with wild-type KRAS was 17.5% vs 10.0% for those with mutant KRAS.
Toxicity
The most frequently occurring AEs in both arms were diarrhoea, nausea, and neutropenia (Table 3). Generally, the incidence of AEs of any grade was similar across the treatment arms. Exceptions included peripheral oedema, which occurred more often in Arm A (20% vs 4% in Arm B; no grade ⩾3), and neutropenia, vomiting, and anaemia, which were more frequent in Arm B (Table 3). Both treatment arms also had a similar incidence of grade ⩾3 AEs (62% in Arm A vs 65% in Arm B) and serious AEs (28% vs 33%), and 12% of patients in each arm discontinued treatment or the study because of AEs. There were six (6%) fatal events in Arm A and three (6%) in Arm B. Of these, metastatic colon/colorectal cancer (Arm A, n=2) and cardiorespiratory arrest (Arm B, n=2) occurred in >1 patient. Other fatal AEs in Arm A were diarrhoea, suicide, pulmonary oedema, and acute myocardial infarction; one patient in Arm B had a fatal AE reported as ‘disability’. None of the fatal AEs were considered by study investigators to be related to study treatment.
The incidence of AEs identified as being of specific interest before the study was initiated (including arterial and venous thromboembolic events, hypertension, and perforations) was generally similar across both treatment arms (Table 4); however, some AEs warrant special mention. There was one gastrointestinal perforation (grade 3 abdominal abscess) and one grade 5 pulmonary oedema (both in Arm A). Additionally, one patient in Arm A had grade 5 acute myocardial infarction, one patient had grade 4 pulmonary embolism, and one patient had grade 4 cerebral venous thrombosis. In Arm B, one patient had grade 4 arterial thrombosis and two patients had grade 4 pulmonary embolism.
Pharmacokinetics
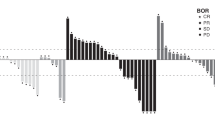
Median (per cent coefficient of variation (CV%)) trebananib Cmax (221 μg ml−1 (69.8%); n=64) and Cmin (15.6 μg ml−1 (56.2%); n=74) following coadministration with FOLFIRI at week 5 were similar to those reported in the first-in-human phase 1 monotherapy study (236 μg ml−1 and 13.7 μg ml−1, respectively) (Herbst et al, 2009). Intensive trebananib pharmacokinetic analysis was performed for seven patients (Figure 3A). Among these patients, mean (CV%) steady-state clearance for trebananib was 1.76 ml h−1 kg−1 (31.0%). At week 5, median (CV%) plasma irinotecan Cmax was similar in Arms A and B (1800 ng ml−1 (54.7%) and 1970 ng ml−1 (37.9%), respectively). Week 5 median (CV%) plasma SN-38 Cmax values were lower in Arm A than Arm B (22.4 ng ml−1 (61.5%) vs 31.6 ng ml−1 (62.3%)). However, variability within each arm was high and the difference was not statistically significant. Median (CV%) steady-state plasma concentrations (Css) for 5-FU were also lower in Arm A than Arm B at both week 1 (542 ng ml−1 (345%) vs 1310 ng ml−1 (306%)) and week 5 (347 ng ml−1 (328%) vs 560 ng ml−1 (151%)). Again, variability within each arm was high and the difference was not statistically significant. Median Css in Arm B at week 1 was higher for women than for men (Figure 3).

(A) Descriptive statistics for the pharmacokinetics of trebananib at week 5 among patients who received trebananib 10 mg kg−1 QW plus FOLFIRI (intensive pharmacokinetic analysis subset). (B) Cmax at week 5 of irinotecan among patients who received trebananib 10 mg kg−1 QW plus FOLFIRI or placebo plus FOLFIRI. (C) Cmax at week 5 of SN-38 among patients who received trebananib 10 mg kg−1 QW plus FOLFIRI or placebo plus FOLFIRI. (D) Baseline 5-FU Css at week 1 among patients who received placebo plus FOLFIRI by patient sex. (E) Css of 5-FU at week 5 among patients who received trebananib 10 mg kg−1 QW plus FOLFIRI or placebo plus FOLFIRI. Cmax=maximum observed concentration; Css=concentration at steady state; CV=coefficient of variation; QW=once weekly.
Anti-trebananib antibodies
Pre-existing nonneutralising anti-trebananib antibodies were detected in 3 out of 90 patients in Arm A, and postbaseline nonneutralising anti-trebananib antibodies developed in 1 out of 85 patients. No patient had anti-trebananib neutralising antibodies.
Biomarkers
Of the eight biomarkers tested in this study two showed a notable pharmacodynamic response. After initiation of treatment, serum PLGF increased above baseline in both Arms A and B; this increase was greater in Arm A from week 1 to week 13 (Supplementary Figure 1). Similarly, serum sVCAM-1 was elevated above baseline in both treatment arms throughout the study period, with a greater increase in Arm A than in Arm B (Supplementary Figure 2). For both PLGF and sVCAM-1, greatest increases above baseline in Arm A were measured at week 1 and 5 postdose assessments. There were limited or no changes from baseline in other biomarkers and no associations between any of the tested biomarkers and clinical outcomes (data not shown). Angiopoietin-1 and -2 could only be measured at baseline due to assay interference from trebananib present in the serum samples. Further analysis showed no association between baseline levels of these two markers and outcomes, specifically PFS (data not shown).
Discussion
In this phase 2 study, the combination of trebananib plus FOLFIRI had acceptable toxicity but did not prolong PFS compared with placebo plus FOLFIRI. In contrast, ORR appeared to favour patients in Arm A vs Arm B, although the proportion of patients with reductions in tumour size from baseline was similar. There were no apparent imbalances in prognostic/predictive factors that might have influenced the PFS results, and there is no clear explanation for the lack of correlation between PFS and ORR. Trebananib pharmacokinetic parameters were consistent with those reported in previous studies (Herbst et al, 2009; Mita et al, 2010; Karlan et al, 2012), but suggested reduced exposure to SN-38 (an irinotecan metabolite) and 5-FU among patients in Arm A. However, because the data were highly variable any contribution of this finding to the efficacy outcomes is difficult to assess.
PFS among patients in Arm B was longer than that reported for patients in other studies who received FOLFIRI following failure of a regimen containing 5-FU and/or oxaliplatin (2.5–4.7 months) (Tournigand et al, 2004; Peeters et al, 2010; Van Cutsem et al, 2011). This is somewhat surprising given that the eligibility criteria required that patients had progressed within 6 months of their most recent chemotherapy dose, which would have been expected to yield a population with relatively poor prognosis. In contrast, no patients in Arm B had an objective response, whereas previous studies have reported ORRs of 4% to 11% for patients receiving FOLFIRI following failure of 5-FU and/or oxaliplatin (Tournigand et al, 2004; Peeters et al, 2010; Van Cutsem et al, 2011). Notably, the estimated ORR in Arm A (14%) was not only higher than the ORR in Arm B but also higher than the historical range.
The incidence of AEs, grade ⩾3 AEs, serious AEs, and AEs leading to discontinuation were similar for both treatment arms. The nature and incidence rate of toxicities in the trebananib arm were consistent with those reported in previous studies of trebananib administered as monotherapy (Herbst et al, 2009) or when combined with chemotherapy (Mita et al, 2010; Karlan et al, 2012); no new toxicity signals were identified. Trebananib had a specific toxicity profile. Peripheral oedema (no grade ⩾3), which occurred more frequently in Arm A than in Arm B, appears to be a toxicity specific to trebananib treatment and has been reported in previous studies (Herbst et al, 2009; Mita et al, 2010; Karlan et al, 2012). Adverse events such as hypertension, haemorrhage, and thromboembolic events did not occur with greater incidence in Arm A than in Arm B. These AEs are of interest because they have been reported in studies of patients with mCRC receiving 5-FU–based chemotherapy plus VEGF pathway inhibitors (Hurwitz et al, 2004; Giantonio et al, 2007; Saltz et al, 2008; Van Cutsem et al, 2011). A distinct toxicity profile for trebananib is consistent with its mechanism of action of blocking the angiopoietin/Tie2 receptor pathway, separate from the VEGF cascade.
Trebananib exposure when coadministered with FOLFIRI was similar to that reported for trebananib 10 mg kg−1 administered as monotherapy (Herbst et al, 2009) or in combination with various chemotherapy regimens (Mita et al, 2010; Karlan et al, 2012). Pharmacokinetic parameters for irinotecan were comparable with and without trebananib administration. SN-38 and 5-FU exposures were lower in Arm A than Arm B; however, the data must be interpreted with caution considering the high pharmacokinetic variability. Given that trebananib is a peptibody and that 5-FU is metabolised by dihydropyrimidine dehydrogenase (DPD) (van Kuilenburg, 2004) and irinotecan undergoes glucuronidation by UGT1A1 (Gupta et al, 1994; Rouits et al, 2004), pharmacokinetic interactions were not anticipated. The data indicated higher plasma 5-FU concentrations in women vs men, which is consistent with previous studies showing that women generally have lower DPD expression, and thus metabolise 5-FU more slowly than men (Milano et al, 1992; Milano and McLeod, 2000; Yamashita et al, 2002; Kubota, 2003). The studies’ findings might also explain the lower plasma 5-FU concentrations measured in Arm A, compared with Arm B, because more male patients were randomised to that arm.
There has been interest in the use of predictive biomarkers to identify patients with mCRC most likely to derive benefit from specific targeted therapies (Deschoolmeester et al, 2010). We tested a panel of eight biomarkers in our study. Given the interdependent nature of angiogenic signalling pathways, the panel included molecules from both the angiopoietin/Tie2 axis and the VEGF pathway as well as molecules that are known to be involved in vascular remodelling (sVCAM), a consequence of angiopoietin signalling. Increases in serum levels of PLGF and sVCAM-1 occurred in both treatment arms; however, there was evidence of an additive effect for trebananib compared with placebo. Some research suggests that PLGF and sVCAM-1 have important roles in the development and progression of colorectal cancer (Velikova et al, 1998; Wei et al, 2005). We hypothesise that the observed changes in PLGF and sVCAM-1 reflect a response of the vasculature to trebananib. Both molecules have been proposed to be prognostic markers in various tumour types, including colorectal cancer (Silva et al, 2006; Okugawa et al, 2008; Willett et al, 2009; Bass et al, 2010). We tested for associations between changes in PLGF and sVCAM-1 and efficacy outcomes; however, none were identified in this study. Angiopoietin-1 and -2 were measured at baseline in each treatment arm but no association with PFS or other outcomes was found. Similarly, there was no evidence that KRAS status influenced outcomes. Additional work aimed at identifying a biomarker for trebananib is currently ongoing. Other molecules that could be tested may include the Tie2 receptor, platelet-derived growth factor, Notch, and molecules involved in vascular remodelling (e.g., intercellular adhesion molecule (ICAM)).
The chief limitation of this study was the relatively small number of patients enroled. Furthermore, evaluation of a higher dose of trebananib could have been of interest. A phase 1 study of trebananib in patients with solid tumours examined doses ranging from 0.3 mg kg−1 QW to 30 mg kg−1 QW. Although a maximum tolerated dose was not reached, pharmacokinetic data suggested that doses of 3–10 mg kg−1 would provide sufficient exposure to achieve antitumour activity (Herbst et al, 2009). In subsequent studies, trebananib doses up to 10 mg kg−1 QW in combination with several chemotherapy and targeted therapy regimens were evaluated (Mita et al, 2010; Eatock et al, 2012; Karlan et al, 2012; Rini et al, 2012). However, data from the phase 2 study of trebananib plus weekly paclitaxel for the treatment of recurrent ovarian cancer indicated a dose-response relationship (Karlan et al, 2012), and an exposure-response analysis of the results suggested that greater improvements in PFS might be achieved in that setting by administering trebananib at concentrations greater than 10 mg kg−1 (Lu et al, 2011). Given these data, assessment of trebananib in the present study at a dose higher than 10 mg kg−1 QW might have yielded different results. Three ongoing phase 3 trials in ovarian cancer are evaluating trebananib 15 mg kg−1 QW in combination with chemotherapy (NCT01204749, NCT01493505, and NCT01281254). Finally, the OS results are not yet mature and there were imbalances in post-progression therapy between the arms. Consequently, these data must be interpreted with caution.
In summary, administration of trebananib plus FOLFIRI in this estimation study did not prolong PFS compared with placebo plus FOLFIRI in patients with previously treated mCRC, but there was a trend toward improved ORR. Pharmacokinetic parameters of trebananib coadministered with FOLFIRI were comparable to those reported for trebananib monotherapy. Although exposures of 5-FU and SN-38 (but not irinotecan) were lower with trebananib coadministration, high data variability limits conclusions about drug–drug interactions. Toxicity of the treatment combination was manageable and AEs, including the distinct toxicity profile of trebananib, were consistent with what has been previously reported for FOLFIRI and trebananib. Although trebananib plus FOLFIRI did not improve PFS in this study, evidence continues to support the concept of antiangiogenesis as a treatment approach in second-line FOLFIRI, including for patients who have previously received angiogenesis inhibitors (Van Cutsem et al, 2011). It is possible that treatment approaches incorporating inhibitors of the angiopoietin/Tie2 axis could have a role if administered at different doses/schedules, in less advanced disease and/or if administered in combination with other targeted agents (e.g., VEGF inhibitors). Trebananib plus bevacizumab as first-line therapy in patients with mCRC is currently being evaluated in a phase 2 study (ClinicalTrials.gov, NCT01249521).
References
Bass MB, Sherman SI, Schlumberger MJ, Davis MT, Kivman L, Khoo HM, Notari KH, Peach M, Hei YJ, Patterson SD (2010) Biomarkers as predictors of response to treatment with motesanib in patients with progressive advanced thyroid cancer. J Clin Endocrinol Metab 95: 5018–5027
Brookmeyer R, Crowley J (1982) A confidence interval for the median survival time. Biometrics 38: 29–41
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473: 298–307
Chung YC, Hou YC, Chang CN, Hseu TH (2006) Expression and prognostic significance of angiopoietin in colorectal carcinoma. J Surg Oncol 94: 631–638
Coxon A, Bready J, Min H, Kaufman S, Leal J, Yu D, Lee TA, Sun JR, Estrada J, Bolon B, McCabe J, Wang L, Rex K, Caenepeel S, Hughes P, Cordover D, Kim H, Han SJ, Michaels ML, Hsu E, Shimamoto G, Cattley R, Hurh E, Nguyen L, Wang SX, Ndifor A, Hayward IJ, Falcon BL, McDonald DM, Li L, Boone T, Kendall R, Radinsky R, Oliner JD (2010) Context-dependent role of angiopoietin-1 inhibition in the suppression of angiogenesis and tumor growth: implications for AMG 386, an angiopoietin-1/2-neutralizing peptibody. Mol Cancer Ther 9: 2641–2651
Deschoolmeester V, Baay M, Specenier P, Lardon F, Vermorken JB (2010) A review of the most promising biomarkers in colorectal cancer: one step closer to targeted therapy. Oncologist 15: 699–731
Eatock M, Tebbutt N, Bampton CL, Strickland A, Valladares-Ayerbes M, Swieboda-Sadlej A, Van Cutsem E, Nanayakkara N, Sun Y-N, Zhong D, Bass MB, Adewoye AH, Bodoky G (2012) Phase 2 randomized, double-blind, placebo-controlled study of AMG 386 in combination with cisplatin and capecitabine in patients with metastatic gastro-oesophageal cancer. Ann Oncol doi:10.1093/annonc/mds502 (in press)
Fujiyama S, Matsubara H, Nozawa Y, Maruyama K, Mori Y, Tsutsumi Y, Masaki H, Uchiyama Y, Koyama Y, Nose A, Iba O, Tateishi E, Ogata N, Jyo N, Higashiyama S, Iwasaka T (2001) Angiotensin AT(1) and AT(2) receptors differentially regulate angiopoietin-2 and vascular endothelial growth factor expression and angiogenesis by modulating heparin binding-epidermal growth factor (EGF)-mediated EGF receptor transactivation. Circ Res 88: 22–29
Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA, Benson AB (2007) Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 25: 1539–1544
Goede V, Coutelle O, Neuneier J, Reinacher-Schick A, Schnell R, Koslowsky TC, Weihrauch MR, Cremer B, Kashkar H, Odenthal M, Augustin HG, Schmiegel W, Hallek M, Hacker UT (2010) Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer 103: 1407–1414
Gupta E, Lestingi TM, Mick R, Ramirez J, Vokes EE, Ratain MJ (1994) Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res 54: 3723–3725
Herbst RS, Hong D, Chap L, Kurzrock R, Jackson E, Silverman JM, Rasmussen E, Sun YN, Zhong D, Hwang YC, Evelhoch JL, Oliner JD, Le N, Rosen LS (2009) Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J Clin Oncol 27: 3557–3565
Huang H, Bhat A, Woodnutt G, Lappe R (2010) Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer 10: 575–585
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342
Karlan BY, Oza AM, Richardson GE, Provencher DM, Hansen VL, Buck M, Chambers SK, Ghatage P, Pippitt CH, Brown JV, Covens A, Nagarkar RV, Davy M, Leath CA, Nguyen H, Stepan DE, Weinreich DM, Tassoudji M, Sun YN, Vergote IB (2012) Randomized, double-blind, placebo-controlled phase II study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian cancer. J Clin Oncol 30: 362–371
Kubota T (2003) 5-fluorouracil and dihydropyrimidine dehydrogenase. Int J Clin Oncol 8: 127–131
Lu J-F, Rasmussen E, Karlan BY, Vergote IB, Navale L, Kuchimanchi M, Melara R, Stepan DE, Weinreich DM, Sun Y-N (2011) Exposure-response relationship of AMG 386 in combination with weekly paclitaxel in recurrent ovarian cancer and its implication for dose selection. Cancer Chemother Pharmacol 69 (5): 1135–1144
Milano G, Etienne MC, Cassuto-Viguier E, Thyss A, Santini J, Frenay M, Renee N, Schneider M, Demard F (1992) Influence of sex and age on fluorouracil clearance. J Clin Oncol 10: 1171–1175
Milano G, McLeod HL (2000) Can dihydropyrimidine dehydrogenase impact 5-fluorouracil-based treatment? Eur J Cancer 36: 37–42
Mita AC, Takimoto CH, Mita M, Tolcher A, Sankhala K, Sarantopoulos J, Valdivieso M, Wood L, Rasmussen E, Sun YN, Zhong ZD, Bass MB, Le N, LoRusso P (2010) Phase 1 study of AMG 386, a selective angiopoietin 1/2-neutralizing peptibody, in combination with chemotherapy in adults with advanced solid tumors. Clin Cancer Res 16: 3044–3056
Newcombe RG (1998) Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med 17: 873–890
Okugawa Y, Miki C, Toiyama Y, Koike Y, Yokoe T, Saigusa S, Tanaka K, Inoue Y, Ohigashi S, Kusunoki M (2008) Serum soluble VCAM-1 as a valuable prognostic marker in colorectal carcinoma (abstract). Presented at: American Society of Clinical Oncology Gastrointestinal Cancers Symposium. San Francisco, CA
Oliner J, Min H, Leal J, Yu D, Rao S, You E, Tang X, Kim H, Meyer S, Han SJ, Hawkins N, Rosenfeld R, Davy E, Graham K, Jacobsen F, Stevenson S, Ho J, Chen Q, Hartmann T, Michaels M, Kelley M, Li L, Sitney K, Martin F, Sun JR, Zhang N, Lu J, Estrada J, Kumar R, Coxon A, Kaufman S, Pretorius J, Scully S, Cattley R, Payton M, Coats S, Nguyen L, Desilva B, Ndifor A, Hayward I, Radinsky R, Boone T, Kendall R (2004) Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 6: 507–516
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, Andre T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28: 4706–4713
Rini B, Szczylik C, Tannir NM, Koralewski P, Tomczak P, Deptala A, Dirix LY, Fishman M, Ramlau R, Ravaud A, Rogowski W, Kracht K, Sun YN, Bass MB, Puhlmann M, Escudier B (2012) AMG 386 in combination with sorafenib in patients with metastatic clear cell carcinoma of the kidney: a randomized, double-blind, placebo-controlled, phase 2 study. Cancer 118 (24): 6152–6161
Rouits E, Boisdron-Celle M, Dumont A, Guerin O, Morel A, Gamelin E (2004) Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin Cancer Res 10: 5151–5159
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26: 2013–2019
Silva HC, Garcao F, Coutinho EC, De Oliveira CF, Regateiro FJ (2006) Soluble VCAM-1 and E-selectin in breast cancer: relationship with staging and with the detection of circulating cancer cells. Neoplasma 53: 538–543
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216
Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, de Gramont A (2004) FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 22: 229–237
Van Cutsem E, Nordlinger B, Cervantes A, Group EGW (2010) Advanced colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann Oncol 21 (Suppl 5): v93–v97
Van Cutsem E, Tabernero J, Lakomy R, Prausova J, Ruff P, Van Hazel G, Moiseyenko V, Ferry D, McKendrick J, Tellier A, Castan R, Allegra C (2011) Intravenous (IV) aflibercept versus placebo in combination with irinotecan/5-FU (FOLFIRI) for second-line treatment of metastatic colorectal cancer (MCRC): results of a multinational phase III trial (EFC10262-VELOUR). Presented at: 13th World Congress on Gastrointestinal Cancer. Barcelona, Spain (22–25 June 2011)
van Kuilenburg AB (2004) Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer 40: 939–950
Velikova G, Banks RE, Gearing A, Hemingway I, Forbes MA, Preston SR, Hall NR, Jones M, Wyatt J, Miller K, Ward U, Al-Maskatti J, Singh SM, Finan PJ, Ambrose NS, Primrose JN, Selby PJ (1998) Serum concentrations of soluble adhesion molecules in patients with colorectal cancer. Br J Cancer 77: 1857–1863
Volkova E, Willis JA, Wells JE, Robinson BA, Dachs GU, Currie MJ (2011) Association of angiopoietin-2, C-reactive protein and markers of obesity and insulin resistance with survival outcome in colorectal cancer. Br J Cancer 104: 51–59
Wei SC, Tsao PN, Yu SC, Shun CT, Tsai-Wu JJ, Wu CH, Su YN, Hsieh FJ, Wong JM (2005) Placenta growth factor expression is correlated with survival of patients with colorectal cancer. Gut 54: 666–672
Willett CG, Duda DG, di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, Lahdenranta J, Chung DC, Fischman AJ, Lauwers GY, Shellito P, Czito BG, Wong TZ, Paulson E, Poleski M, Vujaskovic Z, Bentley R, Chen HX, Clark JW, Jain RK (2009) Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J Clin Oncol 27: 3020–3026
Yamashita K, Mikami Y, Ikeda M, Yamamura M, Kubozoe T, Urakami A, Yoshida K, Kimoto M, Tsunoda T (2002) Gender differences in the dihydropyrimidine dehydrogenase expression of colorectal cancers. Cancer Lett 188: 231–236
Acknowledgements
We thank Michael B. Bass (Amgen Inc.) for the biomarker analysis and Don Zhong (Amgen Inc.) for anti-trebananib antibody analysis; and Chris A. Kirk, PhD, and Ali Hassan, PhD (Complete Healthcare Communications, Inc.), whose work was funded by Amgen Inc., for assistance in the preparation of this manuscript.
Clinical trial registration: ClinicalTrials.gov registration number, NCT00752570
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The results of this study were presented in part at the World Congress on Gastrointestinal Cancer; Barcelona, Spain; June 22–25, 2011.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Peeters, M., Strickland, A., Lichinitser, M. et al. A randomised, double-blind, placebo-controlled phase 2 study of trebananib (AMG 386) in combination with FOLFIRI in patients with previously treated metastatic colorectal carcinoma. Br J Cancer 108, 503–511 (2013). https://doi.org/10.1038/bjc.2012.594
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.594
Keywords
This article is cited by
-
Angiopoietin-1 and Angiopoietin-2 Inhibitors: Clinical Development
Current Oncology Reports (2019)
-
Safety and Tolerability of Anti-Angiogenic Protein Kinase Inhibitors and Vascular-Disrupting Agents in Cancer: Focus on Gastrointestinal Malignancies
Drug Safety (2019)
-
Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges
Nature Reviews Clinical Oncology (2018)
-
Angiopoietin-1 deficiency increases tumor metastasis in mice
BMC Cancer (2017)
-
Pharmacokinetic drug–drug interaction assessment of peptibody trebananib in combination with chemotherapies
Cancer Chemotherapy and Pharmacology (2015)
