
Abstract
Phosphodiesterase-9A (PDE9A) expression is upregulated during cardiac hypertrophy and heart failure. Accumulating evidence suggests that PDE9A might be a promising therapeutic target for heart diseases. The present study sought to investigate the effects and underlying mechanisms of C33(S), a novel selective PDE9A inhibitor, on cardiac hypertrophy in vitro and in vivo. Treatment of neonatal rat cardiomyocytes (NRCMs) with PE (100 μmol/L) or ISO (1 μmol/L) induced cardiac hypertrophy characterized by significantly increased cell surface areas and increased expression of fetal genes (ANF and BNP). Furthermore, PE or ISO significantly increased the expression of PDE9A in the cells; whereas knockdown of PDE9A significantly alleviated PE-induced hypertrophic responses. Moreover, pretreatment with PDE9A inhibitor C33(S) (50 and 500 nmol/L) or PF-7943 (2 μmol/L) also alleviated the cardiac hypertrophic responses in PE-treated NRCMs. Abdominal aortic constriction (AAC)-induced cardiac hypertrophy and ISO-induced heart failure were established in SD rats. In ISO-treated rats, oral administration of C33(S) (9, 3, and 1 mg·kg−1·d−1, for 3 consecutive weeks) significantly increased fractional shortening (43.55%±3.98%, 54.79%±1.95%, 43.98%±7.96% vs 32.18%±6.28%), ejection fraction (72.97%±4.64%, 84.29%±1.56%, 73.41%±9.37% vs 49.17%±4.20%) and cardiac output (60.01±9.11, 69.40±11.63, 58.08±8.47 mL/min vs 48.97±2.11 mL/min) but decreased the left ventricular internal diameter, suggesting that the transition to heart failure was postponed by C33(S). We further revealed that C33(S) significantly elevated intracellular cGMP levels, phosphorylation of phospholamban (PLB) and expression of SERCA2a in PE-treated NRCMs in vitro and in ISO-induced heart failure model in vivo. Our results demonstrate that C33(S) effectively protects against cardiac hypertrophy and postpones the transition to heart failure, suggesting that it is a promising agent in the treatment of cardiac diseases.
Similar content being viewed by others


Introduction
Cardiac hypertrophy is a complex remodeling process of the heart that occurs in response to a variety of extrinsic and intrinsic stimuli1. It is characterized by an increase in cell size and protein synthesis, the higher organization of sarcomere, and the reactivation of fetal genes, including atrial natriuretic peptide (ANF) and brain natriuretic peptide (BNP)2. Cardiac hypertrophy has been considered to be a compensatory mechanism to normalize cardiac function3. However, prolonged cardiac hypertrophy is always associated with increased morbidity and mortality4.
As a ubiquitous intracellular second messenger, cGMP participates in numerous cellular pathophysiological activities5. The protective effects of the cGMP/PKG signaling pathway on the cardiovascular system have been widely reported6,7,8,9. The increasing generation of intracellular cGMP can protect against cardiac hypertrophy that is induced by abdominal aortic constriction (AAC)10. In isolated neonatal rat cardiac myocytes (NRCMs), the direct application of cGMP-permeable analogues protects against hypertrophy by activating PKG and by regulating cardiac calcium flux11. In the heart, cGMP/PKG signaling is able to counter acute and chronic hypertrophic stress12.
The effects of cGMP could be diminished by phosphodiesterases (PDEs)13. Compared with other PDEs, PDE9A has the highest affinity to cGMP14. PDE9A can be detected in all tissues, including the heart15. In the past few years, PDE9A has been regarded as a therapeutic target for the treatment of various diseases16,17,18. A PDE9A inhibitor, PF-04447943(PF-7943), has been reported to elevate central cGMP levels in the brain and cerebrospinal fluid of rodents and has been confirmed to be well tolerated by humans in clinical trials16. When fed with a high fat diet, PDE9A knockdown mice develop a phenotype with reduced insulin resistance and weight, as well as low fat mass, suggesting a role for PDE9A in metabolic diseases19,20. Recently, Lee et al reported that PDE9A expression is upregulated during cardiac hypertrophy and heart failure21. Selective pharmacological inhibitors of PDE9A protect the heart from sustained neurohormonal stimuli and pressure overload by regulating cGMP signaling21. Mechanistically, PDE9A regulates the degradation of cGMP in a natriuretic-peptide-dependent manner rather than in a nitric-oxide-dependent manner21. Based on these findings, it is hypothesized that PDE9A might be a promising therapeutic target for heart diseases.
We previously identified (S)-6-((1-(4-chlorophenyl)ethyl)amino)-1-cyclopentyl-1,5,6,7-tetrahydro-4H-pyrazolo[3,4-day]pyrimidin-4-one [C33(S)] as a novel PDE9A inhibitor with an IC50 value of 11 nmol/L and a higher selectivity for PDE9A versus other PDE isoforms22. In this report, we hypothesized that C33(S) may protect against cardiac hypertrophy by regulating cGMP signaling. To verify our hypothesis, we carried out a series of in vitro and in vivo experiments to gain more insight into the potential effects of C33(S) on cardiac hypertrophy and heart failure.
Materials and methods
Antibodies and reagents
Rabbit anti-PDE9A polyclonal antibody was purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Mouse anti-α-tubulin monoclonal antibody was obtained from Sigma (St Louis, MO, USA). Rabbit polyclonal antibodies against GAPDH, phospholamban and phospho-phospholamban (at Ser-16) were purchased from Cell Signaling Technology Inc (CST, USA). Lipofectamine 2000 was obtained from Invitrogen (Carlsbad, CA). PF-7943 was purchased from MedChem Express (Princeton, NJ, USA). Compound C33(S) was synthesized22 and kindly provided by Prof Hai-bin LUO (School of Pharmaceutical Sciences, Sun Yat-Sen University). The cGMP direct immunoassay kit (colorimetric) was purchased from BioVision, Inc (Milpitas boulevard, Milpitas, CA, USA). Other reagents were from Sigma–Aldrich unless otherwise stated.
Primary culture of neonatal rat cardiomyocytes
As previously described23, NRCMs were isolated from the ventricles of 1- to 3-day-old Sprague-Dawley (SD) rats. Briefly, the hearts were removed immediately after the rats were anesthetized, and the minced ventricles were dispersed at 37°C in 0.08% trypsin solution approximately 12–14 times for 5 min each time. Cell suspensions were collected and, finally, the cells were harvested by centrifugation for 8 min at 1400×g and then suspended in Dulbecco's modified Eagle's medium (DMEM, Gibco, BRL Co, Ltd, USA) supplemented with 10% fetal bovine serum (FBS). The suspensions were plated in culture flasks for 1 h at 37 °C in a humidified atmosphere (5% CO2 and 95% air). Finally, cardiomyocytes were seeded onto culture dishes in DMEM supplemented with 10% FBS and 5-bromodeoxyuridine (0.1 mmol/L). After 48 h, the culture medium was changed to DMEM supplemented with 1% FBS. Cells were pretreated with C33(S) or PF-7943 for 1 h and subsequently stimulated with PE (100 μmol/L) or ISO (1 μmol/L).
Animal model, echocardiography and morphometric measurements
SD rats (male, 180-220 g, SPF grade, Certification No. 4400850000012196) from the Experimental Animal Center of Sun Yat-Sen University (Guangzhou, China) were housed under controlled environmental conditions (a 12-h:12-h light/dark cycle and a room temperature of 21–23°C) and had free access to standard laboratory food and water. The animal experiments were approved by the Research Ethics Committee of Sun Yat-Sen University and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No 85-23, revised 1996).
Abdominal aortic constriction (AAC) surgery was conducted as previously described24. Briefly, rats were randomly divided into two groups (the sham group and the AAC group) and anaesthetized with 10% chloral hydrate (350 mg/kg, ip). Each group contained 10 animals. The adequacy of anesthesia was monitored by evaluating and recording body temperature, cardiac and respiratory rates and patterns, muscle relaxation, and lash reflex. Under sterile conditions, the abdominal aorta above the kidneys was exposed through a midline abdominal incision and constricted at 4-5 cm above the suprarenal artery with a 5-0 silk suture that was tied around both the aorta and a blunted 22-gauge needle. The needle was promptly removed after constriction. The sham group underwent a similar procedure without banding the aorta.
For the ISO-induced cardiac (ventricular) heart failure model, SD rats were randomly divided into five groups: the control group; the ISO-induced heart failure model group; the ISO+high dose C33(S) group (ISO+C33-H); the ISO+medium dose C33(S) group (ISO+C33-M); and the ISO+low dose C33(S) group (ISO+C33-L). Each group contained 10 animals. ISO (2.5 mg·kg−1·d−1, ip) was given to rats for 3 consecutive weeks according to our preliminary experimental results and a previous report25. At the same time, C33(S) was suspended in sodium carboxymethyl cellulose and intragastrically administered to rats at three different dosages (high dose, 9 mg·kg−1·d−1; medium dose, 3 mg·kg−1·d−1; and low dose, 1 mg·kg−1·d−1). Rats in the vehicle control group received normal saline and sodium carboxymethyl cellulose. Two-dimensional guided M-mode echocardiography was conducted with a Technos MPX ultrasound system (ESAOTE, SpAESAOTE SpA, Italy). Afterwards, rats were sacrificed under anesthesia with 10% chloral hydrate. The hearts were rapidly removed, weighed and then carefully crosscut. The weight of the heart was expressed as a ratio relative to body weight. For morphometric measures, histological cross sections of the hearts were fixed with 4% paraformaldehyde and embedded in paraffin; histological cross sections (4–5 μm thickness) of the hearts were stained with hematoxylin and eosin (HE). From five randomly selected fields, 20 cells were chosen the nucleus of which was in the center with a clear border. Thereafter, the cardiomyocyte transverse diameter was measured from the sections under a light microscope at ×400 magnification.
3-(4, 5)-dimethylthiazol(-2-yl)-2,5-diphenytetrazolium bromide (MTT) assay
MTT assay was performed as previously described26. In brief, NRCMs were seeded onto 96-well plates at a density of 2×10−5 followed by incubation for 12 h. The PDE9A inhibitors (C33(S) and PF-3497) were added into the cell culture medium. After 12 h, the culture medium was discarded. MTT was added into the cell cultures at a final concentration of 0.5 mg/mL to incubate with the cells for 4 h at 37 °C. Then, the culture medium was removed, and dimethyl sulfoxide (DMSO) was added into each well to dissolve the formazan crystals. The absorbance was measured at a wavelength of 570 nm using a microplate reader (Bio-Tek, Elx800, USA). Duplicate assays were performed for each group, and the percent viability was defined as the relative absorbance of the treated cells versus the untreated control cells.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
RNA extraction and real-time RT-PCR were conducted as previously described27. In brief, total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. One microgram of total RNA was reverse transcribed into first-strand cDNA using the One-step RT kit (Takara Biotechnology, Dalian, China). The mRNA levels of the target genes were determined using the SYBR-Green Quantitative PCR kit (TOYOBO, Japan) on the iCycler iQ system (iCycler, Bio-Rad, Hercules, CA, USA). Rat-specific primers for ANP, BNP, PDE9A, PDE5A and β-actin (listed in Supplementary Table 1) were synthesized by Sangon (China). β-actin served as an endogenous control.
Western blot
Western blot analyses were performed as previously described23. Proteins of cultured cells or rat left ventricular tissues were harvested using RIPA lysis buffer (Beyotime, Nantong, Jiangsu, China) containing a protease inhibitor cocktail (Sigma-Aldrich, USA) on ice. Protein concentration was determined by a BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA). Equal amounts of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred to polyvinylidene fluoride (PVDF) membranes (EMD Millipore Corporation, Billerica, MA, USA). The membranes were incubated with primary antibodies overnight at 4°C followed by the appropriate secondary antibodies at room temperature for 1 h. Blots were developed with enhanced chemiluminescence reagent (Pierce, Rockford, IL, USA) and detected by the LAS4000 imager (GE Healthcare, Waukesha, WI, USA). The intensities of the blots were quantified by the Quantity One (Bio-Rad) software.
cGMP assay and serum N-terminal pro-BNP assay
Cellular cGMP levels were determined by a commercial cGMP direct immunoassay kit (Biovision, Milpitas, CA, USA) according to the manufacturer's instructions. Serum N–terminal pro-BNP was measured by an ELISA assay kit obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Measurement of cell surface areas
Cardiomyocytes cultured in 48-well plates were fixed with 4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature or overnight at 4°C. After permeabilization with 0.5% (v/v) Triton-X 100 for 10 min, the cells were blocked with goat serum (Boster Biological Technology, Ltd, China) for 1 h at room temperature or overnight at 4°C. After incubation with 0.1% (v/v) rhodamine-phalloidin for 30 min, the cells were washed with PBS 3 times and counterstained with DAPI. Images of the cardiomyocytes were detected by the High Content Screening system (ArrayScanVTI, Thermo Fisher Scientific, Rockford, IL, USA). The cell surface areas from randomly selected fields (50 for each group) were determined using the built-in image analysis software.
RNA interference
Small inference RNA (siRNA) targeting PDE9A and negative control siRNA were obtained from Genepharma (Shanghai, China). The sequences of the siRNAs are shown in Supplementary Table 2. Cardiomyocytes seeded in 35-mm dishes were transfected with 100 pmol of targeted siRNA or negative-control siRNA using 5 μL of lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Cardiomyocytes were transfected with PDE9A siRNA for 48 and 72 h for the determination of mRNA and protein, respectively. The control groups were transfected with negative control sequences.
Statistical analysis
Data were presented as the mean±SEM. Statistical analysis between two groups was performed by Student's t-test. For multiple comparisons, one-way analysis of variance (ANOVA) was used combined with Bonferroni. The analysis was performed using GraphPad Prism 5.0 (GraphPad Inc, La Jolla, CA, USA). In all cases, differences were considered statistically significant at P<0.05.
Results
PDE9A plays an important role in cardiac hypertrophy
The adrenoceptor agonists PE and ISO have been widely used to stimulate cardiomyocyte hypertrophy23. In our study, NRCMs were treated with PE (100 μmol/L) or ISO (1 μmol/L) for the indicated time points. PE- or ISO-induced cardiomyocyte hypertrophy models were successfully established, as indicated by the increased expression of fetal genes (ANF and BNP) and increased cell surface areas (Figure 1A, B and Supplementary Figure S1A, B).

PDE9A increased during cardiac hypertrophy. Knockdown of PDE9A ameliorates hypertrophic responses. (A, B) Cardiomyocytes were treated with PE or ISO and cells surface areas were measured (cell surface area, ×400). (C, D) Primary cardiac myocytes were treated with PE or ISO at different time points then protein changes of PDE9A were measured. (E, F) The protein levels of PDE9A were detected in AAC-induced cardiac hypertrophic model and ISO-induced heart failure model. *P<0.05, **P<0.01 vs control (or Sham). n=3–6. (G, H, I) PDE9A was silenced in cardioyocytes followed by PE treatment and cells surface areas and mRNA changes of fetal genes were measured (cell surface area, ×400). *P<0.05, **P<0.01 vsNC group. #P<0.05 vs NC+PE group. n=4. Data were expressed as mean±SEM.
In the in vivo study, ISO was ip injected into the rats to induce heart failure. Compared with the control group, ISO-treated rats showed a significant reduction of fractional shortening (FS, 32.18%±6.28% vs 45.85%±7.62%, P<0.01), left ventricular ejection fraction (EF, 49.17%±4.20% vs 75.49%±8.37%, P<0.05), and cardiac output (CO, 48.97±2.11 vs 64.88±4.50 mL/min, P<0.05) but an increase in left ventricular internal diameter (LVID, 5.37±0.30 vs 3.81±0.85 mm, P<0.01) (Table 1). In addition, fetal gene expression and serum N-pro-BNP levels were also increased (Figure 3). All of these results indicated the successful induction of heart failure by ISO.

C33(S) maintained cardiac function in compensated stage and postponed transition to heart failure. Rats were subjected to intraperitoneal (ip) injection of ISO and orally administration with C33(S) at three different concentrations for three weeks. At the end of the third week, cardiac functions were measured and hearts were harvested. (A) Representative diagrams of gross hearts under different treatments. (B) Representative diagram with HE-staining for left ventricular cross sections. (C) Representative diagrams with HE-staining for cardiac myocytes (HE, ×400, scale bar, 50 μm). (D) Representative echocardiographic graphs. (E) The heart to body weight ratio. (F, G) The protein levels of ANF and BNP. (H) The serum N-pro-BNP levels were measured using ELISA assay kit. C33-H: C33(S) 9 mg·kg−1·d−1. C33-M: C33(S) 3 mg·kg−1·d−1. C33-L: C33(S) 1 mg·kg−1·d−1. *P<0.05, **P<0.01 vs control group. #P<0.05, ##P<0.01 vs ISO group. n=3–6. Data were expressed as mean±SEM.
Moreover, the successful establishment of AAC-induced cardiac hypertrophy was indicated by the following observations: (1) the hearts in the AAC group were larger than those in the sham group (Supplementary Figure S2A); (2) HE staining results revealed that the cardiomyocytes in the sham group were arranged in a neat, compact and clear structure with little extracellular matrix, whereas the cells in the AAC group were disorderly arranged with hypertrophy, distortion, increased cell gap and increased extracellular matrix surrounding the cells (Supplementary Figure S2B, S2C); (3) echocardiography showed an increase in left ventricular anterior wall thickness and left ventricular posterior wall thickness, as well as a reduction in left ventricular internal diameter in the AAC rats (Supplementary Figure S2D, F, Supplementary Table S3); and (4) both the ratio of heart weight to body weight and the protein expression of ANF and BNP were elevated in the AAC group (Supplementary Figure S2E, S2G).
Subsequently, the expression of PDE9A was detected in these cardiac disease models. In vitro, the protein expression of PDE9A was elevated by ISO or PE stimulation (Figure 1C, D). In vivo, PDE9A protein expression was also increased in both the ISO-induced heart failure model and the AAC model by approximately 1.4- and 1.7-fold, respectively (Figure 1E, F). Consistent results from both in vitro and in vivo studies indicated that PDE9A may play a key role in cardiac hypertrophy and heart failure.
To further investigate the role of PDE9A in cardiac hypertrophy, endogenous PDE9A was knocked down using RNA interference. Among the three PDE9A siRNAs, si-2 could effectively reduce the mRNA and protein levels of PDE9A without influencing the expression of PDE5A (Supplementary Figure 1D, 1E), and thus si-2 was used in the following experiments. Knockdown of PDE9A alleviated PE-induced hypertrophic responses, as indicated by the decreased cell surface areas and the expression of ANF and BNP (Figure 1G, H, I, Figure S1F). These results imply that PDE9A knockdown protects against cardiac hypertrophy. Strategies that inhibit PDE9A might have therapeutic potential for pathological cardiac hypertrophy.
C33(S) alleviated cardiac hypertrophic responses in vitro and postponed the transition to heart failure in vivo
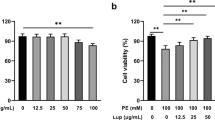
The in vitro study aimed to investigate the potential effects of the novel PDE9A inhibitor C33(S) on PE-induced cardiac hypertrophy. The effects of C33(S) were compared with those of PF-7943, a classical PDE9A inhibitor. MTT results indicated that neither treatment with C33(S) (at concentrations less than 2 μmol/L) nor treatment with PF-7943 at concentrations from 100 nmol/L to 100 μmol/L for 24 h altered the viability of the NRCMs (Supplementary Figure S3). Compared with the PE group, C33(S) at the concentrations of 500 nmol/L and 50 nmol/L significantly decreased cardiomyocyte surface area and the expression of ANF and BNP (Figure 2). The effects of C33(S) were comparable to those of the classical PDE9A inhibitor PF-7943 (Figure 2).

C33(S) could alleviate cardiac hypertrophic responses in vitro. Cardiomyocytes were pre-incubated by C33(S) and PF-7943 for 1 h followed by PE treatment or not. (A, B) Cell surface areas were measured (cell surface area, ×400). (C, D) The mRNA and protein levels of fetal genes were measured. C33(500): C33(S) 500 nmol/L, C33(50): C33(S) 50 nmol/L, PF-7943(2): PF-7943 2 μmol/L. *P<0.05, **P<0.01 vs control group. #P<0.05, ##P<0.01 vsPE group. n=5. Data were expressed as mean±SEM.
Next, we explored the in vivo effects of C33(S) on ISO-induced heart failure. Enlargement of the heart in the ISO-treated rats was reversed by C33(S) treatment at the tested concentrations (9, 3, and 1 mg·kg−1·d−1) (Figure 3A, 3B). Compared with the ISO group, echocardiography results in the C33(S)-treated groups (9, 3, and 1 mg·kg−1·d−1) showed a tendency for increased left ventricular anterior wall thickness, ejection fraction (72.97%±4.64%, 84.29%±1.56%, 73.41%±9.37% vs 49.17%±4.20%), fractional shortening (43.55%±3.98%, 54.79%±1.95%, 43.98%±7.96% vs 32.18%±6.28%) and cardiac output (60.01±9.11, 69.40±11.63, 58.08±8.47 mL/min vs 48.97±2.11 mL/min), as well as a tendency for reduced diastolic internal diameter of the left ventricle at end-diastole (7.61±0.29, 7.11±0.24, 7.06±1.20 mm vs 7.93±0.29 mm) (Figure 3D and Table 1). Additionally, C33(S) not only relieved disordered cell arrangement and enlargement but also decreased the cell gap and extracellular matrix (Figure 3C and Supplementary Figure S4C). Compared with the model group, the ratio of heart weight to body weight and the serum N-pro-BNP levels were both reduced in the C33(S)-treated groups (Figure 3E, H). ANF and BNP protein levels in the heart were also reduced by C33(S) (Figure 3F, 3G). Echocardiography revealed that C33(S) postponed the transition to heart failure by maintaining the heart in the compensation stage.
Taken together, our results show that C33(S) not only exerts protective effects on cardiac hypertrophy in vitro but also improves cardiac function and prevents the development of heart failure in vivo.
C33(S) increased cellular cGMP levels in cardiomyocytes
It is well accepted that cGMP protects against cardiac hypertrophy28,29,30. C33(S) may exert beneficial effects by inhibiting PDE9A22, therefore increasing cGMP levels. Treatment with C33(S) (500 nmol/L and 50 nmol/L) plus PE stimulation significantly elevated the cellular cGMP levels in cardiomyocytes (Figure 4). The effect of C33(S) on intracellular cGMP levels was similar to that of PF-7943 (2 μmol/L) (Figure 4).

C33(S) increased intracellular cGMP levels. Intracellular cGMP levels were measured by cGMP direct immunoassay kit. Primary cardiomyocytes were pre-incubated with C33(S) and PF-7943 for 1 h followed by PE treatment or not. Intracellular cGMP levels were measured. #P<0.05, ##P<0.01 vs PE+DMSO group. n=7. Data were expressed as mean±SEM.
C33(S) reversed the decline of phospholamban (PLB) phosphorylation and the expression of endoplasmic reticulum Ca2+ ATPase (SERCA2a) during cardiac hypertrophy and heart failure.
cGMP exerts its functions via the activation of PKG followed by the phosphorylation of its target proteins, such as PLB31. Thus, the phosphorylation level of PLB could reflect the enzymatic activity of PKG32. PE treatment markedly reduced the phosphorylation of PLB at Ser-16 without influencing total PLB protein expression (Figure 5A, 5B). Phosphorylation of PLB was also decreased in both the ISO-induced heart failure model and the AAC-induced cardiac hypertrophy model (Figure 5C, Supplementary Figure S4A). However, RNA interference of PDE9A or pretreatment with C33(S) (500 nmol/L or 50 nmol/L) or PF-7943 (2 μmol/L) reversed the decrease in PLB phosphorylation (at Ser-16) that was induced by PE stimulation (Figure 5A, 5B). Consistently, C33(S) at the three different concentrations reversed the decrease in PLB phosphorylation (at Ser-16) that was induced by ISO stimulation in vivo (Figure 5C).

C33(S) reversed the declining of PLB phosphorylation at Ser-16. (A) Cardiac myocytes were transfected with PDE9A siRNA then treated with PE. *P<0.05 vs NC group. ##P<0.01 vsNC+PE group. n=4. (B) C33(S) (500 nmol/L and 50 nmol/L) and PF-7943 (2 μmol/L), combined with or without PE treatment. *P<0.05 vs control. #P<0.05, ##P<0.01 vs PE group. n=4. (C) The phosphorylated PLB was measured in ISO induced heart failure model treated with C33(S). The data were displayed by the ratio of p-PLB to t-PLB. C33-H: C33(S) 9 mg·kg−1·d−1. C33-M: C33(S) 3 mg·kg−1·d−1. C33-L: C33(S) 1 mg·kg−1·d−1). **P<0.01 vs control. #P<0.05 vs ISO group. n=3–6. Data were expressed as mean±SEM.
Upregulation of SERCA2a postpones hypertrophic remodeling and improves cardiac function in familial hypertrophy33. Previous studies have also revealed a reduced expression of SERCA2a during cardiac hypertrophy and heart failure34,35,36,37. Thus, the potential changes in SERCA2a expression were investigated. SERCA2a was significantly downregulated by PE stimulation (Figure 6B), AAC operation (Supplementary Figure S4B) and ISO treatment in vivo (Figure 6C). The decrease of SERCA2a protein could be reversed by PDE9A silencing or treatment with C33(S) (Figure 6A–C). Interestingly, the changes in the SERCA2a protein levels were analogous to the changes in the phosphorylation level of PLB. All of these results indicate that the cardiac protective effects of C33(S) are associated with the regulation of PLB phosphorylation and increased SERCA2a expression.

C33(S) could reverse the declining expression of SERCA2a. (A) Cardiac myocytes were transfected with PDE9A siRNA and treated with PE. **P<0.01 vs CON group. #P<0.05 vs PE group. n=4. (B) C33(S) (500 nmol/L and 50 nmol/L) or PF-7943 (2 μmol/L), combined with or without PE treatment. **P<0.01 vs control group. #P<0.05 vs PE group, n=4. (C) The expression of SERCA2a was measured in ISO induced heart failure model treated with C33(S). C33-H: C33(S) 9 mg·kg−1·d−1. C33-M: C33(S) 3 mg·kg−1·d−1. C33-L: C33(S) 1 mg·kg−1·d−1. **P<0.01 vs control group. #P<0.05 vs ISO group. n=3–6 (in vivo). Data were expressed as mean±SEM.
Discussion
The most important finding of this study is that a novel PDE9A inhibitor, C33(S), plays a protective role during cardiac hypertrophy. C33(S) prevents PE-induced cardiomyocyte hypertrophy in vitro, improves cardiac function and postpones the development of heart failure induced by ISO in vivo. Mechanistically, C33(S) elevates cGMP levels by inhibiting PDE9A and relieves the decline of phosphorylated PLB and SERCA2a expression in response to pathological stimuli. These findings shed new light onto the development of C33(S) as a promising therapeutic agent for the treatment of cardiac diseases.
The exploration of PDE inhibitors for the treatment of cardiac diseases began with the success of the PDE5A inhibitor sildenafil in treating cardiac hypertrophy29. However, clinical trials of sildenafil ultimately failed in heart failure patients with preserved ejection fraction (HFpEF)38, probably due to the low bioavailability of nitric oxide in these patients21. PDE9A, with a higher affinity to cGMP than PDE5A (Km 170 nmol/L)15, has been considered to be a potential target for cardiac hypertrophy21.
Cardiac hypertrophy or cardiac remodeling can be induced by hyperactivation of the neurohormonal system39,40, such as α- or β-adrenergic receptor activation, or pressure overload. There are at least six types of adrenoceptors on cardiomyocytes, including three types of the α1-adrenergic receptor (α1A, α1B and α1D) and three types of the β-adrenergic receptor (β1, β2 and β3)41. In the present study, the α-adrenoceptor agonist phenylephrine (PE) was used to induce cardiomyocyte hypertrophy in vitro, whereas in vivo models were induced by chronic treatment of the β-adrenoceptor agonist ISO or by AAC operation. In these cardiac pathological models, an upregulation of PDE9A was observed (Figure 1) while knockdown or inhibition of PDE9A protected against cardiac hypertrophy and heart failure (Figure 1, 2, 3, Supplementary Figure S1), which was in line with a recent report21. Intriguingly, different pathological mechanisms are involved in these models. Chronic treatment of ISO leads to heart failure (characterized by a significant reduction in interventricular diastolic septal wall thickness, fractional shortening and ejection fraction with an increase in left ventricular diastolic diameter and the left ventricular mass ratio of heart-to-body weight42,43,44,45) accompanied by a significant decrease of systolic and diastolic blood pressure, left ventricular peak systolic pressure, left ventricular end-diastolic pressure and heart rate44. Meanwhile, AAC induces an increase in systolic and diastolic blood pressure, developed LV pressure, and early filling deceleration slope46. Although these two in vivo models demonstrate different trends in blood pressure, they both exhibited cardiac remodeling and an upregulation of PDE9A. Thus, these observations suggest that the upregulation of PDE9A is independent of the alteration in blood pressure. Taken together, all of these findings provide solid evidence for the crucial role of PDE9A in regulating cardiac hypertrophy.
As a novel PDE9A inhibitor, C33(S) has a tyrosyl tail and interacts with the hydrophobic pocket of PDE9A, which determines the selectivity to PDE9A over the other PDEs22. In vitro, C33(S) reversed the PE-induced hypertrophic responses at relatively lower concentrations (500 or 50 nmol/L) than the classical PDE9A inhibitor PF-7943 (2 μmol/L) (Figure 2). This is probably attributed to the higher affinity of C33(S) to PDE9A. In vivo, C33(S) (at concentrations of 9, 3 and 1 mg·kg−1·d−1) improved cardiac function and delayed the development of heart failure induced by ISO (Figure 3, Table 1). Thus, C33(S) demonstrates a promising therapeutic advantage for the treatment of cardiac diseases.
The protective effects of the cGMP/PKG pathway on the cardiovascular system have been largely reported6,7,8,9. By inhibiting PDE9A, combined treatment of C33(S) and PE elevated intracellular cGMP levels (Figure 4). However, it seems strange that PE or C33(S) alone did not alter cGMP concentrations compared with the control group (Figure 4). There might be a balance between the synthesis and the degradation of cGMP. As natriuretic peptides (NPs) stimulate the synthesis of cGMP via the activation of membrane-bounded guanylate cyclases47, PE might induce the expression of PDE9A, thereby triggering the degradation of cGMP.
The phosphorylation of PLB is important for the regulation of cardiomyocyte contraction and calcium handling48. The phosphorylation of PLB can be enhanced by PKG31,49, PKA50, and Ca2+-calmodulin-dependent protein kinase (CaMKII)51. By contrast, phosphorylated PLB can also be downregulated in the failing heart, which is probably attributed to the increase of protein phosphatase 1 (PP1) and 2A (PP2A)52. In our study, decreased PLB phosphorylation was observed in both PE-treated cardiomyocytes and hearts from ISO-treated rats (Figure 5). In line with our observations, PLB phosphorylation was decreased in response to long-term ISO stimulation50 or prolonged PE treatment (for 48 h)53. The reduction in PLB phosphorylation induced by α- or β-adrenoceptor agonists might be associated with the downregulation of the kinases PKG, PKA or CaMKII and/or the upregulation of the phosphatases PP1 and PP2A. The PDE9A inhibitor C33(S) reversed the decrease in PLB phosphorylation in vivo and in vitro (Figure 5). This beneficial effect is probably due to the augmented level of cGMP and the downstream activation of PKG. As phosphorylated PLB could activate SERCA2a to regulate Ca2+ handling54, C33(S) might enhance the activity of SERCA2a by reversing the downregulation of phosphorylated PLB.
The present study demonstrated that C33(S) also reversed the decreased expression of SERCA2a induced by PE (in vitro) or ISO (in vivo). The exact mechanisms by which C33(S) alters the expression of SERCA2a are still unknown. C33(S) might activate the cGMP/PKG pathway by inhibiting PDE9A and increasing PLB phosphorylation, ultimately enhancing SERCA2a activity and rectifying Ca2+ dysregulation54. Cardiac hypertrophy and heart failure are characterized by the dysfunction of calcium handling, which is responsible for the activation of multiple transcriptional pathways that are involved in the transcription of fetal genes55. Phosphorylated PLB could activate SERCA2a54, which is responsible for calcium transport to the sarcoplasmic reticulum to preserve cardiomyocyte calcium homeostasis56. Thus, by reversing the decrease in PLB phosphorylation by PE or ISO treatment, C33(S) might enhance the activity of SERCA2a to maintain calcium homeostasis, ultimately preventing the activation of the fetal gene network. It has been reported that SERCA2a could be downregulated by factors that induce the expression of fetal genes in cardiomyocytes34 and that SERCA2a mRNA levels were reduced during human hypertrophic cardiomyopathy35. Therefore, it is very likely that the upregulation of SERCA2a results from improved hypertrophic gene regulation by C33(S).
In conclusion, the novel PDE9A inhibitor C33(S) is a promising therapeutic agent for the treatment of cardiac diseases such as cardiac hypertrophy and heart failure. The cardioprotective mechanisms of C33(S) are associated with an increase in cGMP levels and the activation of downstream PLB, as well as the upregulation of SERCA2a.
Author contribution
Shao-rui CHEN and Pei-qing LIU designed the study; Pan-xia WANG, Si-dong CAI, Jing-yan LI and Guo-shuai FENG conducted the experiments; Hai-bin LUO provided C33(S); Pan-xia WANG, Ping HE and Yi HUANG analyzed the data; and Pan-xia WANG and Zhuo-ming LI wrote the manuscript.
References
Liu R, Molkentin JD . Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs). J Mol Cell Cardiol 2016.
Cotecchia S, Del Vescovo CD, Colella M, Caso S, Diviani D . The alpha1-adrenergic receptors in cardiac hypertrophy: signaling mechanisms and functional implications. Cell Signal 2015; 27: 1984–93.
Takano H, Zou Y, Akazawa H, Toko H, Mizukami M, Hasegawa H, et al. Inhibitory molecules in signal transduction pathways of cardiac hypertrophy. Hypertens Res 2002; 25: 491–8.
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP . Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322: 1561–6.
Francis SH, Blount MA, Corbin JD . Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 2011; 91: 651–90.
Inserte J, Barba I, Poncelas-Nozal M, Hernando V, Agullo L, Ruiz-Meana M, et al. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J Mol Cell Cardiol 2011; 50: 903–9.
Inserte J, Hernando V, Vilardosa U, Abad E, Poncelas-Nozal M, Garcia-Dorado D . Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc 2013; 2: e005975.
Wang Y, Li ZC, Zhang P, Poon E, Kong CW, Boheler KR, et al. Nitric oxide-cGMP-PKG pathway acts on Orai1 to inhibit the hypertrophy of human embryonic stem cell-derived cardiomyocytes. Stem Cells 2015; 33: 2973–84.
Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, et al. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A 2002; 99: 11363–8.
Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF . Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic cons-triction on mouse hearts. J Biol Chem 2003; 278: 47694–9.
Ritchie RH, Irvine JC, Rosenkranz AC, Patel R, Wendt IR, Horowitz JD, et al. Exploiting cGMP-based therapies for the prevention of left ventricular hypertrophy: NO* and beyond. Pharmacol Ther 2009; 124: 279–300.
Zhang M, Koitabashi N, Nagayama T, Rambaran R, Feng N, Takimoto E, et al. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signalling 2008; 20: 2231–6.
Tsai EJ, Kass DA . Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther 2009; 122: 216–38.
Bender AT, Beavo JA . Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006; 58: 488–520.
Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB . Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem 1998; 273: 15559–64.
Verhoest PR, Fonseca KR, Hou X, Proulx-Lafrance C, Corman M . Helal CJ, et al. Design and discovery of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (PF-04447943), a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J Med Chem 2012; 55: 9045–54.
Almeida CB, Traina F, Lanaro C, Canalli AA, Saad ST, Costa FF, et al. High expression of the cGMP-specific phosphodiesterase, PDE9A, in sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD neutrophils. Br J Haematol 2008; 142: 836–44.
Hanna CB, Yao S, Wu X, Jensen JT . Identification of phosphodiesterase 9A as a cyclic guanosine monophosphate-specific phosphodiesterase in germinal vesicle oocytes: a proposed role in the resumption of meiosis. Fertility Sterility 2012; 98: 487–95.e1.
Omar B, Banke E, Ekelund M, Frederiksen S, Degerman E . Alterations in cyclic nucleotide phosphodiesterase activities in omental and subcutaneous adipose tissues in human obesity. Nutr Diabetes 2011; 1: e13.
Deninno MP, Andrews M, Bell AS, Chen Y, Eller-Zarbo C, Eshelby N, et al. The discovery of potent, selective, and orally bioavailable PDE9 inhibitors as potential hypoglycemic agents. Bioorg Med Chem Lett 2009; 19: 2537–41.
Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015; 519: 472–6.
Huang M, Shao Y, Hou J, Cui W, Liang B, Huang Y, et al. Structural asymmetry of phosphodiesterase-9A and a unique pocket for selective binding of a potent enantiomeric inhibitor. Mol Pharmacol 2015; 88: 836–45.
Wang J, Liu Z, Feng X, Gao S, Xu S, Liu P . Tumor suppressor gene ING3 induces cardiomyocyte hypertrophy via inhibition of AMPK and activation of p38 MAPK signaling. Arch Biochem Biophys 2014; 562: 22–30.
Yu SS, Cai Y, Ye JT, Pi RB, Chen SR, Liu PQ, et al. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. Br J Pharmacol 2013; 168: 117–28.
Li P, Luo S, Pan C, Cheng X . Modulation of fatty acid metabolism is involved in the alleviation of isoproterenol-induced rat heart failure by fenofibrate. Mol Med Reports 2015; 12: 7899–906.
Liu M, Li Z, Chen GW, Li ZM, Wang LP, Ye JT, et al. AG-690/11026014, a novel PARP-1 inhibitor, protects cardiomyocytes from Ang II-induced hypertrophy. Mol Cell Endocrinol 2014; 392: 14–22.
Feng XJ, Gao H, Gao S, Li Z, Li H, Lu J, et al. The orphan receptor NOR1 participates in isoprenaline-induced cardiac hypertrophy by regulating PARP-1. Br J Pharmacol 2015; 172: 2852–63.
Perera RK, Sprenger JU, Steinbrecher JH, Hubscher D, Lehnart SE, Abesser M, et al. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ Res 2015; 116: 1304–11.
Patrucco E, Domes K, Sbroggio M, Blaich A, Schlossmann J, Desch M, et al. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci U S A 2014; 111: 12925–9.
Mokni W, Keravis T, Etienne-Selloum N, Walter A, Kane MO . Schini-Kerth VB, et al. Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PLoS One 2010; 5: e14227.
Frantz S, Klaiber M, Baba HA, Oberwinkler H, Volker K, Gabetaner B, et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I. Eur Heart J 2013; 34: 1233–44.
Gorbe A, Giricz Z, Szunyog A, Csont T, Burley DS, Baxter GF, et al. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res Cardiol 2010; 105: 643–50.
Pena JR, Szkudlarek AC, Warren CM, Heinrich LS, Gaffin RD, Jagatheesan G, et al. Neonatal gene transfer of SERCA2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 2010; 49: 993–1002.
Satoh N, Suter TM, Liao R, Colucci WS . Chronic alpha-adrenergic receptor stimulation modulates the contractile phenotype of cardiac myocytes in vitro. Circulation 2000; 102: 2249–54.
Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, et al. Genotype-dependent and -independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation 2016; 134: 1738–48.
Dong J, Gao C, Liu J, Cao Y, Tian L . TSH inhibits SERCA2a and the PKA/PLN pathway in rat cardiomyocytes. Oncotarget 2016; 7: 39207–15.
Kim JH, Trilk JL, Smith R, Asif I, Maddux PT, Ko YA, et al. Cardiac structure and function in elite para-cyclists with spinal cord injury. Med Sci Sports Exerc 2016; 48: 1431–7.
Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309: 1268–77.
Watanabe H, Iwanaga Y, Miyaji Y, Yamamoto H, Miyazaki S . Renal denervation mitigates cardiac remodeling and renal damage in Dahl rats: a comparison with beta-receptor blockade. Hypertens Res 2016; 39: 217–26.
Emdin M, Fatini C, Mirizzi G, Poletti R, Borrelli C, Prontera C, et al. Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: how useful, how feasible? Clin Chim Acta 2015; 443: 85–93.
Zhang Y, Yan J, Chen K, Song Y, Lu Z, Chen M, et al. Different roles of alpha1-adrenoceptor subtypes in mediating cardiomyocyte protein synthesis in neonatal rats. Clin Exp Pharmacol Physiol 2004; 31: 626–33.
Wang JJ, Rau C, Avetisyan R, Ren S, Romay MC, Stolin G . Genetic dissection of cardiac remodeling in an isoproterenol-induced heart failure mouse model. PLoS Genet 2016; 12: e1006038.
Zhang X, Cheng HJ, Zhou P, Kitzman DW, Ferrario CM, Li WM, et al. Cellular basis of angiotensin-(1-7)-induced augmentation of left ventricular functional performance in heart failure. Int J Cardiol 2017; 236: 405–12.
Li H, Lu ZZ, Chen C, Song Y, Xiao H, Zhang YY . Echocardiographic assessment of beta-adrenoceptor stimulation-induced heart failure with reduced heart rate in mice. Clin Exp Pharmacol Physiol 2014; 41: 58–66.
Badenhorst D, Veliotes D, Maseko M, Tsotetsi OJ, Brooksbank R, Naidoo A, et al. Beta-adrenergic activation initiates chamber dilatation in concentric hypertrophy. Hypertension 2003; 41: 499–504.
Perlini S, Chung ES, Aurigemma GP, Meyer TE . Alterations in early filling dynamics predict the progression of compensated pressure overload hypertrophy to heart failure better than abnormalities in midwall systolic shortening. Clin Exp Hypertens 2013; 35: 401–11.
Cerra MC, Pellegrino D . Cardiovascular cGMP-generating systems in physiological and pathological conditions. Curr Med Chem 2007; 14: 585–99.
Kuo IY, Kwaczala AT, Nguyen L, Russell KS, Campbell SG, Ehrlich BE . Decreased polycystin 2 expression alters calcium-contraction coupling and changes beta-adrenergic signaling pathways. Proc Natl Acad Sci U S A 2014; 111: 16604–9.
Inserte J, Hernando V, Ruiz-Meana M, Poncelas-Nozal M, Fernandez C, Agullo L, et al. Delayed phospholamban phosphorylation in post-conditioned heart favours Ca2+ normalization and contributes to protection. Cardiovasc Res 2014; 103: 542–53.
Huang B, Wang S, Qin D, Boutjdir M, El-Sherif N . Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of beta-adrenergic pathway, G(i) protein, phosphodiesterase, and phosphatases. Circ Res 1999; 85: 848–55.
Moon MR, Aziz A, Lee AM, Moon CJ, Okada S, Kanter EM, et al. Differential calcium handling in two canine models of right ventricular pressure overload. J Surg Res 2012; 178: 554–62.
Sande JB, Sjaastad I, Hoen IB, Bokenes J, Tonnessen T, Holt E, et al. Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc Res 2002; 53: 382–91.
Jeong MY, Walker JS, Brown RD, Moore RL, Vinson CS, Colucci WS, et al. AFos inhibits phenylephrine-mediated contractile dysfunction by altering phospholamban phosphorylation. Am J Physiol Heart Circ Physiol 2010; 298: H1719–26.
Colyer J . Phosphorylation states of phospholamban. Ann N Y Acad Sci 1998; 853: 79–91.
Zarain-Herzberg A, Fragoso-Medina J, Estrada-Aviles R . Calcium-regulated transcriptional pathways in the normal and pathologic heart. IUBMB life 2011; 63: 847–55.
Zarain-Herzberg A, Estrada-Aviles R, Fragoso-Medina J . Regulation of sarco(endo)plasmic reticulum Ca2+-ATPase and calsequestrin gene expression in the heart. Can J Physiol Pharmacol 2012; 90: 1017–28.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No 81473205 and 81673433 to Dr Pei-qing LIU), the Major Project of Guangdong Provincial Department of Science and Technology (No 2013B090700010 to Dr Pei-qing LIU), the Major Project of Platform Construction Education Department of Guangdong Province (No 2014GKPT002 to Dr Pei-qing LIU), the Guangdong Province Science and Technology Foundation (No 2015B020232009 to Dr Pei-qing LIU), the Guangdong Province Science and Technology Plan project-Public Research and Capacity Construction (2015 (No 3), to Dr Pei-qing LIU), the Guangzhou Science and Technology Projects (No 201604020121 and 201509030006); and the Medical Scientific Research Foundation of Guangdong Province (No A2015220 to Dr Shao-rui CHEN); as well as funding from the Guangdong Provincial Engineering Laboratory of Druggability and New Drugs Evaluation and the Guangzhou Key Laboratory of Druggability Assessment for Biologically Active compounds.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information are available on the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Table S1
Primers used for RT-qPCR (DOC 37 kb)
Supplementary Table S2
Sequences of siRNAs for PDE9A (DOC 31 kb)
Supplementary Table S3
Echocardiographic parameters of rats subjected to abdominal aortic constriction. (DOC 36 kb)
Supplementary Figure S1
Cardiomyocytes were treated with PE or ISO for 24 hours, then ANF and BNP protein levels were measured (A and B). (DOC 2860 kb)
Supplementary Figure S2
AAC induced cardiac hypertrophy. (DOC 1233 kb)
Supplementary Figure S3
Cell viability was measured by MTT assay, and data were presented as percentage of the control (A and B). (DOC 1692 kb)
Supplementary Figure S4
The phosphorylation and total level of PLB and the protein level of SERCA2a were measured in AAC-induced cardiac hypertrophic model. (DOC 265 kb)
Rights and permissions
About this article

Cite this article
Wang, Px., Li, Zm., Cai, Sd. et al. C33(S), a novel PDE9A inhibitor, protects against rat cardiac hypertrophy through upregulating cGMP signaling. Acta Pharmacol Sin 38, 1257–1268 (2017). https://doi.org/10.1038/aps.2017.38
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.38
Keywords
This article is cited by
-
Renal and cardiac effects of the PDE9 inhibitor BAY 73–6691 in 5/6 nephrectomized rats
Pflügers Archiv - European Journal of Physiology (2024)
-
Epigenome-wide association study on asthma and chronic obstructive pulmonary disease overlap reveals aberrant DNA methylations related to clinical phenotypes
Scientific Reports (2021)
