
Abstract
Aim:
To investigate the effects and underlying mechanisms of plumbagin, a naphthoquinone derived from medicinal plant Plumbago zeylanica, on human gastric cancer (GC) cells.
Methods:
Human gastric cancer cell lines SGC-7901, MKN-28, and AGS were used. The cell viability was examined using CCK-8 viability assay. Cell proliferation rate was determined using both clonogenic assay and EdU incorporation assay. Apoptosis was detected via Annexin V/propidium iodide double-labeled flow cytometry. Western blotting was used to assess the expression of both NF-κB-regulated gene products and TNF-α-induced activation of p65, IκBα, and IKK. The intracellular location of NF-κB p65 was detected using confocal microscopy.
Results:
Plumbagin (2.5–40 μmol/L) concentration-dependently reduced the viability of the GC cells. The IC50 value of plumbagin in SGC-7901, MKN-28, and AGS cells was 19.12, 13.64, and 10.12 μmol/L, respectively. The compound (5–20 μmol/L) concentration-dependently induced apoptosis of SGC-7901 cells, and potentiated the sensitivity of SGC-7901 cells to chemotherapeutic agents TNF-αand cisplatin. The compound (10 μmol/L) downregulated the expression of NF-κB-regulated gene products, including IAP1, XIAP, Bcl-2, Bcl-xL, tumor factor (TF), and VEGF. In addition to inhibition of NF-κB p65 nuclear translocation, the compound also suppressed TNF-α-induced phosphorylation of p65 and IKK, and the degradation of IκBα.
Conclusion:
Plumbagin inhibits cell growth and potentiates apoptosis in human GC cells through the NF-κB pathway.
Similar content being viewed by others

Introduction
Gastrointestinal cancers comprise a large percentage of malignancies and cancer-related deaths, among which gastric and colorectal cancers are most common1. Approximately 700 000 annual deaths make gastric cancer (GC) the second leading cause of mortality in the world2. Nearly two-thirds of gastric cancer mortality occurs in developing countries. In China, the death rate is 42%2. Despite the rapid progress in surgical techniques and chemotherapies, prognosis for patients with GC is usually poor. Therefore, it is urgent to find an effective therapy against this malignant disease.
Recently, dietary components and phytochemicals were shown to be effective in treating various human diseases3, 4. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) is a quinonoid constituent derived from the roots of the medicinal plant Plumbago zeylanica. This plant has been safely used for centuries in traditional ayurvedic and Chinese medicine5. Plumbagin has been reported to exhibit anti-microbial properties6, anti-atherosclerotic effects7, and anticancer activities both in vitro and in vivo5. Previous findings suggest that plumbagin can inhibit cell proliferation and induce apoptosis in breast carcinomas8, suppress promotion and metastasis in prostate cancer5, and induce cell cycle arrest and apoptosis in melanoma cells9. The results of these studies have shown that plumbagin has enormous potential as an anticarcinogenic agent. However, the effectiveness of plumbagin in the treatment of GC, as well as its mechanism of action, has not been investigated.
Although it is clear from previous studies that plumbagin acts as an anticancer agent by inducing apoptosis and halting cell proliferation, how plumbagin mediates these effects is not fully understood. Evidence in the current literature suggests that plumbagin can suppress the activation of nuclear factor-κB (NF-κB) in A549 human lung cancer cells10. Plumbagin can also modulate the activation of p65 and IκBα kinase, leading to the activation of apoptosis in several cancer cell lines11. Plumbagin may also inhibit the expression of NF-κB-regulated gene products, such as B-cell lymphoma-2 (Bcl-2)12, Bcl-xL12, and vascular endothelial growth factor (VEGF)11. Therefore, it is possible that plumbagin exerts its anticarcinogenic action by regulating the NF-κB pathway.
NF-κB is a ubiquitous and evolutionarily conserved transcription factor that is activated in a wide variety of tumors13 and plays a pivotal role in tumorigenesis13, 14. The NF-κB proteins, and other proteins associated with the NF-κB pathway, have been linked to cellular transformation, proliferation, apoptosis suppression, and angiogenesis14. In GC cells, NF-κB was shown to be constitutively activated15, and the activation of NF-κB has been touted as a prognostic parameter in gastric carcinoma16. Thus, agents that suppress NF-κB activation have great potential to be effective in the prevention and treatment of GC. Two NF-κB-suppressing compounds include paeoniflorin17 and wogonin18, which were shown to cause GC cell apoptosis by regulating the activation of NF-κB.
Because plumbagin has been reported to inhibit the activation of NF-κB, and NF-κB plays a pivotal role in gastric carcinoma, we hypothesized that plumbagin may exert its anticarcinogenic effects in human GC cells by blocking the NF-κB pathway.
Materials and methods
Materials
Plumbagin, dimethyl sulfoxide (DMSO), and propidium iodide (PI) were purchased from Sigma-Aldrich (St Louis, MO, USA). Plumbagin was dissolved in DMSO to a concentration of 200 mmol/L and stored in a dark-colored bottle at -20 °C. This stock solution was further diluted in cell culture medium immediately before use. Tumor necrosis factor-α (TNF-α) was purchased from Pepro Tech Inc (Rocky Hill, NJ, USA). The antibodies against Bcl-2, VEGF, tumor factor (TF), β-actin, and Cy3-labeled goat anti-rabbit IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The IAP1 antibody was provided by Epitomic, Inc (Burlingame, CA, USA). The Anti-Bcl-xL, anti-XIAP, and anti-histone H2B antibodies were purchased from Bioworld Technology (Minneapolis, MN, USA). The antibodies against IκB kinase α (IKKα), NF-κB p65, phospho-p65 (Ser 536), IκBα, phospho-IκBα (Ser 32), goat anti-mouse-HRP conjugate, and goat anti-rabbit-HRP conjugate were purchased from Cell Signaling Technology (Beverly, MA, USA). The penicillin, streptomycin, RPMI-1640 medium, and fetal bovine serum (FBS) were obtained from GIBCO BRL Life Technologies (Grand Island, NY, USA).
Cell culture
Human gastric cancer cell lines, SGC-7901, MKN-28, and AGS, were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C.
Cell viability and proliferation assay
Cell viability was assessed using the Cell Counting Kit-8 assay (CCK-8, Dojindo Laboratories, Kumamoto, Japan)19. Briefly, GC cells were grown in 96-well plates (10 000 cells/well) overnight without treatment. The cells were then incubated with varying concentrations of plumbagin and collected at several different time points. At the time of collection, 10 μL of kit reagent, WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfonyl)-2H-tetrazolium], was added to each well and incubated for 4 h at 37 °C. Thereafter, the optical density (OD) was measured at 450 nm using a 96-well multiscanner autoreader (Thermo Electron Corp, Waltham, MA, USA).
The effect of plumbagin on cell proliferation was determined using both a clonogenic assay and an EdU (5-ethynyl-2′-deoxyuridine) incorporation assay. Briefly, SGC-7901 cells were cultured in 30 mm dishes and treated with plumbagin at various concentrations (0, 5, 10, and 20 μmol/L) for 6 h. After treatment, the medium was removed and replaced with fresh medium. Cells were grown for 10 d at 37 °C to allow for colony formation. Subsequently, the cells were washed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde for 15 min at room temperature and stained with 0.5% crystal violet. The images were collected, and the number of colonies in each well were counted.
Proliferating cells were stained with EdU using the Cell-Light EdU DNA Cell Proliferation Kit (RIBOBio Co, Guangzhou, China)20. The cells were seeded in 96-well culture plates and exposed to media with or without plumbagin. All cells were treated with 50 μmol/L of EdU for 4 h at 37 °C. After being fixed with 4% paraformaldehyde for 15 min, the cells were treated with 0.5% Triton X-100 for 20 min and rinsed with PBS three times. Thereafter, the cells were exposed to 100 μL of 1×Apollo® reaction cocktail for 30 min and incubated with 5 μg/mL of Hoechst 33342 to stain the cell nuclei for 30 min. Images were captured using a fluorescent microscope (Olympus, Tokyo, Japan).
Apoptotic cell detection
An Annexin-V-FITC kit (Bender Medsystems, Burlingame, CA, USA) was used according to the manufacturer's instructions. Briefly, the plumbagin-treated GC cells were resuspended in 500 μL binding buffer, followed by staining with an Annexin-V-FITC and PI solution for 30 min at room temperature in the dark. The samples were analyzed immediately using the FACSCalibur flow cytometer (Becton, Dickinson and Co, San Jose, CA, USA).
Western blot analysis
Cells seeded in 10 cm dishes were treated with 10 μmol/L or 5 μmol/L of plumbagin. Total cell extracts were prepared using M-PER mammalian protein extraction reagent and protease inhibitors (Pierce, Rorkford, IL, USA). The cytoplasmic and nuclear extracts were prepared using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Jiangsu, China). Protein concentrations were determined using the BCA protein assay (Pierce, Rorkford, IL, USA). Equivalent amounts of protein (30 μg) were loaded per lane, resolved by SDS-polyacrylamide gel electrophoresis (8%–12%), and transferred to PVDF membranes. After being blocked with a 5% skim milk solution for 1 h, the membranes were incubated with their respective primary antibodies overnight at 4 °C, followed by secondary antibody incubations for 1 h at room temperature. The proteins were visualized using an enhanced chemiluminescence system (ECL, Beyotime Institute of Biotechnology, Jiangsu, China).
Immunofluorescence for NF-κB p65 localization
The effect of plumbagin on the nuclear translocation of p65 was examined by immunofluorescence21. Briefly, GC cells were washed in PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. The fixed cells were permeabilized with 0.5% Triton-X 100 in PBS for 10 min and blocked with a 5% bovine serum albumin in PBS. The anti-NF-κB p65 antibody was diluted 1:100 and incubated overnight at 4 °C. The cells were then incubated with Cy3-labeled secondary antibodies for 1 h and mounted with Hoechst 33342 stain. Images were captured using an A1Si confocal laser-scanning microscope (Nikon, Japan).
Statistical analysis
The data are presented as the mean±SD. The Student's t-test was used to determine the significance between groups. P values of less than 0.05 were considered to be significant. All statistical analyses were performed using the SPSS (Statistical Package for the Social Sciences) 13.0 software.
Results
Plumbagin decreased viability and inhibited the proliferation of GC cells
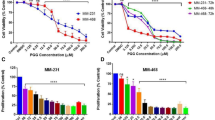
Cell viability was assayed by treating GC cell lines, including SGC-7901, MKN-28, and AGS cells, with various concentrations of plumbagin followed by analysis using the CCK-8 viability assay. We observed that cellular viability was suppressed by plumbagin in a dose-dependent manner in all three of the GC cell lines (Figure 1A). The IC50 values of plumbagin in SGC-7901, MKN-28, and AGS cells were 19.12 μmol/L, 13.64 μmol/L, and 10.12 μmol/L, respectively.

Plumbagin decreased viability and inhibited proliferation of GC cells. (A) Cell viability in plumbagin-treated SGC-7901, MKN-28, and AGS cells. The cells were treated with indicated concentrations (0–40 μmol/L) of plumbagin for 24 h. (B–C) Plumbagin inhibited the cellular DNA replication in SGC-7901 cells (magnification×200). Cells were incubated with 5 μmol/L plumbagin for 12 h. The EdU-labeled replicating cells were examined under a fluorescent microscope. The red and blue cells were counted in a blind manner. (D–E) Plumbagin decreased the number of colony-forming cells. Cells were treated with indicated concentrations of plumbagin (0, 5, 10, and 20 μmol/L) for 6 h, and then the medium was replaced by fresh medium. Cells were allowed to grow for 10 d. The formed cell clones were counted in a blind manner. The data shown are the mean from three independent experiments.
The EdU incorporation assay was performed to detect whether plumbagin could affect the number of proliferating cells. We determined that the number of EdU-positive cells in the plumbagin group was reduced compared to the control group. This indicated that plumbagin inhibited the proliferation of SGC-7901 cells in vitro (Figures 1B and 1C).
To determine the effect of the long-term antiproliferative activity of plumbagin, we used clonogenic assays. The clonogenicity of SGC-7901 cells in the plumbagin groups was reduced in a concentration-dependent manner (Figure 1D). We observed an inhibition of more than 30% for colony formation (Figure 1E).
Plumbagin enhanced the cell apoptosis of GC cells
The amount of apoptotic cell death was quantified with Annexin V-FITC/PI double-labeled flow cytometry. The SGC-7901 cells were pretreated with varying concentrations of plumbagin. This led to an increase in the amount of apoptosis in this cell line (Figure 2A). The total apoptosis rates were 1.77%±0.31%, 8.00%±1.67%, 30.57%±1.25%, and 35.33%±1.31% at plumbagin concentrations of 0 μmol/L, 5 μmol/L, 10 μmol/L, and 20 μmol/L of plumbagin, respectively.

Plumbagin enhanced cell apoptosis of GC cells. (A) Plumbagin induced apoptosis of SGC-7901 cells. Cells were incubated with 0, 5, 10, and 20 μmol/L plumbagin for 12 h. The apoptosis was analyzed by Annexin V-FITC/PI double-staining assay. (B) The degree of apoptotic cell death was quantified. Data represented the mean±SD of three individual experiments (bP<0.05).
Plumbagin suppressed the expression of NF-κB-regulated gene products
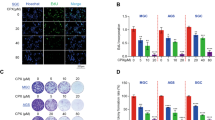
NF-κB is known to regulate the expression of IAP1, XIAP, Bcl-2, and Bcl-xL, all of which are associated with cancer cell survival22, 23, 24. To investigate whether plumbagin inhibits the expression of these proteins, whole-cell protein extracts were prepared and analyzed by Western blotting with the specific antibodies. Plumbagin decreased the expression of these proteins in a time-dependent manner (Figure 3A).

Plumbagin suppressed the expression of NF-κB-regulated gene products. (A) Plumbagin decreased the expression of NF-κB-regulated anti-apoptotic proteins. (B) Plumbagin suppressed the expression of VEGF and TF. SGC-7901 cells were incubated with 10 μmol/L plumbagin for different time periods (0, 2, 4, 8, 12, and 24 h). Whole-cell extracts were prepared and measured by Western blot analysis using the relevant antibodies.
We also determined the effect of plumbagin on the NF-κB-dependent gene products that are involved in angiogenesis and metastasis. We found that plumbagin downregulated the expression of both VEGF and TF (Figure 3B).
Plumbagin inhibited TNF-α-induced phosphorylation and nuclear translocation of NF-κB p65
We investigated the effect of plumbagin on p65 nuclear translocation and its phosphorylation status. In general, p65 is located in the cytoplasm in untreated cells, and TNF-α-induced p65 is detected in the nuclei. In cells pretreated with plumbagin, the TNF-α-induced nuclear translocation of p65 was almost completely suppressed (Figure 4A).

Plumbagin inhibited TNF-α-induced phosphorylation and nuclear translocation of NF-κB p65. (A) Plumbagin inhibited TNF-α-induced p65 localization by immunofluorescence analysis. SGC-7901 cells were either pretreated or untreated with 5 μmol/L plumbagin for 4 h and then exposed to 0.1 nmol/L TNF-α for 30 min (magnification×400). (B) Plumbagin inhibited TNF-α-induced phosphorylation and nuclear translocation of p65. SGC-7901 cells were first treated or untreated with 5 μmol/L plumbagin for 4 h and then exposed to 0.1 nmol/L TNF-α for indicated times. Nuclear extracts (NE) and cytoplasmic extracts (CE) were prepared and analyzed by Western blot with antibodies against p65 and phospho-p65. The antibodies of anti-β-actin and anti-histone H2B were used as controls.
Modifications of p65, such as phosphorylation, play an important role in NF-κB transcriptional activity25. Therefore, we examined the effect of plumbagin on the phosphorylation and expression of p65 in both nuclear extracts (NE) and cytoplasmic extracts (CE) by Western blot. In the nuclear protein extracts from the TNF-α-treated cells, the accumulation of both total and phosphorylated p65 increased in a time-dependent manner. Concurrently, the expression of p65 in cytoplasmic extracts decreased gradually upon TNF-α stimulation. The p65 nuclear translocation and phosphorylation induced by TNF-α were markedly suppressed in the plumbagin-pretreated GC cells (Figure 4B).
Plumbagin inhibited both TNF-α-induced degradation of IκBα and the phosphorylation of IκBα and IKKα
It is well known that after the phosphorylation, ubiquitination, and proteolytic degradation of IκBα, the p50-p65 heterodimer is released and translocates to the nucleus13. To determine whether plumbagin inhibited p65 nuclear translocation was due to the degradation and phosphorylation of IκBα, we exposed the cells (pretreated with or without plumbagin) to TNF-α for varying amounts of time. IκBα was degraded in the control cells; however, in the plumbagin pretreated cells, TNF-α failed to induce the degradation of IκBα. Furthermore, TNF-α increased IκBα phosphorylation in the control cells but had no effect on the plumbagin-pretreated cells (Figure 5). These results indicated that plumbagin suppressed both TNF-α-induced IκBα degradation and the activation of IκBα, which occurs prior to p65 nuclear translocation.

Plumbagin inhibited TNF-α-induced degradation of IκBα, phosphorylation of IκBα and IKKα. Plumbagin inhibited TNF-α induced IκBα degradation, phosphorylation of IκBα and IKKα. SGC-7901 cells were first exposed to 5 μmol/L plumbagin for 4 h and then treated with 0.1 nmol/L TNF-α for the indicated times and analyzed by Western blotting using various antibodies.
The phosphorylation of IκBα required activation of IKK. To examine the effect of plumbagin on IKKα activation, we investigated whether plumbagin could influence TNF-α-induced IKKα phosphorylation. Indeed, plumbagin suppressed TNF-α-dependent IKKα phosphorylation. Both the TNF-α and plumbagin treatments had a weak effect on IKKα protein expression (Figure 5).
Plumbagin potentiated apoptosis induced by TNF-α as well as cisplatin
Because TNF-α and the chemotherapeutic agent cisplatin are two anticancer agents that are commonly used in the clinic, we investigated whether plumbagin affects TNF-α- and/or cisplatin-induced apoptosis. The results displayed an increase in the cytotoxic effect induced by either TNF-α or cisplatin in the presence of plumbagin. This suggests that plumbagin potentiated the apoptotic effects of both TNF-α and cisplatin (Figure 6).

Plumbagin potentiated apoptotic effects of TNF-α and cisplatin. (A–B) SGC-7901 cells were pretreated with 10 μmol/L plumbagin for 2 h, and then treated with 0.1 nmol/L TNF-α and cisplatin for 24 h. Cell viability was then analyzed by the CCK-8 method. Data represented the mean±SD of three individual experiments (bP<0.05 compared to control group. eP<0.05 compared with TNF-α or cisplatin treatment only).
Discussion
The aim of this study was to investigate whether plumbagin has an anticancer effect on human GC cells via inhibition of the NF-κB pathway. We identified, for the first time, that GC cells treated with plumbagin underwent growth inhibition, apoptosis and increased chemosensitivity in a dose-dependent manner. We also observed that the protein expression levels of various anti-apoptotic proteins were downregulated by plumbagin in a time-dependent manner. Furthermore, we determined that plumbagin suppressed both NF-κB activation and the expression of NF-κB-regulated gene products. The effects of plumbagin on the NF-κB pathway largely explained how plumbagin increased the chemosensitivity of GC cells.
It is interesting to note that plumbagin caused apoptosis in human GC cell lines, including the well-differentiated MKN-28 cells, and the partially-differentiated SGC-7901 and AGS cell lines. The effective concentration of plumbagin necessary to induce GC cell death is comparable with several existing natural products that have been tested on human GC cells, such as wogonin and curcumin18, 26. The downregulated anti-apoptotic proteins that we observed upon treatment with plumbagin, including IAP1, XIAP, Bcl-2, and Bcl-xL, are the main reasons for increased apoptosis. Because Bcl-2 and Bcl-xL are key members in the mitochondrial apoptotic pathway, we conclude that mitochondria may be one of the pathways for apoptosis.
We determined that plumbagin was able to suppress the expression of proteins involved in anti-apoptosis (ie, IAP1, XIAP, Bcl-2, and Bcl-xL), angiogenesis (VEGF), and metastasis (TF)27, 28, all of which are linked to, and regulated by, NF-κB. Based on our findings, we hypothesized that plumbagin may have an effect on the NF-κB pathway in GC cells. Indeed, plumbagin blocked the nuclear translocation of NF-κB p65. Plumbagin also caused the downregulation of both the expression and phosphorylation of NF-κB p65 in nucleus induced by TNF-α.
Exposing cancer cells to TNF-α initiates numerous intracellular signaling cascades, most of which activate the IKK complex, which consists of IKKα, IKKβ, and IKKγ29. In the current study, plumbagin inhibited TNF-α-induced phosphorylation of IKKα. IκBα is phosphorylated at serine residues 32 and 34 by IKKs, and this phosphorylation triggers the degradation of IκBα, which allows for the release and translocation of p65 into the nucleus to activate the transcription of target genes29. We observed that plumbagin inhibited TNF-α-induced degradation of IκBα by suppressing its phosphorylation. Similar to our data, several studies have shown that some natural constituents, such as berberine, berbamine, and curcumin, can suppress the activation of NF-κB and its target genes and cause apoptosis in various cancer cells30, 31, 32. Whether plumbagin is a selective inhibitor of other transcription factors in GC cells remains to be elucidated.
Chemotherapy is widely used in the treatment of numerous malignancies and is a potent therapeutic method; however, chemoresistance remains a major obstacle in the chemotherapeutic treatment of cancer. Overexpression of transcription factors, such as NF-κB, contributes to enhanced cancer cell survival and chemoresistance33. Interestingly, we found that plumbagin potentiated apoptosis induced by TNF-α or cisplatin. Plumbagin, in combination with TNF-α or cisplatin, was more effective than the single agent alone. This might be explained by the fact that the apoptosis induced by TNF-α or cisplatin can be suppressed by the activation of NF-κB34, 35, which is inhibited by plumbagin. This again suggests that plumbagin could be a useful therapeutic strategy in human GC.
Given its strong anticancer abilities, the toxicity of plumbagin has been extensively evaluated. In rodents, plumbagin had dose-related toxic side effects, including diarrhea, skin rashes, and hepatic and reproductive toxicity11. Plumbagin did not exhibit significant toxicity on normal tissues at a dose of 2 mg·kg−1·d−1 9. Additional preclinical and prospective randomized clinical trials are required to determine the full potential of this agent.
In summary, our findings strongly indicate that plumbagin acts as an anticancer agent by inhibiting cell proliferation, inducing apoptosis, and potentiating the chemosensitivity of GC cells. These effects are, in part, mediated by suppressing both NF-κB activation and the expression of NF-κB-regulated gene products. Based on the evidence provided here, plumbagin should be strongly considered as a basis for the development of novel pharmaceutical agents that target human GC cells to improve the treatment of this disease.
Author contribution
Jing LI, You QIN, Rui CHEN, and Jia LI performed the experiments. Jing LI, Lin SHEN, and Fu-rong LU participated in the design of the study and prepared the manuscript. Yan LI, Han-zi ZHAN, and Yuan-qiao HE contributed to the analyses and interpretation of the data and performed the statistical analysis. Han-zi ZHAN revised the manuscript.
References
Qiao L, Wong BC . Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist Update 2009; 12: 55–64.
Parkin DM, Bray F, Ferlay J, Pisani P . Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108.
Surh YJ . Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 2003; 3: 768–80.
Lu FR, Shen L, Qin Y, Gao L, Li H, Dai Y . Clinical observation on trigonella foenum-graecum L total saponins in combinaiton with sulphanylureas in the treatment of type 2 diabetes mellitus. Chin J Integr Med 2008; 14: 56–60.
Aziz MH, Dreckschmidt NE, Verma AK . Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone — refractory prostate cancer. Cancer Res 2008; 68: 9024–32.
Mossa JS, El-Feraly FS, Muhammad I . Antimycobacterial constituents from Juniperus procera, Ferula communis and Plumbago zeylanica and their in vitro synergistic activity with isonicotinic acid hydrazide. Phytother Res 2004; 18: 934–37.
Ding YX, Chen ZJ, Liu S, Che D, Vetter M, Chang CH . Inhibition of Nox-4 activity by plumbagin, a plant-derived bioactive naphthoquinone. J Pharm Pharmacol 2005; 57: 111–6.
Kuo PL, Hsu YL, Cho CY . Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol Cancer Ther 2006; 5: 3209–21.
Wang CC, Chiang YM, Sung SC, Hsu YL, Chang JK, Kuo PL . Plumbagin induces cell cycle arrest and apoptosis through reactive oxygen species/c-Jun N-terminal kinase pathways in human melanoma A375.S2 cells. Cancer Lett 2008; 259: 82–98.
Shieh JM, Chiang TA, Chang WT, Chao CH, Lee YC, Huang GY, et al. Plumbagin inhibits TPA-induced MMP-2 and u-PA expressions by reducing binding activities of NF-kappaB and AP-1 via ERK signaling pathway in A549 human lung cancer cells. Mol Cell Biochem 2010; 335: 181–93.
Sandur SK, Ichikawa H, Sethi G, Ahn KS, Aggarwal BB . Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem 2006; 281: 17023–33.
Hsu YL, Cho CY, Kuo PL, Huang YT, Lin CC . Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induce apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase–mediated phosphorylation at serine 15 in vitro and in vivo. J Pharmacol Exp Ther 2006; 318: 484–94.
Aggarwal BB . Nuclear factor-kappaB: the enemy within. Cancer Cell 2004; 6: 203–8.
Gupta SC, Kim JH, Prasad S, Aggarwal BB . Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metast Rev 2010; 29: 405–34.
Li Q, Yu YY, Zhu ZG, Ji YB, Zhang Y, Liu BY, et al. Effect of NF-kappaB constitutive activation on proliferation and apoptosis of gastric cancer cell lines. Eur Surg Res 2005; 37: 105–10.
Yamanaka N, Sasaki N, Tasaki A, Nakashima H, Kubo M, Morisaki T, et al. Nuclear factor-kappaB p65 is a prognostic indicator in gastric carcinoma. Anticancer Res 2004; 24: 1071–5.
Wu H, Li W, Wang T, Shu Y, Liu P . Paeoniflorin suppress NF-kappaB activation through modulation of I kappaB alpha and enhances 5-fluorouracil-induced apoptosis in human gastric carcinoma cells. Biomed Pharmacother 2008; 62: 659–66.
Zhao Q, Wang J, Zou MJ, Hu R, Zhao L, Qiang L, et al. Wogonin potentiates the antitumor effects of low dose 5–fluorouracil against gastric cancer through induction of apoptosis by down-regulation of NF-kappaB and regulation of its metabolism. Toxicol Lett 2010; 197: 201–10.
Park HH, Lee KY, Kim SH, Lee YJ, Koh SH . L-DOPA-induced neurotoxicity is reduced by the activation of the PI3K signaling pathway. Toxicology 2009; 265: 80–6.
Lv L, Xiao XY, Gu ZH, Zeng FQ, Huang LQ, Jiang GS . Silencing USP22 by asymmetric structure of interfering RNA inhibits proliferation and induces cell cycle arrest in bladder cancer cells. Mol Cell Biochem 2011; 346: 11–21.
Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB . Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF–induced apoptosis through modulation of the nuclear factor–kappaB signaling pathway. Blood 2007; 110: 3517–25.
Li C, Yang Z, Zhai C, Qiu W, Li D, Yi Z, et al. Maslinic acid potentiates the anti–tumor activity of tumor necrosis factor alpha by inhibiting NF–kappaB signaling pathway. Mol Cancer 2010; 9: 73.
Olsen LS, Hjarnaa PJ, Latini S, Holm PK, Larsson R, Bramm E, et al. Anticancer agent CHS 828 suppresses nuclear factor-kappa B activity in cancer cells through downregulation of IKK activity. Int J Cancer 2004; 111: 198–205.
Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem 1999; 274: 8531–8.
Yang F, Tang E, Guan K, Wang CY . IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol 2003; 170: 5630–5.
Cai XZ, Wang J, Li XD, Wang GL, Liu FN, Cheng MS, et al. Curcumin suppresses proliferation and invasion in human gastric cancer cells by downregulation of PAK1 activity and cyclin D1 expression. Cancer Biol Ther 2009; 8: 1360–8.
Ma YY, He XJ, Wang HJ, Xia YJ, Wang SL, Ye ZY, et al. Interaction of coagulation factors and tumor–associated macrophages mediates migration and invasion of gastric cancer. Cancer Sci 2010; 102: 336–42.
Xie TX, Aldape KD, Gong W, Kanzawa T, Suki D, Kondo S, et al. Aberrant NF-kappaB activity is critical in focal necrosis formation of human glioblastoma by regulation of the expression of tissue factor. Int J Oncol 2008; 33: 5–15.
Ducut Sigala JL, Bottero V, Young DB, Shevchenko A, Mercurio F, Verma IM . Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science 2004; 304: 1963–7.
Pandey MK, Sung B, Kunnumakkara AB, Sethi G, Chaturvedi MM, Aggarwal BB . Berberine modifies cysteine 179 of IkappaBalpha kinase, suppresses nuclear factor-kappaB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res 2008; 68: 5370–9.
Liang Y, Xu RZ, Zhang L, Zhao XY . Berbamine, a novel nuclear factor kappaB inhibitor, inhibits growth and induces apoptosis in human myeloma cells. Acta Pharmacol Sin 2009; 12: 1659–65.
Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB . Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol Pharmacol 2006; 69: 195–206.
Dhandapani KM, Mahesh VB, Brann DW . Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFkappaB transcription factors. J Neurochem 2007; 102: 522–38.
Wang X, Chen W, Lin Y . Sensitization of TNF-induced cytotoxicity in lung cancer cells by concurrent suppression of the NF-kappaB and Akt pathways. Biochem Biophys Res Commun 2007; 355: 807–12.
Giri DK, Aggarwal BB . Constitutive activation of NF-kappaB causes resistance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells. Autocrine role of tumor necrosis factor and reactive oxygen intermediates. J Biol Chem 1998; 273: 14008–14.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No 81072945 and No 81001670). The authors are grateful to the Department of Central Laboratory, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China, for allowing us to use their experimental facilities and providing us their technical support.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, J., Shen, L., Lu, Fr. et al. Plumbagin inhibits cell growth and potentiates apoptosis in human gastric cancer cells in vitro through the NF-κB signaling pathway. Acta Pharmacol Sin 33, 242–249 (2012). https://doi.org/10.1038/aps.2011.152
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.152
Keywords
This article is cited by
-
KIF15 facilitates gastric cancer via enhancing proliferation, inhibiting apoptosis, and predict poor prognosis
Cancer Cell International (2020)
-
Interleukin-13 Gene Modification Enhances Grafted Mesenchymal Stem Cells Survival After Subretinal Transplantation
Cellular and Molecular Neurobiology (2020)
-
IFN-γ regulates human dental pulp stem cells behavior via NF-κB and MAPK signaling
Scientific Reports (2017)
-
A Comprehensive Review on Pharmacotherapeutics of Three Phytochemicals, Curcumin, Quercetin, and Allicin, in the Treatment of Gastric Cancer
Journal of Gastrointestinal Cancer (2017)
