
Abstract
Aim:
To elucidate how krüppel-like factor 4 (KLF4) activates mitofusin 2 (mfn-2) expression in all-trans retinoic acid (ATRA)-induced vascular smooth muscle cell (VSMC) differentiation.
Methods:
The mfn-2 promoter-reporter constructs and the KLF4 acetylation-deficient or phosphorylation-deficient mutants were constructed. Adenoviral vector of KLF4-mediated overexpression and Western blot analysis were used to determine the effect of KLF4 on mfn-2 expression. The luciferase assay and chromatin immunoprecipitation were used to detect the transactivation of KLF4 on mfn-2 gene expression. Co-immunoprecipitation and GST pull-down assays were used to determine the modification of KLF4 and interaction of KLF4 with p300 in VSMCs.
Results:
KLF4 mediated ATRA-induced mfn-2 expression in VSMCs. KLF4 bound directly to the mfn-2 promoter and activated its transcription. ATRA increased the interaction of KLF4 with p300 by inducing KLF4 phosphorylation via activation of JNK and p38 MAPK signaling. KLF4 acetylation by p300 increased its activity to transactivate the mfn-2 promoter.
Conclusion:
ATRA induces KLF4 acetylation by p300 and increases the ability of KLF4 to transactivate the mfn-2 promoter in VSMCs.
Similar content being viewed by others


Introduction
The Krüppel-like factor 4 (KLF4/GKLF) is a pleiotropic zinc finger transcription factor involved in both proliferation and differentiation in several tissues, as a tumor suppressor or an oncogene depending on the molecular context1, and also plays an essential role in reprogramming differentiated somatic cells into pluripotent stem cells2, 3. As a negative regulator of the cell cycle, KLF4 activates numerous genes that encode negative regulators of the cell cycle as well as suppresses expression of genes that promote cell cycle progression4, 5. In the gut, KLF4 activates the expression of intestinal alkaline phosphatase (IAP) and regulates gut epithelium differentiation6. In the eye, KLF4 activates Krt12 and Aqp 5 promoters and plays a critical role in postnatal ocular surface maturation and maintenance7. In vascular smooth muscle cells (VSMCs), KLF4 can function as an anti-proliferation factor or a prodifferentiation factor depending on the interaction partner8. We have recently demonstrated that all-trans retinoic acid (ATRA) induced the expression of KLF4 and mitofusin 2 (mfn-2), a member of the mitofusin family, in VSMCs9, 10. However, the role of KLF4 in the regulation of mfn-2 expression has not been well characterized.
Mfn-2 [also named hyperplasia suppressor gene (HSG)] is a mitochondrial fusion protein that is expressed mainly in tissues with high energetic requirements, such as skeletal muscle and heart. Several lines of evidence indicate that mfn-2 modulates mitochondrial metabolism by regulating mitochondrial membrane potential, fuel oxidation, and the oxidative phosphorylation system11, 12. Recent evidence also shows that mfn-2 inhibits tumor cell proliferation and increases their sensitivity to chemotherapy and radiotherapy even above p5313. In addition, recent study has shown that mfn-2 exhibits a profound anti-proliferative effect on VSMCs in vitro and in vivo via inhibition of the Ras-Raf-MEK-ERK1/2 MAPK signaling pathway14, and promotes VSMC and cardiomyocyte apoptosis via the mitochondrial death pathway15, 16. These observations suggest that mfn-2 is a multi-function protein with regulatory and structural nature. Thus, it is important to reveal the molecular mechanisms by which mfn-2 expression is regulated. It has been recently demonstrated that mfn-2 can be induced directly by PPARδ activated by its selective ligand (GW0742) in the heart17. However, little is known about how mfn-2's expression is regulated in VSMCs. Our recent studies demonstrate that ATRA induces KLF4 and mfn-2 expression in VSMCs9, 10. However, the actual relationship between KLF4 and mfn-2 in VSMC biology is not fully understood.
In this study, we investigate the action of KLF4 in the context of ATRA-induced mfn-2 expression and demonstrate that ATRA-induced mfn-2 expression is dependent on KLF4 in VSMCs.
Materials and methods
Cell culture and treatment
VSMCs were isolated from the thoracic aorta of 90–100 g male Sprague-Dawley rats as previously described18. VSMCs were maintained and passaged in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (HyClone). In all experiments, only cell passages 3–5 were used. All experiments were initiated when the cells reached 70% confluence, except if stated otherwise. Cultured VSMCs were growth-arrested by incubation in serum-free DMEM for 24 h and then stimulated by 10 μmol/L ATRA (Sigma-Aldrich) as indicated time. For the inhibitor studies, cells were pretreated with inhibitors SP600125 (Promega) or SB203580 (Promega) in DMSO for 2 h before the application of ATRA.
Western blot analysis
The cells were lysed with 150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.5, 1% Nonidet P-40, 0.5% sodium deoxycholic acid, and Complete Protease Inhibitor Cocktail Tablets (Roche), and then the protein was isolated as previously described19, 20. The protein content was determined with the Bio-Rad Bradford protein assay. Equal amounts of protein were separated by SDS-PAGE, transferred to a PVDF membrane (Millipore) and analyzed by Western blotting using standard protocols. The membranes were blocked with 5% milk in TTBS for 2 h at room temperature and incubated overnight at 4 °C using the following primary antibodies: 1:500 rabbit anti-mfn-2 (Santa Cruz), 1:500 rabbit anti-KLF4 (Santa Cruz), 1:500 mouse anti-acetylated lysine (anti-Ac-Lys) (Santa Cruz), 1:400 rabbit anti-p300 (Santa Cruz), 1:800 rabbit anti-phospho-p38 (Cell Signaling), 1:1000 rabbit anti-p38 (Cell Signaling), 1:800 rabbit anti-phospho-JNK (Cell Signaling), 1:500 rabbit anti-JNK (Cell Signaling), and 1:1000 mouse anti-β-actin (Santa Cruz). The membranes were then incubated for 1 h at room temperature with a 1:5000 dilution of anti-rabbit/horseradish peroxidase or anti-mouse/horseradish peroxidase (Santa Cruz) and developed with the Chemiluminescence Plus Western blot analysis kit (Santa Cruz).
RNA preparation and quantitative real-time RT-PCR
Total RNA from cultured VSMCs was isolated using Trizol reagent (Invitrogen) according to the manufacturer's protocol. RNA (2 μg) was reverse-transcribed using PrimeScriptTM RT reagent Kit (DRR037S, TaKaRa Bio, Japan), and the single stranded cDNA was amplified by quantitative real-time RT-PCR using SYBR® Premix Ex TaqTM Kit (DRR041S, TaKaRa Bio, Japan) on an ABI7300 Sequence Detection PCR System (Applied Biosystems, USA). The following primers were used: KLF4, 5′-AGAGGAGCCCAAGCCAAAGA-3′ (sense) and 5′-CAGTCACAGTGGTAAGGTTTCTC-3′ (antisense); mfn-2, 5′-CTCAGGAGCAGCGGGTTTATTGTCT-3′ (sense) and 5′-TGTCGAGGGACCAGCATGTCTATCT-3′ (antisense); GAPDH, 5′-GCTGAGTATGTCGTGGAGTCT-3′ (sense) and 5′-GTTCACACCCATCACAAACAT-3′ (antisense). The mRNA expression of the target gene was normalized to GAPDH.
Adenovirus expression vector and plasmids
The full-length rat KLF4 cDNA was cloned into the pAd/CMV/V5-DEST vector (Invitrogen) to create the KLF4 adenovirus pAd-KLF410. KLF4 small interfering RNA (KLF4-siRNA) was synthesized by Sigma, as previously described10. The resulting constructs were packaged in A293 cells (ATCC) by transfection with Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Culture supernatants from individual A293 cells were used to infect VSMCs. The cells were splited after 24 h infection and selected with 0.3 g/L G418 for 14 d. For reporter gene assay, various fragments of the 5' flanking region of the mfn-2 gene were amplified and subcloned into the pGL3-basic reporter gene vector (Promega). Expression plasmid of GFP-KLF4 has been previously described10. The plasmid encoding p300 was a gift from Dr WR HARRISON (Baylor College of Medicine). The plasmid encoding HDAC2 was a gift from Dr Y KIM (Sungkyunkwan University School of Medicine). p300-HAT gene (1195–1810) was amplified and subcloned into the pGEX-4T vector. The acetylation-deficient mutants of GST- and GFP- tagged KLF4 K 225 to R (KLF4 K225R), KLF4 K 229 to R (KLF4 K229R), and KLF4 K 225/229 to R (KLF4 K225/229R) and phosphorylation deficient mutants in the three zinc fingers of KLF4, GFP-KLF4 (S415A), GFP-KLF4 (S444A) and GFP-KLF4 (S470A), were constructed using the QuikChange site-directed mutagenesis kit (Stratagene), following the manufacturer's instructions. Each mutation was verified by DNA sequence analysis.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as described previously21, 22. In brief, VSMCs at 80% confluence were crosslinked with 1% formaldehyde for 10 min, lysed and sonicated 5–10 times each for 10 s at 4 °C to reduce the average DNA length to 0.4–0.5 kb. The samples were diluted 10-fold, and then pre-cleared with protein A-agarose/salmon sperm DNA for 30 min at 4 °C followed by an overnight incubation at 4 °C with 1:500 anti-KLF4 or 1:500 anti-mouse IgG (as a negative control). The immune complexes were precipitated with protein A-agarose for 1 h. The samples were heated at 65 °C for 4 h, treated with proteinase K, and the DNA was extracted with phenol/chloroform. PCR was performed with specific primers for the mfn-2 promoter region: 5′-GGCGCACGCAAACCTCTAATC-3′ and 5′-GCCTGGTAACCCGTGTCGTCC-3′.
Reporter gene assays
A293 cells were transfected with mfn-2 promoter-reporter constructs and expression plasmid for KLF4 or other plasmids using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. The cells were harvested after 24 h, and the activities of both firefly luciferase and Renilla luciferase were measured in the LB 955 Luminometer system using the Dual-Luciferase Reporter System (Promega), according to the manufacturer's recommendations. The activity of firefly luciferase was normalized to that of Renilla luciferase. A minimum of three independent transfections was performed for each experimental group.
Co-immunoprecipitation assay
Co-immunoprecipitation was performed as described previously23. Briefly, cell lysates were first pre-cleared with 20 μL of protein A-agarose (50% v/v). The supernatants were immunoprecipitated with 2 μg of anti-KLF4, anti-phosphoserine, anti-acetylated lysine, or anti-p300 antibodies for 1 h at 4 °C, followed by incubation with protein A-agarose overnight at 4 °C. Protein A-agarose-antigen-antibody complexes were collected by centrifugation at 12,000×g for 20 s at 4 °C. The pellets were washed four times with 500 μL IPH buffer (50 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% Nonidet P-40, 0.1 mmol/L PMSF) for 20 min each time at 4 °C. Bound proteins were resolved by SDS-PAGE, followed by Western blotting with the anti-p300 or anti-KLF4 antibodies. The experiments were replicated three times at least.
GST pull-down assays
Glutathione S-transferase (GST)-KLF4, GST-KLF4 (K225R), GST-KLF4 (K229R), and GST-KLF4 (K225/229R) fusion proteins were expressed in E coli and purified with GST beads (glutathione-agarose from Sigma)22. The cell lysates were incubated with equal amounts of GST beads containing GST-KLF4, GST-KLF4 mutants or GST proteins for 2 h at 4 °C in the following buffer: 20 mmol/L Tris-HCl, pH 7.5, 300 mmol/L NaCl, 1 mmol/L dithiothreitol, 0.4% Nonidet P-40, 0.5 g/L bovine serum albumin (BSA), and 1 mmol/L PMSF. Protein complexes were washed three times with the above-described buffer and then once with the same buffer containing 150 mmol/L NaCl but lacking BSA. After washing, bound proteins were resolved by SDS-PAGE and detected by Western blotting with the anti-p300 or anti-GST antibodies.
In vitro acetylation reactions
Purified GST-KLF4 or GST-KLF4 mutants (300 ng) were incubated with GST-p300-HAT (100 ng) (1195–1810) in 30–100 μL HAT buffer (50 mmol/L Tris-HCl, pH 8.0, 10% glycerol, 0.1 mmol/L EDTA, 1 mmol/L dithiothreitol, 5 μmol/L trichostatin A, and 20 μmol/L acetyl-CoA) at 30 °C for 30 min, and acetylation of KLF4 was analyzed by Western blotting using anti-Ac-lys antibody24, 25.
Statistical analysis
All experiments were performed in triplicate. Results were expressed as the means±SEM. Differences between two groups were assessed using analysis of variance followed by a Student's t-test.
Results
KLF4 up-regulates mfn-2 expression in VSMCs
Since ATRA is known to induce the expression of KLF4 and mfn-29, 10, we sought to determine whether KLF4 is responsible for mfn-2 expression in VSMCs treated by ATRA. We examined the effect of ATRA on the expression of KLF4 and mfn-2. ATRA induced the expression of mfn-2 and KLF4 in a time-dependent manner (Figure 1A). In response to ATRA, KLF4 and mfn-2 expression began to increase within 12 h, and increased by 1.7-fold and 2.8-fold at 24 h, respectively, reached peak at 48 h, and still stayed at a higher level until 72 h, which was consistent with their mRNA levels as measured by quantitative real-time RT-PCR (Figure 1B). To further determine whether ATRA-induced mfn-2 expression is due to the increased expression of KLF4, we transfected VSMCs with KLF4 expression vector (pAd-KLF4) or pAd (as a control). KLF4 expression dramatically increased in VSMCs infected with pAd-KLF4 (Figure 1C). Moreover, overexpression of KLF4 increased protein and mRNA levels of mfn-2 in a time-dependent manner, as shown by Western blotting and quantitative real-time RT-PCR, respectively (Figure 1D and 1E). In contrast, RNA interference was used to silence the expression of KLF4 and to assess its effects on ATRA-induced mfn-2 expression. Off-target activity of the rat-specific KLF4 siRNA was examined using a non-silencing control siRNA (NS-siRNA). As shown in Figure 1F, when VSMCs were transfected with rat KLF4-specific siRNA to block the endogenous KLF4 expression induced by ATRA, ATRA-elicited increase in mfn-2 expression was significantly reduced. The non-silencing control siRNA had no impact on the expression of mfn-2. These results suggest that KLF4 mediates mfn-2 expression induced by ATRA.

KLF4 up-regulates mfn-2 expression in VSMCs. (A) VSMCs were serum-starved for 24 h and then treated with ATRA (10 μmol/L) for different times (12, 24, 48, and 72 h). Cells were collected and Western blotting was performed using anti-KLF4 and anti-mfn-2 antibodies to examine the expression of KLF4 and mfn-2, respectively. β-Actin was used as a control for equal protein loading. (B) KLF4 and mfn-2 expression induced by ATRA was detected by quantitative real-time RT-PCR. VSMCs were treated with ATRA for different times (6, 12, 24, and 48 h), and total RNA was isolated from VSMCs and subjected to quantitative real-time RT-PCR analysis by using specific primers for KLF4 and mfn-2. GAPDH was used as an internal control. The bars represent the means±SEM from three independent experiments. bP<0.05 vs control (0 h of ATRA). (C) Adenovirus-mediated KLF4 overexpression in VSMCs. VSMCs were infected with pAd-KLF4 or pAd for 24 h before harvesting. Western blotting was performed using antibodies against KLF4 or β-actin (as a loading control). (D) VSMCs were infected with pAd-KLF4 for the indicated times. The cell lysates were subjected to Western blotting with antibodies against mfn-2 and β-actin. (E) VSMCs were infected with pAd-KLF4 for the indicated times. Total RNA was isolated from VSMCs and subjected to quantitative real-time RT-PCR analysis by using specific primers for mfn-2. GAPDH was used as an internal control. The bars represent the means±SEM. from three independent experiments. bP<0.05 vs control (0 h of pAd-KLF4). (F) The effects of KLF4 knockdown of by siRNA on ATRA-induced mfn-2 gene expression. VSMCs were transfected with KLF4-specific siRNA (KLF4-siRNA) or non-silencing control siRNA (NS-siRNA) for 24 h. Cells were treated with ATRA (10 μmol/L) for 24 h, and then harvested and subjected to Western blotting to determine the levels of KLF4 and mfn-2. β-Actin was used as a control for equal protein loading.
KLF4 binds directly to the mfn-2 promoter and regulates its transcription
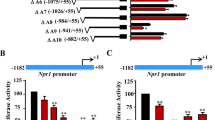
To further identify whether KLF4 activates the mfn-2 promoter, A293 cells were transiently co-transfected with the mfn-2 promoter fused to the pGL3-Basic luciferase vector (pGL3-mfn-2-luc) and KLF4 expression plasmid. The data showed KLF4 overexpression increased the mfn-2 promoter activity in a dose-dependent manner (Figure 2A). To identify the KLF4-responsive elements involved in KLF4-induced mfn-2 expression, progressive 5' deletion constructs of the mfn-2 promoter fused to the luciferase reporter gene were generated and transiently transfected into A293 cells. As shown in Figure 2B, progressive deletion of the promoter sequence from −1333 to −441 did not affect the mfn-2 promoter activity induced by KLF4. Further deletion between −441 and −273 markedly attenuated the promoter activity to that of the promoter-less plasmid, indicating that the region between −441 and −273 is critical for conferring the capacity to respond to KLF4. The sequence analysis of the mfn-2 promoter region between −1333 and +15 revealed the presence of four putative KLF4-binding boxes. To evaluate which is responsible for KLF4 binding and thereby regulates the promoter activity, the four boxes including −1152GTGGG−1148(box1), −840CACCC−836(box2), −356CACCC−352(box3), and−122GTGGG−118(box4) were mutated, respectively. The results from luciferase activity assay showed the inactivation of a single box 1, box 2, or box 4 had no effect on the mfn-2 promoter activity compared with wild-type promoter (Figure 2C). However, the inactivation of the box 3 completely blocked the response of the mfn-2 promoter to KLF4, suggesting that box 3, which is located in the promoter region between −441 and −273, plays a critical role in the KLF4-mediated transcriptional activation of mfn-2. To investigate whether KLF4 binds to the mfn-2 promoter within intact chromatin, ChIP assays were performed. The DNA-protein complex was immunoprecipitated with anti-KLF4 antibody, and recovered DNA was analyzed by PCR using specific primers for the promoter region containing the KLF4-binding box 3. As shown in Figure 2D, DNA fragments containing the box 3 sequence could be detected in the immunoprecipitates pulled down by anti-KLF4 antibody, indicating that KLF4 binds directly to the mfn-2 promoter. Taken together, these data have demonstrated that the mfn-2 promoter is a direct transcriptional target of KLF4.

Identification of the KLF4-responsive elements of the mfn-2 promoter. (A) Effect of KLF4 on the mfn-2 promoter activity. A293 cells were transfected in triplicate with increasing amounts of pEGFP-KLF4 and the indicated amounts of pRL-TK, pGL3-mfn-2-luc, and pGL3-Basic. Cell lysates were subjected to luciferase activity assays using the dual-luciferase reporter assay system, and the luciferase activity was normalized to pRL-TK activity. The bars represent the means±SEM from three independent experiments. bP<0.05 vs control (0 μg of pEGFP-KLF4). (B) A series of 5′-deletion mutants of the mfn-2 promoter linked to luciferase reporter gene were schematically represented on the left of the figure. These mfn-2 promoter-reporter constructs were transfected into A293 cells along with pEGFP-KLF4, and luciferase activities were measured with a dual-luciferase reporter assay system. The bars represent the means±SEM from three independent experiments. bP<0.05 vs the promoter region starting at positions −1333, −949, and −441, respectively. (C) The mutations of KLF4-binding sites were indicated on the left of the figure. These mfn-2 promoter-reporter constructs were transfected into A293 cells along with pEGFP-KLF4, and luciferase activities were measured as indicated above. The bars represent the means±SEM from three independent experiments. bP<0.05 vs the wildtype promoter. (D) ChIP assays. Chromatin fragments were isolated from VSMCs transfected with or without pAd-KLF4 and immunoprecipitated using an antibody against KLF4 or an irrelevant antibody (Ig, nonimmune IgG). Input DNA was taken from the sample prior to immunoprecipitation. The input and immunoprecipitated DNA was used as PCR templates to amplify the mfn-2 promoter region containing the KLF4-binding sites. The results are representative of three independent experiments with similar results.
ATRA induces acetylation of KLF4
To determine the specificity of anti-Ac-lys antibody, we used cross-CoIP to detect the the level of acetylated-KLF4 in ATRA-induced VSMCs. The immunoprecipitates pulled down with anti-KLF4 antibody were detected by anti-Ac-lys antibody and the immunoprecipitates pulled down with anti-Ac-lys antibody were detected by anti-KLF4 antibody. The results showed that the acetylated KLF4 was detected by both methods at the predicted molecular mass, which proved the specificity of anti-Ac-lys antibody (Figure 3A). Previous study has shown that KLF4 is acetylated by p300, which is associated with regulation of its transactivation function 26. We speculated that acetylation of KLF4 could also be important for its binding to the mfn-2 promoter. To test the hypothesis, we determined the levels of KLF4 acetylation in VSMCs treated with ATRA compared to treatment with ethanol (as a control). The result revealed that ATRA enhanced the levels of KLF4 acetylation within 24 h after ATRA treatment in a time-dependent manner, quantitative results showing a 2.2-fold increase at 1 h and a 5-fold increase at 24 h after ATRA treatment (Figure 3B), suggesting ATRA treatment induced KLF4 acetylation. We further sought to determine whether ATRA-induced KLF4 acetylation is caused by the interaction of KLF4 with p300. Co-IP assay showed that treatment with ATRA for 1 h enhanced p300 levels in the precipitates pulled down with anti-KLF4 antibody. Likewise, the level of KLF4 pulled down with anti-p300 antibody also markedly increased in cells treated with ATRA (Figure 3C), suggesting that ATRA treatment promotes the interaction of KLF4 with p300. To determine whether the increase in KLF4 acetylation induced by ATRA results from its interaction with p300, we performed an in vitro acetylation assay. Recombinant p300-HAT and GST-KLF4 or its acetylation-deficient mutants were incubated with cold acetyl-CoA, and KLF4 acetylation was measured by Western blotting with an anti-Ac-lys antibody. As shown in Figure 3D, the acetylation of KLF4 was strongly increased in the presence of p300, but acetylation-deficient mutants KLF4-K225R, KLF4-K229R, and KLF4-K225/229R could not be acetylated by p300. To identify whether these acetylation site mutations affect the ability of KLF4 to interact with p300, we incubated the lysates from VSMCs treated with and without ATRA with GST-KLF4 or its acetylation-deficient mutants, and found that both single point mutants K225R, K229R and double point mutant K225/229R could still interact with p300 in ATRA-treated cells (Figure 3E), consistent with previous findings26. To further determine the effect of KLF4 acetylation on the mfn-2 promoter activation, A293 cells were co-transfected with the mfn-2 promoter reporter plasmids, along with different combinations of expression plasmids for KLF4, KLF4 mutant, p300, or deacetylase HDAC2, and luciferase activity was detected. As expected, the strongest activation of the mfn-2 promoter was observed when KLF4 expression plasmids and p300 were co-transfected into the cells, with an approximately 10-fold luciferase activity relative to baseline. When deacetylase HDAC2, which makes KLF4 deacetylated, was co-expressed with KLF4, the mfn-2 promoter activity was reduced to 10% of that transfected with KLF4 alone. Meanwhile, acetylation-deficient mutant of KLF4 (K225/229R) could not enhance the promoter activity, indicating that KLF4 acetylation by p300 is required for the activation of the mfn-2 promoter (Figure 3F).

Acetylation of KLF4 by p300 enhances its binding to the mfn-2 promoter. (A) Identification of anti-Ac-lys antibody. VSMCs were treated with 10 μmol/L ATRA for 2 h, cell lysates were immunoprecipitated using an irrelevant antibody (nonimmune IgG) or the antibodies against KLF4 and acetylated lysine, the precipitates were detected via Western blotting using anti-KLF4 or anti-acetylated lysine antibodies. (B) ATRA induced KLF4 acetylation. VSMCs were treated with 10 μmol/L ATRA for the indicated times. Cell lysates were immunoprecipitated with anti-KLF4 antibody, and acetylated KLF4 was detected via Western blotting using anti-Ac-lys antibody. Blots for total KLF4 are also shown. (C) ATRA increased the interaction of KLF4 with p300. VSMCs were treated with ATRA for 1 h. Cell lysates were immunoprecipitated with anti-KLF4 or anti-p300 antibodies as indicated (IP). The precipitates were analyzed by Western blotting (IB) with anti-p300 and anti-KLF4 antibodies, respectively. (D) KLF4 was acetylated by p300 in vitro. KLF4 or its acetylation-deficient mutants (300 ng) and p300 (100 ng) were incubated with acetyl-CoA for 30 min at 30 °C, and reaction products were separated by SDS-PAGE, and then acetylated KLF4 was determined by Western blotting using anti-Ac-lys antibody. Blots for total KLF4 are also shown. (E) The interaction of p300 with KLF4 and its acetylation-deficient mutants. The lysates of VSMCs treated with or without ATRA were incubated with GST, GST-KLF4, GST-KLF4 (K225R), GST-KLF4 (K229R), or GST-KLF4 (K225/229R). After pull down with GST beads, p300 was detected via Western blotting using anti-p300 antibody, and KLF4 was detected with anti-GST antibody. (F) Acetylation of KLF4 enhanced the mfn-2 promoter activity. Luciferase assay was performed in A293 cells transfected with the mfn-2 promoter-reporter plasmid (containing nucleotides −441 to +15 of the mfn-2 promoter), along with different combinations of expression plasmids for KLF4 or its acetylation-deficient mutant (K225/229R), p300, and deacetylase HDAC2. The bars represent the means±SE from three independent experiments. bP<0.05 vs the control.
ATRA promotes the interaction of KLF4 with p300 by inducing KLF4 phosphorylation
To understand whether ATRA-induced interaction of KLF4 with p300 depends on KLF4 phosphorylation, we detected the levels of phospho-KLF4 in VSMCs treated with ATRA. As shown in Figure 4A, ATRA stimulation rapidly induced the phosphorylation of KLF4 within 0.5 h. KLF4 phosphorylation reached a maximum at 1 h and remained constant level for at least 2 h. Because the activation of JNK and p38 MAPK signaling is crucial for many fundamental cellular processes27, 28, we sought to determine whether the activation of JNK and p38 MAPK mediates KLF4 phosphorylation induced by ATRA. Thus, we examined the effect of ATRA on the phosphorylation of JNK and p38 MAPK by Western blotting using phospho-specific antibodies. ATRA increased the levels of phospho-JNK and phospho-p38 in a time-dependent manner without affecting the levels of total JNK and total p38 (Figure 4B). These results demonstrate that the JNK and p38 MAPK signaling play key roles in ATRA-induced KLF4 phosphorylation in VSMCs. Next, we investigated whether KLF4 phosphorylation affects its interaction with p300. The effects of SP600125, a JNK inhibitor and SB203580, a p38 MAPK inhibitor on ATRA-induced KLF4 phosphorylation and ATRA-increased interaction of KLF4 with p300 in cells treated with ATRA were detected. As shown in Figure 4C, both SP600125 and SB203580 blocked ATRA-induced KLF4 phosphorylation and thereby abolished the interaction of KLF4 with p300 induced by ATRA treatment (Figure 4D), suggesting that the activation of JNK and p38 signaling pathways is involved in ATRA-induced KLF4 phosphorylation, and that KLF4 phosphorylation is necessary for its interaction with p300. To further understand whether KLF4 acetylation depends on its phosphorylation and its interaction with p300, the effects of SP600125 and SB203580 on KLF4 acetylation induced by ATRA treatment were detected. As shown in Figure 4E, inhibition of JNK and p38 activation by their respective inhibitors abrogated ATRA-induced KLF4 acetylation (Figure 4E). These data suggest that ATRA promotes the interaction of KLF4 with p300 by inducing KLF4 phosphorylation via JNK and p38 signaling pathways. To further demonstrate that the phosphorylation of KLF4 is critical for its interaction with p300, we mutated three phosphorylation sites (S415A, S444A, and S470A), which are located within the domain of KLF4 interaction with other proteins, and examined the significance of these three sites for KLF4 interaction with p300. When the Ser470 was mutated to alanine, KLF4 phosphorylation induced by ATRA was reduced to 30% of the wild-type KLF4 or the S415A and S444A mutants (Figure 4F). Furthermore, the mutation of phosphorylation site (Ser470) decreased ATRA-induced interaction of KLF4 with p300 and subsequent KLF4 acetylation to 50% of the wild-type KLF4 (Figure 4G and H). These results indicated that the phosphorylation site (Ser470) is important for the interaction of KLF4 with p300. We also evaluated the effect of the mutation at KLF4 phosphorylation site (Ser470) on ATRA-induced mfn-2 promoter activity. As shown in Figure 4I, the coexpression of the S470A mutant of KLF4 with p300 could not activate the mfn-2 promoter compared with the coexpression of the wild-type KLF4 with p300 (Figure 3F). Similarly, ATRA-elicited increase in the mfn-2 promoter activity was abrogated by treatment with SP600125 and SB203580 or by transfection with the S470A mutant. These results further suggest that ATRA stimulates the interaction of KLF4 with p300 by inducing KLF4 phosphorylation, which in turn increases KLF4 acetylation and its transactivation to the mfn-2 promoter.

ATRA promotes KLF4 phosphorylation and its interaction with p300 by activating p38 MAPK and JNK signaling. (A) VSMCs were treated with 10 μmol/L ATRA for the indicated times, cell lysates were immunoprecipitated with antibody to phosphoserine and immunoblotted with antibody to KLF4. (B) VSMCs were treated with 10 μmol/L ATRA for the indicated times, cell lysates were immunoblotted using phospho-specific antibodies to detect phosphorylated JNK and phosphorylated p38, respectively. Blots for total protein (JNK or p38) are also shown. (C) VSMCs were pretreated with JNK inhibitor SP600125 (20 μmol/L) or p38 inhibitor SB203580 (20 μmol/L) for 2 h before exposure to ATRA (10 μmol/L) for 1 h. The phosphorylated KLF4 was assessed as described above. (D) VSMCs were pretreated with SP600125 (20 μmol/L) or SB203580 (20 μmol/L) for 2 h before exposure to ATRA (10 μmol/L) for 1 h. Interaction of KLF4 with p300 was examined by CoIP and Western blot analysis with the indicated antibodies as described above. (E) VSMCs were pretreated with SP600125 (20 μmol/L) or SB203580 (20 μmol/L) for 2 h before exposure to ATRA (10 μmol/L) for 1 h. Cell lysates were immunoprecipitated with anti-KLF4 antibody, and acetylated KLF4 was detected by Western blotting using anti-Ac-lys antibody. Blots for total KLF4 are also shown. (F) VSMCs were transfected with expression vectors for GFP-N1, GFP-KLF4, or its phosphorylation-deficient mutants, GFP-KLF4 (S415A), GFP-KLF4 (S444A), and GFP-KLF4 (S470A), after 24 h of transfection, cells were treated with or without ATRA (10 μmol/L) for 1 h. Cell lysates were immunoprecipitated with antibody to phosphoserine and immunoblotted with antibody to KLF4. (G) VSMCs were transfected with expression vectors for GFP-N1, GFP-KLF4, or GFP-KLF4 (S470A), after 24 h of transfection, cells were treated with or without ATRA (10 μmol/L) for 1 h. Interaction between KLF4 and p300 was examined by CoIP and Western blot analysis with the indicated antibodies as described above. (H) VSMCs were transfected with expression vectors for GFP-N1, GFP-KLF4, or GFP-KLF4 (S470A), after 24 h of transfection, cells were treated with or without ATRA (10 μmol/L) for 1 h. Cell lysates were immunoprecipitated with anti-KLF4 antibody, and acetylated KLF4 was detected by Western blotting using anti-Ac-lys antibody. Blots for total KLF4 are also shown. (I) Luciferase activity assays. A293 cells were transfected with the mfn-2 promoter-reporter plasmid (containing nucleotides −441 to +15 of the mfn-2 promoter), along with different combinations of expression plasmids for GFP-N1, GFP-KLF4 or its phosphorylation-deficient mutant GFP-KLF4 (S470A), and p300 for 24 h, and then treated with ATRA (10 μmol/L) for 24 h in the presence or absence of SP600125 (20 μmol/L) or SB203580 (20 μmol/L). The bars represent the means±SEM from three independent experiments. bP<0.05 vs GFP-N1 group.
Discussion
We have previously shown that ATRA inhibits VSMC proliferation by up-regulating expression of p53 gene and induces VSMC differentiation by increasing expression of SM22α and SMα-actin mediated by KLF410. We also found that mfn-2 is involved in VSMC differentiation induced by ATRA9. These findings suggest that both KLF4 and mfn-2 play an important role in regulating proliferation and differentiation of VSMCs. This prompted us to hypothesize the presence of a directly relationship between KLF4 and mfn-2 in the regulation of VSMC biology. The major purpose of the present study was to examine KLF4's involvement in ATRA-induced mfn-2 expression and the action mechanism of ATRA whereby ATRA exerts its regulatory effect on KLF4 and mfn-2 genes.
In this study, we showed that KLF4 mediated ATRA-induced mfn-2 expression in VSMCs. To determine whether KLF4 could directly regulate mfn-2 gene transcription, we constructed mfn-2 promoter-reporter plasmids spanning different regions of the promoter and various KLF4-binding site mutants and analyzed their activities in A293 cells. The results indicate that KLF4-binding site 3 lain between −441 bp and −273 bp is required for mfn-2 transcriptional activation by KLF4. ChIP assays showed that KLF4 bound directly to the mfn-2 promoter. These results suggest that the mfn-2 promoter is a direct target of KLF4.
Recent studies have shown that regulation of KLF family activity can occur on multiple levels, including interaction with cofactors and modifications (eg, phosphorylation, acetylation, etc), which results in additional regulation affecting the specificity of actions of KLFs. For example, the activity of KLF5 is increased via its interaction with the coactivator/acetylase p30029, and is decreased via its interaction with deacetylase (histone deacetylase 1)21. Our earlier observation showed that treating VSMCs with ATRA leads to the increase in KLF4 acetylation by inducing HDAC2 phosphorylation and its dissociation from KLF430. However, it remains unclear whether ATRA increases KLF4 acetylation by inducing the interaction of KLF4 with p300 in ATRA-treated VSMCs. Here, we showed that ATRA treatment rapidly and strongly promotes the KLF4 acetylation by increasing the interaction of KLF4 with p300. In order to further investigate whether the KLF4 acetylation affects its transcriptional activation function, we performed a luciferase activity assay. The results showed that coexpression of KLF4 and p300 markedly increased the mfn-2 promoter activity, while KLF4 acetylation site mutant K225/229R almost completely lost the function of activation of the mfn-2 promoter. These results suggest that the acetylation of KLF4 is necessary for the activation of the mfn-2 promoter by KLF4, which is consistent with previous observations that acetylation of KLF4 is responsible for KLF4-mediated induction of intestinal alkaline phosphase and p2126.
Protein phosphorylation is an important mechanism by which a transcription factor recruits coactivators, such as phosphorylation of cAMP response element-binding protein (CREB) at Ser133 leading to the recruitment of CREB-binding protein (CBP)31, phosphorylation of KLF5 at the CBP interaction region enhancing its interaction with CBP and its transactivation function32. Based on these observations, we hypothesized that the ATRA-induced increase in the interaction of KLF4 with p300 might be related to KLF4 phosphorylation. Indeed, we demonstrated that ATRA rapidly and strongly induced KLF4 phosphorylation in a time-dependent manner. Previous studies have demonstrated that the JNK and p38 MAPK signaling pathways play important roles in VSMC proliferation and differentiation27, 28. To further define the signaling pathways leading to ATRA-induced KLF4 phosphorylation, we examined the effects of JNK and p38 MAPK inhibitors on KLF4 phosphorylation induced by ATRA. The results revealed that inhibition of JNK or p38 MAPK by their inhibitors almost completely blocked KLF4 phosphorylation induced by ATRA. Meanwhile, ATRA-induced interaction of KLF4 with p300 was reduced, subsequently leading to the decrease in KLF4 acetylation. It has been proved that a phosphorylation-acetylation switch regulates STAT1 signaling33. Phosphorylation of Fli1 promotes its interaction with p300/CBP-associated factor and subsequent acetylation in response to TGF-β34. Our data showed that KLF4, as a transcription factor regulated by multiple signaling pathways, can be subjected to an unexpected cross-regulation between phosphorylation and acetylation. ATRA-induced KLF4 phosphorylation promotes its interaction with p300, which in turn enables it to be acetylated by p300. In addition, it has been reported that the interaction of KLF5 with RARα was reduced when serine in the second zinc finger of KLF5 was mutated to alanine23. These results provided possibilities that the serine in the zinc finger of KLF4 may also be important for KLF4 phosphorylation and its interaction with p300. Indeed, we found that serine mutation in the third zinc finger of KLF4 significantly reduced the level of KLF4 phosphorylation and its interaction with p300. As a result, the activity of KLF4 to transactivate the mfn-2 promoter was attenuated.
In summary, KLF4, as a target of ATRA-mediated signaling, plays an important role in the regulation of mfn-2 expression in VSMCs. We show that ATRA-induced KLF4 acetylation depends on KLF4 phosphorylation mediated by JNK and p38 MAPK signaling activated by ATRA. These results describe a novel mechanism by which ATRA up-regulates the expression and phosphorylation of KLF4 to ultimately activate the mfn-2 promoter and further expand the network of KLF4 actions in VSMCs.
Author contribution
Jin-kun WEN and Mei HAN designed research and revised the manuscript; Rui ZHANG performed research and analyzed data; Bin ZHENG, Ying-jie LI, and Ya-nan SHU performed research.
References
Rowland BD, Bernards R, Peeper DS . The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol 2005; 7: 1074–82.
Okita K, Ichisaka T, Yamanaka S . Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448: 313–7.
Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007; 448: 318–24.
Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC . Gut-enriched Krüppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res 2000; 28: 2969–76.
Wei D, Kanai M, Jia Z, Le X, Xie K . Krüppel-like factor 4 induces p27Kip1 expression in and suppresses the growth and metastasis of human pancreatic cancer cells. Cancer Res 2008; 68: 4631–9.
Hinnebusch BF, Siddique A, Henderson JW, Malo MS, Zhang W, Athaide CP, et al. Enterocyte differentiation marker intestinal alkaline phosphatase is a target gene of the gut-enriched Krüppel-like factor. Am J Physiol Gastrointest Liver Physiol 2004; 286: G23–30.
Swamynathan SK, Katz JP, Kaestner KH, Ashery-Padan R, Crawford MA, Piatigorsky J . Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema, and loss of conjunctival goblet cells. Mol Cell Biol 2007; 27: 182–94.
Wassmann S, Wassmann K, Jung A, Velten M, Knuefermann P, Petoumenos V, et al. Induction of p53 by GKLF is essential for inhibition of proliferation of vascular smooth muscle cells. J Mol Cell Cardiol 2007; 43: 301–7.
Jiang GJ, Han M, Zheng B, Wen JK . Hyperplasia suppressor gene associates with smooth muscle alpha-actin and is involved in the redifferentiation of vascular smooth muscle cells. Heart Vessels 2006; 21: 315–20.
Wang C, Han M, Zhao XM, Wen JK . Krüppel-like factor 4 is required for the expression of vascular smooth muscle cell differentiation marker genes induced by all-trans retinoic acid. J Biochem 2008; 144: 313–21.
Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 2003; 278: 17190–7.
Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 2005; 14: 1405–15.
Wu L, Li Z, Zhang Y, Zhang P, Zhu X, Huang J, et al. Adenovirus-expressed human hyperplasia suppressor gene induces apoptosis in cancer cells. Mol Cancer Ther 2008; 7: 222–32.
Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol 2004; 6: 872–83.
Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP . Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res 2007; 101: 1113–22.
Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem 2007; 282: 23354–61.
Li Y, Yin R, Liu J, Wang P, Wu S, Luo J, et al. Peroxisome proliferator-activated receptor delta regulates mitofusin 2 expression in the heart. J Mol Cell Cardiol 2009; 46: 876–82.
Zheng B, Wen JK, Han M . hhLIM is involved in cardiomyogenesis of embryonic stem cells. Biochemistry (Mosc) 2006; 71 Suppl 1: S71–6.
Liu B, Han M, Wen JK . Acetylbritannilactone inhibits neointimal hyperplasia after balloon injury of rat artery by suppressing nuclear factor-{kappa}B activation. J Pharmacol Exp Ther 2008; 324: 292–8.
Zheng B, Han M, Bernier M, Zhang XH, Meng F, Miao SB, et al. Krüppel-like factor 4 inhibits proliferation by platelet-derived growth factor receptor beta-mediated, not by retinoic acid receptor alpha-mediated, phosphatidylinositol 3-kinase and ERK signaling in vascular smooth muscle cells. J Biol Chem 2009; 284: 22773–85.
Zheng B, Han M, Wen JK, Zhang R . Human heart LIM protein activates atrial-natriuretic-factor gene expression by interacting with the cardiac-restricted transcription factor Nkx2.5. Biochem J 2008; 409: 683–90.
Li AY, Han M, Zheng B, Wen JK . Roscovitine inhibits ERK1/2 activation induced by angiotensin II in vascular smooth muscle cells. FEBS Lett 2008; 582: 243–8.
Zhang XH, Zheng B, Han M, Miao SB, Wen JK . Synthetic retinoid Am80 inhibits interaction of KLF5 with RAR alpha through inducing KLF5 dephosphorylation mediated by the PI3K/Akt signaling in vascular smooth muscle cells. FEBS Lett 2009; 583: 1231–6.
Dornan D, Shimizu H, Perkins ND, Hupp TR . DNA-dependent acetylation of p53 by the transcription coactivator p300. J Biol Chem 2003; 278: 13431–41.
Hansson ML, Popko-Scibor AE, Saint Just Ribeiro M, Dancy BM, Lindberg MJ, Cole PA, et al. The transcriptional coactivator MAML1 regulates p300 autoacetylation and HAT activity. Nucleic Acids Res 2009; 37: 2996–3006.
Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C . Krüppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem 2007; 282: 33994–4002.
Hayashi K, Takahashi M, Kimura K, Nishida W, Saga H, Sobue K . Changes in the balance of phosphoinositide 3-kinase/protein kinase B (Akt) and the mitogen-activated protein kinases (ERK/p38MAPK) determine a phenotype of visceral and vascular smooth muscle cells. J Cell Biol 1999; 145: 727–40.
Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, et al. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol 2003; 23: 795–801.
Miyamoto S, Suzuki T, Muto S, Aizawa K, Kimura A, Mizuno Y, et al. Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol Cell Biol 2003; 23: 8528–41.
Meng F, Han M, Zheng B, Wang C, Zhang R, Zhang XH, et al. All-trans retinoic acid increases KLF4 acetylation by inducing HDAC2 phosphorylation and its dissociation from KLF4 in vascular smooth muscle cells. Biochem Biophys Res Commun 2009; 387: 13–8.
Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH . Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993; 365: 855–9.
Zhang Z, Teng CT . Phosphorylation of Krüppel-like factor 5 (KLF5/IKLF) at the CBP interaction region enhances its transactivation function. Nucleic Acids Res 2003; 31: 2196–208.
Kramer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Guhrs KH, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 2009; 23: 223–35.
Asano Y, Trojanowska M . Phosphorylation of Fli1 at threonine 312 by protein kinase C delta promotes its interaction with p300/CREB-binding protein-associated factor and subsequent acetylation in response to transforming growth factor beta. Mol Cell Biol 2009; 29: 1882–94.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No 30971457, 90919035), the Special Found for Preliminary Research of Key Basic Research Project of Ministry of Science and Technology of China (No 2008CB517402), the Natural Science Foundation of Hebei Province (No C2008001049).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, R., Han, M., Zheng, B. et al. Krüppel-like factor 4 interacts with p300 to activate mitofusin 2 gene expression induced by all-trans retinoic acid in VSMCs. Acta Pharmacol Sin 31, 1293–1302 (2010). https://doi.org/10.1038/aps.2010.96
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.96
