
Abstract
Hyperdiverse tropical rainforests, such as the aseasonal forests in Southeast Asia, are supported by high annual rainfall. Its canopy is dominated by the species-rich tree family of Dipterocarpaceae (Asian dipterocarps), which has both ecological (e.g., supports flora and fauna) and economical (e.g., timber production) importance. Recent ecological studies suggested that rare irregular drought events may be an environmental stress and signal for the tropical trees. We assembled the genome of a widespread but near threatened dipterocarp, Shorea leprosula, and analyzed the transcriptome sequences of ten dipterocarp species representing seven genera. Comparative genomic and molecular dating analyses suggested a whole-genome duplication close to the Cretaceous-Paleogene extinction event followed by the diversification of major dipterocarp lineages (i.e. Dipterocarpoideae). Interestingly, the retained duplicated genes were enriched for genes upregulated by no-irrigation treatment. These findings provide molecular support for the relevance of drought for tropical trees despite the lack of an annual dry season.
Similar content being viewed by others

Introduction
Average annual rainfall is the highest in tropical rainforests, which harbor hotspots of biodiversity. Southeast Asian tropical rainforests are commonly aseasonal, without distinct intra-annual dry seasons, and are characterized by the dominant canopy tree family of Dipterocarpaceae1,2,3. Recent research has pursued the importance of rainfall variation and drought for promoting species distribution4 and for triggering reproduction5,6,7,8 in tropical forests, although ecologists have long-viewed light and soil characteristics as the main drivers of environmental filtering and species distributions in ever-wet tropical forests9. Drought events in this system are often associated with irregular supra-annual El Niño Southern Oscillations (ENSO), and climate models project more frequent and severe ENSO events10,11,12. These increased drought patterns could alter synchronous general flowering5,6,7,8,13, reduce plant growth and carbon sequestration14, increase tree mortality15,16, and shift species composition17.
To complement the existing ecological studies, genomic studies may elucidate the potential importance of the inter-annual drought on plants. One of the major limitations of tropical plant studies is the paucity of genetic and genomic data for species of environmental and forestry relevance in contrast to crop and commodity-producing species (cacao18, rubber tree19, oil palm20, and durian21). Nonetheless, several molecular studies using real-time PCR or de novo transcriptome approaches of Dipterocarpaceae suggested that expression levels of phenology- and stress-related genes7,8 were associated with ENSO-related fluctuations in drought or temperature. This premises that a genome assembly would be valuable to test the relevance of drought in tropical trees.
The dominant tree family, Dipterocarpaceae (comprised of >500 species) has the center of diversity in tropical Southeast Asia, where 488 species of the subfamily Dipterocarpoideae are found1,2. Their evolutionary origin remains enigmatic. While many dipterocarp researchers have proposed an ancient origin of the family in Gondwanaland (e.g., >120 Ma (million years ago))3, molecular dating studies have suggested a much younger date22,23. In support of the importance of inter-annual drought events, dipterocarp species appear to have maintained a functional response to drought at the community level, which promotes species coexistence24 and diversity25 and synchronizes reproduction5,6,7,8. Besides their ecological importance, Asian dipterocarps lead the international tropical timber market, therefore playing an important role in the economy of many countries within the region26. They are critically important as keystone species and serve as active carbon sink3. Despite the research activities of Asian dipterocarps dating back to 182527, the main issue in tropical tree breeding and improvement are the complexity and cost of the breeding programs as well as the long breeding cycles. Additionally, many of the dipterocarp species are now categorized as near threatened or endangered as a result of exploitation and massive population reduction28, further indicating the need of the genomic resources for strengthening research related to genetic conservation of dipterocarps29,30.
Here, we report a draft genome assembly of Shorea leprosula, a species that has been used as a representative of Dipterocarpaceae to assess genetic diversity by allozymes, nuclear SSR, AFLPs, and chloroplast loci31,32,33,34,35. It is locally known as Meranti Tembaga, and is internationally traded under the Light Red Meranti timber group. This species is widely distributed throughout aseasonal tropical rainforests of Southeast Asia (Peninsular Malaysia, Borneo, and Sumatra)1,36, but is classified as a near-threatened category under the IUCN Red List37. We showed that an ancient whole-genome duplication (WGD) event coincided with the Cretaceous–Paleogene (K-Pg) boundary using the genome-wide data of 19 distribution-wide S. leprosula individuals as well as of 10 species from seven genera of Dipterocarpaceae. Genes that were upregulated by no-irrigation treatment were significantly enriched in the retained duplicated genes. Climate data supported that S. leprosula is distributed in the environments with irregular drought despite the lack of annual dry season. The availability of the genome assembly of a dipterocarp is of great utility for genetic conservation and plant breeding in facing global changes.
Results
Genome assembly
Whole-genome sequencing of S. leprosula (Fig. 1) was performed on Illumina HiSeq platform, using paired-end and mate-pair libraries with various insert sizes ranging from 170 bp to 17 kb, with over 380-fold coverage of its haploid genome (n = 7)38,39 (Supplementary Table 1). The contig and scaffold N50 lengths obtained from the ALLPATHSLG40 assembly were 7.8 kb (spanning the longest 71,752 contigs) and 2.07 Mb (with 2913 scaffolds above 1 kb), respectively. The total size of the assembly of scaffolds was 340.5 Mb (Table 1). Thus, the scaffolds covered ~85% and ~87% of the estimated genome ∼402 Mb by flow cytometry41 and ~391 Mb by k-mer distribution42, respectively. K-mer Analysis Toolkit (KAT)43 analysis revealed two peaks (Supplementary Fig. 1), confirming the genome of S. leprosula is heterozygous. The frequency of the k-mers in the assembly confirmed that the assembly is haploid (i.e., only one of the two heterozygous variants is present).

a Tree trunk. b Flowers. c Mature winged fruits.
To validate the genome assembly, we mapped all paired-end and mate-pair reads to the assembled genome and found that the vast majority of the reads (93.35%) aligned (Supplementary Table 1). To assess the completeness of our assembly, we compared it to 1440 core genes in the Embryophyta lineage using BUSCO44, finding that 93.3% of them were present (79.7% in a single copy, 13.6% in two copies), with only 2.5% and 4.2% fragmented or missing, respectively, comparable to available assemblies of cacao (95.8%)18 and durian (90.3%)21 in Malvales. We also confirmed that the vast majority of RNA-seq reads of seven organs of S. leprosula (namely leaf buds, flower bud, flower, inner bark, small seed, large seed, and calyx) obtained from the sequenced individual were mapped on the assembly (~86%) (Supplementary Table 2).
Genome annotation
To annotate the S. leprosula assembly, we first identified transposable elements and non-genic repeated sequences. We found that about 132 Mb of sequence (corresponding to 33% of the assembly) were attributed to transposable elements and repeats (Table 1 and Supplementary Table 3). Gene prediction with AUGUSTUS45 and the RNA-seq reads of seven organs described above resulted in 60,563 protein-coding gene models (Supplementary Table 4). In a further evaluation, the S. leprosula models were compared with the protein-coding genes of Theobroma cacao18 (cacao, Malvaceae, which is distantly related in Malvales and is still the closest well-characterized relative of Dipterocarpaceae without lineage-specific genome duplication) and Arabidopsis thaliana, and we found that 43,868 genes were supported by homology. Moreover, out of the 43,868 genes with homology, 20,690 genes showed synteny with the T. cacao assembly by using MCScanX. Based on these empirical supports, we classified the predicted genes into three categories: category A for the 20,690 genes with synteny; category B for the 23,178 genes with homology with either T. cacao and/or A. thaliana but without synteny; category C for the 16,695 genes without clear homology (Supplementary Fig. 2 and Supplementary Tables 4 and 5). Category C was composed mostly of predicted genes shorter than 80 or 50 amino acids (aa) (mean length ~122 aa compared to those in the categories A and B being 414 and 458 aa on average, respectively (Supplementary Table 5).
To test whether genes in the A and B categories are also present in the individuals from different populations and other dipterocarp species, we analyzed the resequencing data of 19 S. leprosula individuals covering the distribution range (Borneo, Sumatra, and Peninsular Malaysia, Supplementary Table 6), obtaining 673,772 SNPs. The resequencing of three dipterocarp species Shorea platycarpa, Neobalanocarpus heimii, and Dryobalanops aromatica (Supplementary Table 7) showed relatively high mapping rate (73–92%), allowing the identification of homologs. We found that 30,677 (70%) out of 43,868 genes of categories A and B were present in all the studied individuals and species (Supplementary Table 5). Using the 30,677 genes that were found in all samples (Supplementary Table 4), genome-wide average nucleotide diversity (π), Watterson’s theta (θw), and Tajima’s D values were estimated as 0.0072, 0.0095, and −0.9801, respectively (Supplementary Table 8 and Supplementary Fig. 3), which was comparable to a previous study that used fewer nuclear loci46. Admixture analysis of 19 individuals of S. leprosula from Peninsular Malaysia, Sumatra, and Borneo based on the cross-validation error plot suggests the presence of two subpopulations (K = 2) (Supplementary Fig. 4); where the samples from Borneo were split from those of Peninsular Malaysia and Sumatra (Supplementary Fig. 5). Because of the empirical support in closely related species and populations, and longer protein sequences, we considered that the gene models in the categories A and B (43,868 genes) were of high-confidence genes.
Ancient whole-genome duplication (WGD)
In order to understand the genome evolution in Dipterocarpaceae, we assessed synteny between S. leprosula and T. cacao. As visualized in a dotplot (Fig. 2a and Supplementary Table 9), most T. cacao genomic regions were syntenic to two genomic regions of S. leprosula. This suggested that the entire genome of S. leprosula duplicated after its divergence from the lineage of T. cacao. Among the 20,690 S. leprosula genes (category A) that had syntenic homologs in T. cacao, more than half (12,886 genes, 62%) were retained as duplicates in the collinear blocks of S. leprosula (Supplementary Tables 4 and 10). We then estimated the expected number of synonymous substitutions per synonymous site (Ks) among the S. leprosula collinear duplicates. The Ks distribution showed a single and distinct peak around Ks = 0.3 (Fig. 2b). This result further supports that these genes duplicated around the same time, most probably via a single WGD event. In addition, the Ks estimates between the S. leprosula and T. cacao orthologs were considerably larger than those between the S. leprosula collinear duplicates (hereafter, referred to as “the WGD-retained duplicates”) (Fig. 2b). This is consistent with the WGD being specific to the lineage of S. leprosula and not shared with the lineage of T. cacao, and also suggests that the WGD is considerably younger than the divergence of Dipterocarpaceae and Malvaceae.

a Collinearity dotplot between Theobroma cacao chromosomes and Shorea leprosula scaffolds. Dots with different colors represent different collinear blocks. Chromosomes and scaffolds sequence are separated by gray line. Source data on the order and the orientation of the S. leprosula scaffolds used for the dotplot are found in Supplementary Table 9. Dotplot was generated based on the results of MCScanX using VGCS2.0. Red and blue lines correspond to the two sets of the S. leprosula scaffolds (set 1 and 2 in Supplementary Table 9). b Ks distribution of S. leprosula paralogs in collinear blocks (n = 4513), orthologs of S. leprosula and T. cacao (n = 11,239), and orthologs of S. leprosula and Vatica umbonata (n = 10,280) are shown in red, green, and blue, respectively. Note that the average ratio of the Ks of the S. leprosula–T. cacao orthologs and the Ks of the V. umbonata–T. cacao orthologs was 1.00, suggesting that the rates of synonymous substitutions in S. leprosula and V. umbonata are highly similar. Source data are provided as Supplementary Data 1. The Ks distribution of the orthologs of S. leprosula and the remaining Dipterocarpoideae species are shown in Supplementary Fig. S6.
To test whether the WGD can be observed in (is shared with) the other dipterocarp species, we examined the Ks distributions of the duplicated genes (between 4004 to 7108 genes) obtained by transcriptome assembly of 10 other species from seven different genera (Supplementary Tables 4 and 11). The Ks distributions of all species also had single peaks around Ks = 0.3 (Supplementary Fig. 6a), suggesting that the WGD event occurred before the split of the examined species in Dipterocarpoideae. To validate this finding further, we also checked the Ks distributions of ortholog pairs between S. leprosula and the other Dipterocarpoideae species (Fig. 2b, Supplementary Fig. 6b, and Supplementary Data 1). In all the studied species, the peak of Ks estimates for orthologous genes was lower than the peak corresponding to the WGDs. Taken together, these results place the WGD event after the split from T. cacao, but before the divergence of the examined Dipterocarpoideae species.
The WGD event coincided with the K-Pg boundary, as in other plant lineages
To further understand when the WGD event occurred, we estimated the timing of the WGD event by focusing on the WGD-retained duplicates in S. leprosula that have syntenic homologs in the T. cacao genome. To obtain an age estimate of the WGD, phylogenetic dating was performed (Supplementary Data 2) using a Bayesian evolutionary analysis framework previously described47 for 204 orthologous groups with cleaned alignment lengths of at least 100 aa. For each of these orthologous groups, the dates for each node were estimated by incorporating fossil calibrations and the dates obtained from previous studies (i.e., secondary calibrations) as prior information to account for the uncertainty in the ages of the calibrations. Using two different calibration settings (Supplementary Table 12), we estimated the timing of the WGD event as 66.9 Ma (95% CI, 61.3–69.3 Ma) and 69.7 Ma (95% CI, 67.7–75.3 Ma). Likewise, the divergence between the Dipterocarpaceae and Malvaceae was estimated to be ~86–98 Ma, whereas the divergence between the different dipterocarp lineages represented by the nodes 4 and 5 were estimated to be ~42–50 and ~36–40 Ma, respectively (Fig. 3, Supplementary Figs. 7–9 and, Supplementary Table 13). These results suggest that the ancestral dipterocarp lineage underwent a WGD close to the Cretaceous–Paleogene (K-Pg) extinction event of ~66 Ma, as in many other angiosperm plant linegaes47,48.

a Representative gene tree used for phylogenetic dating and the estimated ages of each node, which are the modes of the kernel density estimates of the age distributions shown in (b), based on the parameter Setting 1. The red bars correspond to the 95% confidence intervals that were obtained by calculating the mode of 1000 bootstrap density estimates of the ages of each family shown in (b). Only the confidence intervals with a range of >1 million years are shown. Note that these are not posterior uncertainty intervals and does not take into account the posterior uncertainty in each individual family (see Supplementary Table S12 for the high posterior density of each family). Node 3 corresponds to the dipterocarp WGD. Source data are shown in Supplementary Table S13. b Age distribution of the divergence of the nodes based on the parameter Setting 1. Source data are shown in Supplementary Table S12.
Characterization of duplicated genes in dipterocarps
We next characterized the WGD-retained duplicates in the S. leprosula genome. First, we focused on their overall evolutionary trends. Previous studies suggest that genes retained as duplicates after WGD tend to show slower evolutionary rates at nonsynonymous sites than genes not retained as duplicates during the long rediploidization processes (loss of some gene duplicates after WGD)49,50. To test whether a similar trend is observed in the WGD-retained duplicates of S. leprosula, we estimated Ka, Ks, and Ka/Ks using the orthologs between S. leprosula and T. cacao and compared the results between the WGD-retained duplicates and the genes that lost the syntenic duplicates derived from the WGD event (“the non-retained genes”). Our analysis showed that Ka and Ka/Ks estimates for the WGD-retained duplicates were significantly lower than those for the non-retained genes (Supplementary Fig. 10), indicating slower evolutionary rates of the WGD-retained duplicates at nonsynonymous sites. In contrast, such a significant difference was not observed for the Ks estimates between the WGD-retained duplicates and the non-retained genes (Supplementary Fig. 10). Therefore, changes in substitution rates were specific to the nonsynonymous sites.
Gene retention and loss are shown to be nonrandom with respect to gene function in ancient polyploids51,52,53,54,55,56. Hence, we examined the common functions of the 12,886 WGD-retained duplicates using a gene ontology (GO) enrichment test against the GO terms of A. thaliana orthologs. A large number of genes related to transcriptional regulation, signal transduction, and development were retained (Supplementary Table 14), consistent with previous findings reported for A. thaliana and other plants51,52,53,54,55. In addition to these terms commonly enriched in retained duplicated genes, drought-related terms, such as “response to salt stress” and “response to abscisic acid” were also found. To test whether the retention of the drought-related genes is specific to S. leprosula or a common feature among the Dipterocarpoideae species, we investigated the retention of these duplicated genes in the resequencing data obtained from the population and the interspecific samples (Supplementary Tables 6 and 7). Of the 12,886 WGD-retained duplicates, most of them (87%, 11,250 genes) had both copies of the corresponding homologs (Supplementary Table 15). A GO enrichment test of this conserved gene set also yielded similar results including “response to abscisic acid” (Supplementary Table 16). These data suggest that the retention of the drought-related duplicated genes is a common feature among the Dipterocarpoideae species in aseasonal tropics, rather than being a lineage-specific character of S. leprosula.
We also examined the common functions of tandemly duplicated genes in the S. leprosula genome by GO enrichment test. We found that 1212 genes in the category A had tandemly duplicated copies (Supplementary Table 4), and that their enriched functions were not overlapped with those of the WGD-retained duplicates (Supplementary Table 17).
Functional analysis of drought-responsive genes via no-irrigation treatment
Although we obtained results showing that drought-related genes were significantly enriched in the WGD-retained duplicates using the GO terms assigned based on the homologies to the A. thaliana orthologs, homology to functionally verified A. thaliana genes does not ensure that the S. leprosula homologs also have a role in response to drought. Therefore, we characterized drought-responsive genes of S. leprosula by performing a no-irrigation treatment of S. leprosula seedlings (Supplementary Table 18). Leaf samples were collected for RNA-seq analysis at the beginning of the treatment and at the 7th day, which was slightly before the 9th day when the typical wilting symptom (withered and brown leaves) was observed (Fig. 4a, b). Under this water stress condition, we conducted an expression analysis using genes from all three categories. Differential expression analysis identified 1200 upregulated and 914 downregulated genes in total, of which the A category had 829 and 658 genes, respectively (Supplementary Fig. 11 and Supplementary Tables 19 and 20). In the upregulated gene list, the highest-ranking GO terms were similar to those known to be involved in the drought response, such as “response to water deprivation”, “response to abscisic acid”, and “response to salt stress” (Supplementary Table 21). In addition, the enriched categories encompassed “response to chitin” and “response to oxidative stress”, which may be attributable to the crosstalk of the signaling of abscisic acid, wounding, and defense facing the high pressure of pathogens in the tropics57,58. GO terms related to photosynthesis, light, and biosynthetic processes (starch, chlorophyll, glycogen, and amylopectin) were enriched among the downregulated genes (Supplementary Table 22).

a S. leprosula seedlings at 0th day of treatment. b On the 9th day of the treatment. Seedlings with no-irrigation treatment had brown withered leaves, while the control seedlings with 50 mL of water daily had green leaves. c, d Mosaic plots to check enrichment of upregulated (c) and downregulated (d) drought-response genes in the S. leprosula WGD-retained duplicates. An asterisk in (c) shows significant enrichment of the upregulated genes in the S. leprosula WGD-retained duplicates (P-value after Bonferroni correction: 0.0004). Up: upregulated genes, Non-up: non-upregulated genes. The source data are shown in Table 2.
Using the genes that responded to the no-irrigation treatment, we tested whether these are significantly enriched in the WGD-retained duplicates. Fisher’s exact test showed significant enrichment of upregulated genes in the WGD-retained duplicates (Bonferroni corrected P = 0.0004, Table 2, and Fig. 4c), in contrast to non-significant enrichment for the downregulated genes (Bonferroni corrected P = 1.0000, Table 2 and Fig. 4d). This result is consistent with that obtained in the GO analysis described above, and indicates that the observed enrichment of drought-response genes is not likely due to artifacts in the GO enrichment test based on the homologies to the A. thaliana orthologs. These WGD-retained drought-up genes also showed slower evolutionary rates at nonsynonymous sites, compared with the non-retained genes (Supplementary Fig. 10). On the other hand, such significant enrichments of drought-response genes were not found in the tandemly duplicated genes (Supplementary Table 23), similarly to the results of the GO enrichment test (Supplementary Table 17). We found that the list of WGD-retained drought-up genes encompassed genes involved in diverse molecular roles in drought stress-response pathways (Supplementary Table 19), including a homolog of ABI1 (encoding a receptor component of plant hormone abscisic acid), DREB2C (encoding a key transcriptional factor in dry treatment), and TIP1 and TIP3 (encoding water-transport aquaporins) (Supplementary Fig. 11). These results support the hypothesis that some of the WGD-retained duplicates in the Dipterocarpoideae species tend to function in drought response.
Irregular drought instead of annual dry season
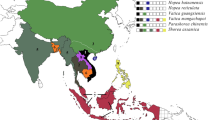
To examine whether the populations of S. leprosula experience dry environments, we analyzed multiple datasets of precipitation across the range of S. leprosula. First, we extracted the precipitation of the driest month across the spatial range of S. leprosula from the WorldClim data. Despite a broad variation, most localities (173) across the range of S. leprosula had greater than 100 mm of rainfall in the driest month (i.e., the driest month still met the evapotranspiration demands at the site). These values showed little overlap with species in seasonal forests (Shorea roxburghii is provided as an example from seasonal forests; Supplementary Fig. 12a, b). These data indicate few sites across the range of S. leprosula have an annual dry season. Second, we analyzed average 30-day cumulative rainfall from 2001 to 2014 measured at two localities within the distribution of S. leprosula (Pasoh Forest Reserve, Peninsular Malaysia, and Danum Valley Field Centre, Borneo). We found it fell below 100 mm roughly 20% in Pasoh and roughly 5% in Danum site (Supplementary Fig. 12c–f). The latter site was wetter but there were still supra-annual drought events (below 100 mm in 2002 and 2010). The combination of these modeled and observed climate data suggest that S. leprosula is distributed in the environments with irregular drought events even if they lack annual dry season.
Discussion
We sequenced the genome of S. leprosula using Illumina paired-end and mate-pair sequencing strategy, yielding sequence dataset of ~388-fold genome coverage. K-mer analysis, BUSCO analysis, and high-read-mapping rate indicated the completeness and accuracy of our genome assembly. We annotated 43,868 high-confidence genes showing homology to T. cacao and A. thaliana proteomes.
Our comparative genomic and molecular dating results, together with many recent studies on angiosperm evolution, allow us to propose the following scenario regarding the evolution and biogeography of Dipterocarpaceae. First, the Dipterocarpaceae lineage split from the lineage of Malvaceae in the Late Cretaceous, followed by a WGD in the common ancestor of the Dipterocarpoideae species close to the K-Pg boundary, after which the Dipterocarpoideae lineages diverged during the Eocene. Thus, Dipterocarpaceae provides another example that has been observed across many plant groups where the diversification occurred following a WGD around the K-Pg extinction event59,60,61. This timeline contrasts with the scenario hypothesized by many dipterocarp researchers which posits that the Dipterocarpaceae originated on Gondwanaland >120 Ma3 or >135 Ma62, and that Dipterocarpaceae and related lineages distributed in South America, Africa, Madagascar, Seychelles, and Asia diverged due to the breakup of the Gondwanan landmasses3,63, i.e., Gondwanan vicariance. However, the Gondwanan origin of Dipterocarpaceae is clearly not consistent with the generally accepted timeline of the divergence of various angiosperm/eudicot clades, in particular, that the origin of the eudicots should not be much older than ∼130 Ma22,64. Moreover, molecular dating studies of many different Tropical and Southern Hemisphere plant groups that show “Gondwanan” trans-continental distributions have reported much younger divergence date estimates that are not consistent with a strict Gondwanan vicariance scenario65,66,67,68,69. Our estimates of the divergence between Dipterocarpaceae and Malvaceae (86–98 Ma) are much younger than the proposed dates assuming a Gondwanan origin, as expected considering our priors, although it is worth noting that they are slightly older than previous estimates based on molecular dating (~70–80 Ma22,63). In addition, we obtained much younger estimates for the divergence of the Dipterocarpoideae lineages that are not consistent with the assumption that the separation of India and the Seychelles caused the divergence of certain lineages (see also “Methods”)3,63.
The main hypothesis to explain the inconsistencies between the molecular dating results and vicariance scenarios in many plant lineages is that long-distance and trans-oceanic dispersals are much more common than thought before65,66,67,68,69. For Dipterocarpaceae, such dispersals have been considered highly unlikely because their seeds lack dormancy, show salt-intolerance, and have low dispersal capacity3,63. Yet, results indicating long-distance dispersal have also been obtained for plant groups, such as the Nothofagus species, which show trans-oceanic distributions despite having poor dispersal capacity65. It is also worth noting that the exact timing and nature of the Gondwanan breakup is debatable, and that there may have been connected landmasses that enabled overland dispersal after the various proposed dates of the separation of landmasses66,67,70. Thus, while our results, combined with various recent findings, suggest that dispersal played a key role in the trans-continental distribution of Dipterocarpaceae, its exact mechanism remains an open question that is relevant also to many other plant groups.
It is known that the aseasonal tropical rainforests of Southeast Asia region (where dipterocarps dominate) receives high annual rainfalls. S. leprosula is a typical species in aseasonal tropical rainforests, and the precipitation of the driest month in its habitat is clearly higher than those in the habitat of S. roxburghii, which is a species in seasonal tropics (Supplementary Fig. 12a, b). Although S. leprosula inhabits regions with no annual dry season, our results showed that the drought-up genes are preferentially retained after the WGD event in this species (Table 2 and Fig. 4), and these WGD-retained drought-up genes are likely to conserve their functions because of their slower evolutionary rates at nonsynonymous sites (Supplementary Fig. 10). It is yet to be shown whether these substitution rate differences are biologically relevant. Nevertheless, the WGD-retained duplicates were conserved among the three species in different genera (Shorea, Dryobalanops, and Neobalanocarpus) inhabiting aseasonal tropics as well as among the 19 S. leprosula individuals of different populations (Supplementary Tables 15 and 16). The observed conservation suggests that these WGD-retained drought-related genes have been functionally important, not only at the WGD event, but also during the subsequent period in dipterocarp species in aseasonal tropics. At the WGD event, the genome duplication and duplicated drought-related genes might allow the ancestral dipterocarp species to develop tolerance to harsh environments during the mass-extinction period of the K-Pg boundary because contemporary polyploids often show enhanced environmental tolerance71,72,73. After the period around the WGD event, paleoclimate studies suggest that Asian dipterocarps lived in climates with dry seasons74,75,76, which might have contributed to the retention of the WGD-derived drought-related genes. In the present-day condition, aseasonal tropics in Southeast Asia receive high annual rainfalls and also suffers from occasional drought mostly due to ENSO. Although such drought conditions rarely occur, the irregular supra-annual drought (Supplementary Fig. 12) may be the basis for the preferential retention of drought-related duplicated genes in the Asian dipterocarps of aseasonal tropics. The observed preferential retention of the WGD-derived drought-related genes does not contradict the recent ecological studies that showed the relevance of inter-annual drought events in dipterocarp species in aseasonal tropical rainforests in Southeast Asia5,6,7,8,24,25. Nevertheless, it is still difficult to reveal the significance of an additional copy of a drought-related gene. We note that the enrichment of retained drought-related genes in Dipterocarpaceae was originated by WGD (Supplementary Tables 14 and 16) rather than by tandem duplication (Supplementary Table 17), in contrast to lineage-specific tandem duplication of stress-related genes reported in e.g., A. thaliana77.
In 2015, Malaysia and Indonesia contributed over 37.8% (93.7 million m3) of the total global production of tropical saw and veneer log, and more than 70% (4.8 million m3) of the total global export of plywood26. The growing demand for timber and timber products requires that tree breeders accelerate the improvement of germplasm. The lack of improved planting materials and knowledge of genetic and genomic resources such as the availability of high-density markers or even genetic maps for any dipterocarps hinders the success of forestry plantation. Our data of genome assembly, genome-wide polymorphisms, and divergence between 10 additional dipterocarp species will serve as a solid basis for establishing a molecular breeding program for Dipterocarpaceae. Here, we identified 673,772 SNPs by the resequencing of 19 individuals throughout the distribution range. The population structure analysis showed the split of Bornean populations from those of Peninsular Malaysia and Sumatra, which informs the design of breeding and association studies. Our findings support the hypothesis stating that canopy trees35,78,79 and other terrestrial organisms80,81,82 in Sundaland were divided into two clusters from the drowning of Sunda Shelf after the Last Glacial Maximum83.
Dipterocarp species are keystones in Asian tropical ecosystems. The biomass estimates of natural Asian dipterocarp forests range from 205 to 496 Mg per ha84,85,86, with biomass values 30–60% higher than those of the corresponding forest in Amazonia87,88,89, which highlights their high carbon storage value3. Presently, a large number of dipterocarp species have and are currently being planted and monitored in the Sabah Biodiversity Experiment and FRIM’s Common Garden Experiment sites, and thus would provide opportunities for establishing genome-wide association studies, genomic selection, and ecological genomics analyses29,30. Considering the critical contribution of tropical forests to the earth systems, it is urgent to fill the gap of molecular knowledge about tropical trees to a level that is comparable to that of temperate regions.
Methods
Sequencing of Shorea leprosula genome
Sample collection
Leaf samples of S. leprosula were obtained from a reproductively mature (diameter at breast height, 50 cm) diploid tree B1_19 (DNA ID 214) grown in the Dipterocarp Arboretum, Forest Research Institute Malaysia (FRIM).
DNA extraction
Genomic DNA was extracted from leaf samples using the 2% cetyltrimethylammonium bromide (CTAB) method90 and purified using a High Pure PCR Template Purification kit (Roche).
Library preparation and sequencing
Paired-end (170, 500, and 800 bp) and mate-pair (2 kb) genomic libraries were prepared using a TruSeq DNA Library Preparation kit (Illumina) and a Mate Pair Library Preparation kit (Illumina), respectively. Mate-pair libraries with larger insert sizes were constructed using a Nextera Mate Pair Library Preparation kit (Illumina). Ten micrograms of genomic DNA were tagmented in a 400 μl reaction and fractionated using SageELF, with the recovery of 11 fractions with 3–16+ kb. Each fraction was circularized and fragmented with a Covaris S2. Biotin-containing fragments were purified using Dynabeads M-280 streptavidin beads. Sequencing adapters (KAPA TruSeq Adapter kit) were attached using a KAPA Hyper Prep kit. The libraries were amplified for 10–13 cycles and purified with 0.8× AMpure XP. DNA libraries were then sequenced (~388× coverage) using Illumina HiSeq2000 (TruSeq libraries) and HiSeq2500 (Nextera libraries) at the Functional Genomics Center Zurich (FGCZ), University of Zurich, Switzerland (Supplementary Table 1).
Genome assembly
Adapters and low-quality bases for all paired-end and mate-pair reads were removed using Trimmomatic91. The filtered paired-end reads of the 170 bp library were used to identify the genome size using k-mer distribution generated by Jellyfish92 that was implemented in the scripts by Joseph Ryan42. The raw R1 reads from paired-end 170 and 800 bp libraries (clipped at 95 bp, representing about 70 genome equivalents) were used to estimate the heterozygosity using KAT43 with a k-mer size of 23 nt. De novo genome assembly of all reads was performed using ALLPATHSLG assembler v5248840.
Assembly verification and assessment of the assembled genome
Assembly validation
To validate the genome assembly, we mapped (i) the short reads used for the genome assembly, (ii) scanned the assembly for the presence of single-copy orthologs, and (iii) mapped transcriptome sequences obtained from seven organs.
Assembly verification by mapping of short reads
For each library used for genome assembly, all trimmed reads were aligned to the assembled S. leprosula genome using Burrows–Wheeler Aligner (BWA) v0.7.1293. Then, mapping ratio was calculated for each BAM file using Samtools94 with “flagstat” command.
Identification of highly conserved single-copy orthologs
BUSCO v3.1.042 was run with the Embryophyta dataset and Arabidopsis as the species for AUGUSTUS prediction (see subsection below “Protein-coding gene prediction”).
Assembly verification by mapping transcriptome sequences
For mapping transcriptome sequences, samples of seven organs (leaf bud, flower bud, flower, inner bark, small seed, large seed, and calyx) were obtained from the S. leprosula individual used for the genome sequencing (Supplementary Table 2). Total RNA was extracted from each sample using RNeasy Plant Mini Kit (Qiagen) and it was treated with Turbo DNase I (Takara). Library preparation was carried out using a TruSeq RNA Library Preparation kit (Illumina). Paired-end sequencing was conducted for all the libraries using Illumina HiSeq2000 at the FGCZ, University of Zurich, Switzerland. Adapters and low-quality bases for all paired-end reads were removed using Trimmomatic. The trimmed sequences of each library were mapped onto the assembled genome using STAR aligner v2.4.2a95, and mapping ratio was obtained from the output file of STAR.
Genome annotation
Repeat sequence analysis
Both homology-based and de novo prediction analyses were used to identify the repeat content in the S. leprosula assembly. For the homology-based analysis, we used Repbase (version 20120418) to perform a TE search with RepeatMasker (4.0.5) and the WuBlast search engine. For the de novo prediction analysis, we used RepeatModeler to construct a TE library. Elements within the library were then classified by homology to Repbase sequences (see subsection below “Preparation of repeat sequences for evidence-based gene prediction”).
Protein-coding gene prediction
S. leprosula protein-coding genes were predicted by AUGUSTUS v3.245. For ab initio gene prediction, we used a pre-trained A. thaliana metaparameter implemented in AUGUSTUS. For the evidence-based gene prediction, we used the information of exon, intron and repeat sequences of S. leprosula as hints for the AUGUSTUS gene prediction. The details of the preparation of the hints were described in the following subsections.
Preparation of repeat sequences for evidence-based gene prediction
We used RepeatModeler to construct a de novo library of repeated sequences in the S. leprosula assembly. Then, using RepeatMasker, we generated a file containing the information of the positions of repeat sequences in the S. leprosula genome based on the RepeatModeler library. Elements within the library were then classified by homology to Repbase sequences. Finally, the hint file for repeat sequences in GFF format was prepared using the two scripts, “10_makeGffRm.pl” and “12_makeTeHints.pl”, stored in https://gitlab.com/rbrisk/ahalassembly.
Preparation of the exon and intron information for evidence-based gene prediction
To obtain the exon and intron hints, we used the mapping data of RNA-seq obtained from seven organs of the sequenced S. leprosula individual as described above. First, we merged all the mapping data stored in different BAM files into a single BAM file using SAMtools. Then, we prepared the intron hint file in GFF format using the, “bam2hints” script of AUGUSTUS. The exon hint file was also generated from the merged BAM file using the two AUGUSTUS scripts, “bam2wig” and “wig2hints.pl”. To conduct evidence-based gene prediction with AUGUSTUS, the three hint files (repeat sequences, intron and exon) described above were merged into a single file in GFF format.
BUSCO analysis
Genome annotation completeness were assessed with BUSCO v3.1.044 using the Embryophyta odb9 dataset composed of 1440 universal Embryophyta single-copy genes. We referred to these 1440 genes as core genes in the main text.
Comparison with the proteome of Theobroma cacao
T. cacao’s gene models18 were downloaded from Phytozome 11 (https://phytozome.jgi.doe.gov/pz/portal.html). Then, comparison was conducted with BLASTP96 using the T. cacao proteomes as the BLAST database (E-value cutoff: 1.0E-10). Only the best hit was stored for each gene. We considered these best hits of the T. cacao genes as orthologs of the S. leprosula genes. When the T. cacao orthologs were identified by the BLASTP search, the orthologs of A. thaliana were defined based on the T. cacao-A. thaliana orthologous information provided by Phytozome 11 (Supplementary Table 4). When the T. cacao orthologs were not identified, the orthologs of A. thaliana were searched by BLASTP (E-value cutoff: 1.0E-10) using the A. thaliana proteomes obtained from TAIR 10 (https://www.arabidopsis.org) as the BLAST database.
Synteny analysis
Based on the result of the above BLASTP searches, we assessed synteny between the S. leprosula scaffolds and the T. cacao chromosomes using MCScanX97. Genome information of T. cacao in GFF format was also obtained from Phytozome 11 as described above, which was used as an input file for MCScanX.
Assessment of the genome assembly
Population data and other dipterocarp species
To assess whether the genome assembly could be used as a reference for the S. leprosula individuals from various populations, we checked mapping ratio, SNP positions, and admixture using the distribution-wide S. leprosula samples. Similarly, to assess whether the S. leprosula assembly could be used as a reference for aligning data from closely related species and determining their mapping ratios. For interspecific analysis, the following three Dipterocarpoideae species: S. platycarpa, D. aromatica, and N. heimii were used (Supplementary Table 7).
Sample collection and DNA extraction
Leaf samples of 19 S. leprosula individuals from different populations and three other dipterocarp species (S. platycarpa, D. aromatica, and N. heimii) were used as described in Supplementary Tables 6 and 7. Genomic DNA was extracted using the same method as described above.
Library preparation and sequencing
Paired-end genomic libraries (200 bp) were prepared using a TruSeq DNA Library Preparation kit (Illumina). DNA libraries were then sequenced (~16× coverage each) using Illumina HiSeq2000.
Mapping and SNP calling
Adapters and low-quality bases from resequencing reads were removed using Trimmomatic. All trimmed reads were then mapped and aligned to the S. leprosula assembly using BWA. Variants were called using GATK v3.598. Duplicated reads were marked using Picard 2.6.0. Within GATK, HaplotypeCaller was used to identify variants for each sample by generating an intermediate genomic variant call format (gVCF). Subsequently, gVCF files were merged using GenotypeGVCFs to produce a raw VCF file containing SNPs and INDELs. Low-quality variants were removed from the raw VCF file by applying the hard filters implemented in GATK. Variants with genotype quality (GQ) < 20 were discarded, to capture confident genotypes with 99% accuracy. INDELs were discarded and only biallelic SNPs were retained for subsequent analysis.
Conservation of the predicted genes in population samples and other dipterocarp species
To check whether the predicted genes are conserved, we used the variant data obtained by resequencing the population samples and three dipterocarp species described above. After variant calling and quality filtering, Beagle v4.199 was used for genotype phasing and imputing missing genotypes. Using in-house scripts, we aligned all genes from the phased data with reference to our predicted genes (.gff3 format). After the alignment, if a gene in a sample had less than 30% of ambiguous regions (missing data or less than 5× coverage), we considered that the gene existed in the sample. Then, if the gene was present in all the sequenced samples, it was considered as conserved.
Estimation of nucleotide diversity, Watterson’s theta and Tajima’s D for the predicted genes
To quantify genome-wide polymorphisms of S. leprosula, two measures were calculated: π, nucleotide diversity, i.e., the average number of pairwise nucleotide differences per site between sequences in a sample100; and θw, intraspecific diversity, which is based on the number of polymorphic sites in a sample of sequences but is independent of their frequency101. The analyses were implemented using the Compute program from the libsequence package102. We also calculated Tajima’s D (D), an index of frequency spectrum101.
Admixture analysis
For genetic admixture analysis, we used the raw VCF file obtained from GATK as described above. VCFtools103 was used for additional variant filtration. First, we retained variants that were successfully genotyped in 50% of individuals and had a minimum quality score of 30, a minor allele count of 3, and a minimum depth for a genotype call of 3. Subsequently, we restricted the set to variants that were called in a high percentage of individuals (95%), a set mean depth of genotypes of 20, and a minor allele frequency of 0.05. Only biallelic SNPs were retained for subsequent analysis. PLINK v1.9104 was used to convert the filtered VCF format into the PLINK format (.bed/.bim/.fam) as input for ADMIXTURE v1.3105.
Assessment of whole-genome duplication (WGD)
Dotplot analysis
Collinearity dotplot between T. cacao chromosomes and S. leprosula scaffolds (Fig. 2a) were generated by VGCS v2.0106. To visualize two sets (set 1 and 2) of the collinear blocks along the T. cacao chromosomes, we changed the order of the S. leprosula scaffolds and their orientation based on the results of MCScanX under the assumption that there is complete collinearity between the two species and that each S. leprosula scaffold was used only once for the analysis (Supplementary Table 9).
Ks analysis between duplicated genes and between orthologs
To conduct Ks analysis, we first identified duplicated genes and orthologs. Based on the collinear blocks and collinear genes obtained by MCScanX, groups of genes showing a 1:1 or 1:2 relationship between T. cacao and S. leprosula were identified as orthologs. In this study, the two S. leprosula genes within each 1:2 orthologous group were identified as duplicated genes (paralogs) created by the WGD (Supplementary Tables 4 and 10), which we referred to as “WGD-retained duplicates”. In contrast, the S. leprosula genes showing a 1:1 orthologous relationship was defined as “Non-retained genes”. To understand the timing of duplications, we estimated Ks between the duplicates using the S. leprosula genome data and transcriptome data from 10 other dipterocarp species. Furthermore, to understand the timing of the divergence of the species, we estimated the Ks between orthologs using the T. cacao and data from the other dipterocarp species. The details are described in the following subsections.
Sample collection, RNA extraction, and sequencing for the 10 dipterocarp species
We collected calyxes of fruits of the following 10 dipterocarp species in FRIM: Dipterocarpus costulatus, D. aromatica, Dryobalanops oblongifolia, Hopea wightiana, N. heimii, Shorea kunstleri, Shorea sumatrana, Upuna borneensis, Vatica odorata, and Vatica umbonata (Supplementary Table 11). The calyx samples were immersed in RNAlater (Ambion) immediately after harvesting and stored at −20 °C. RNA was extracted using the CTAB method90. DNA was removed with Turbo DNase I (Takara). Purification was conducted using the RNeasy Plant Mini Kit (Qiagen). Paired-end sequencing was conducted for all the libraries using Illumina HiSeq2000.
Transcriptome assembly for the 10 dipterocarp species
Before the assembly of the transcriptome, sequences with low-quality bases were removed using Trimmomatic with a parameter set to “HEADCROP:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36”. Using the trimmed sequences, de novo transcriptome assembly by Trinity assembler (version r20140413p1)107 was conducted for each species with a default parameter. The numbers of reads before and after trimming, and those of the obtained contigs by assembly are found in Supplementary Table 11.
Identification of orthologs for the 10 dipterocarp species
Protein sequences of the genes for the 10 dipterocarp species were obtained with TransDecoder. The reciprocal BLASTP best hits (E-value cutoff: 1.0E-10) between the predicted S. leprosula and each dipterocarp species’ proteins were identified as orthologs.
Estimation of Ks
The Ks between each homologous (orthologous or paralogous) gene pair was estimated as follows. For each gene pair, first, the amino acid sequences were aligned using BLASTP. Then, the alignments were edited by retaining the aligned positions only if the three aligned positions both upstream and downstream did not contain any alignment gaps. Alignments were also retained only if they were longer than 150 aa and covered at least half of the length of both amino acid sequences. When estimating the Ks between orthologous gene pairs in orthologous groups with a 1:2 relationship, the orthologous gene pair producing the longer alignment was used. Nucleotide alignments of the coding sequences were created using the amino acid alignment as a guide, and the Ks was estimated using the coding sequences by CODEML with the Yang and Nielsen model from the PAML package108 with the following parameters: model = 0, NSsites = 0, fix_alpha = 1, alpha = 0, fix_kappa = 0, RateAncestor = 0, CodonFreq = 2. For each of the 10 other Dipterocarpaceae species, Ks was estimated between the hits identified by all against all BLASTP according to the criteria outlined above. When the pairwise Ks are estimated between all paralogs, if a particular gene is duplicated multiple times, the Ks of the same duplication events will be estimated multiple times. As such, when obtaining Ks distributions for the 10 other Dipterocarpaceae species, single Ks estimates representing each duplication event were obtained by clustering the paralogs into gene families based on the Ks estimates as previously described48.
Time estimation of the WGD event
Preparation of a gene set for phylogenetic dating of WGD
Based on the orthologs and paralogs identified above, 204 orthologous gene families were created for each S. leprosula WGD duplicate pair. Starting with S. leprosula WGD duplicate pairs with Ks = 0.2–0.6, a T. cacao ortholog was added if the Ks between the T. cacao and S. leprosula orthologs was 0.5–1.2. For both S. leprosula genes, one ortholog from either D. aromatica or D. oblongifolia, and one ortholog from U. borneensis, V. odorata, or V. umbonata were added if the Ks between the orthologs was 0.05–0.30. If multiple orthologs were present, the gene with the lowest amino acid divergence (Ka, estimated together with the Ks as described above with its S. leprosula ortholog was chosen. Finally, Gossypium raimondii genes identified as collinear orthologs with T. cacao by the PLAZA database109 were added only if the T. cacao gene corresponded to one or two G. raimondii genes. If there were two G. raimondii genes, the gene with a lower amino acid divergence with the T. cacao ortholog based on the amino acid alignment was chosen, as the alignments are more likely to be reliable. Thus, all orthologous gene families contained two S. leprosula genes, one T. cacao gene, one G. raimondii gene, two duplicates of either D. aromatica or D. oblongifolia, and two duplicates of U. borneensis, V. odorata, or V. umbonata (see Fig. 3a). For each orthologous gene family, the amino acid sequences of each gene were aligned using MAFFT version 7110 with the alignment option linsi. The alignments were cleaned by removing poorly aligned positions and divergent regions using Gblocks version 0.9b111, and gene families with a remaining alignment length of at least 100 aa were retained for further phylogenetic dating.
Phylogenetic dating of WGD
Phylogenetic dating was performed on each orthologous gene family using the BEAST package v1.8112 following the method previously described47. Briefly, an uncorrelated relaxed clock model that assumes an underlying log-normal distribution (UCLD) was used, whereas the Le-Gascuel (LG) substitution model113 with gamma-distributed rate heterogeneity across sites using four rate categories114 was set as the underlying evolutionary model. A Yule pure birth process115 was specified for the underlying tree model, and a uniform prior between 0 and 100 for the Yule birth rate was used. An exponential prior with mean 0.5 on the rate heterogeneity parameter, mean 1/3 on the standard deviation of the UCLD clock model, and a diffuse gamma prior with shape 0.001 and scale 1000 on the mean of the UCLD clock model were used. The BEAST files (.xml) that were used to run without data under the two different calibration settings (see below) are provided as Supplementary Data 3. The MCMC analysis for each orthologous gene family was run for 10 million generations while sampling every 1000 generations, resulting in a total of 10,000 samples per family. The topology was fixed according to the widely accepted phylogenetic relationship shown in Fig. 3a. The calibrations and constraints are described in detail below.
The resulting files of each family were processed with LogAnalyser, which is part of the BEAST package, with a burn-in of 1000 samples, and only those with a minimum effective sample size (ESS) of at least 200 for all statistics were retained. For the files retained, the median ages were used to represent the age of each node. Although one family in Setting 2 (FamilyID 85 in Supplementary Table 12) was removed as it had very low (<200) ESS for multiple statistics, all the remaining families had an ESS of more than 200 for all statistics. Subsequently, for nodes 1–5, age distributions of the median age estimates of each family were obtained. Then, the kernel density estimate (KDE) of the ages of all the families was calculated using the R density function, and the mode was used as the consensus age of each node. Finally, in order to obtain 95% CIs of the consensus age of each node, 1000 bootstrap datasets of the age estimates of each family were created, and the mode of the KDE was calculated for each bootstrap dataset, as described in a previous study47. Then, the modes of the 26th and 974th bootstrap density estimate (ranked in order of increasing value of their mode) were taken as the lower and higher 95% CI boundary, respectively.
Calibrations and constraints for phylogenetic dating of WGD
The nodes corresponding to the divergence of T. cacao and G. raimondii (node 2) and the divergence of Shorea and Dryobalanops (node 5) were both constrained based on fossil records. A minimum age of 55.8 Ma was assigned to the T. cacao–G. raimondii node (node 2) based on the fossil from the middle-to-late Paleocene that has been attributed to the Eumalvoideae22,116. A minimum age of 34 Ma was assigned to the Shorea–Dryobalanops node based on fossils from the late Eocene attributed to Shorea74, which, to our knowledge, were the earliest fossils that could be confidently attributed to Shorea. Log-normal prior distributions with the means equal to the minimum fossil age plus 10% were assigned to the T. cacao-G. raimondii and Shorea-Dryobalanops nodes. These correspond to 61.38 Ma for T. cacao–G. raimondii (mean = 1.719, offset = 55.8) and 37.4 Ma for Shorea–Dryobalanops (mean = 1.22, offset = 34), with a standard deviation of 1 for these two nodes63. Although there were no appropriate fossil calibrations that could be assigned to the root node, the divergence between Dipterocarpaceae and Malvaceae has been estimated as ∼70–80 Ma by previous phylogenetic dating studies22,64. To incorporate this information, a normal prior distribution with a mean of 75 Ma and a standard deviation of 8 was assigned to this node as a secondary calibration.
Considering the various uncertainties associated with the fossil and secondary calibrations, an alternative set of calibrations (Setting 2) was used to perform phylogenetic dating. In particular, we considered that the settings described above (Setting 1) may be slightly biased toward producing younger age estimates; therefore, we applied a setting that allows each node to explore age estimates that are older. For instance, the true divergence date can potentially be a lot older than the fossil records used as lower bounds. In addition, a fossil from, e.g., the late Eocene can be anywhere between ∼34 and ∼41 Ma. Thus, assigning a prior distribution with a mean that is only a few million years older than the youngest possible date of the fossil can be argued as being rather restrictive. Similarly, we considered the possibility that previous estimates of the divergence dates between Dipterocarpaceae and Malvaceae are underestimated due to the limited sampling of Dipterocarpaceae and/or the low substitution rates of woody lineages such as Dipterocarpaceae117. In fact, the posterior age distribution of the root node from Setting 1 was a lot older than the prior age distribution. As such, a log-normal prior distribution with the mean age corresponding to 70.7 Ma and a standard deviation of 0.25 (offset = 55.8, mean = 2.7), based on the phylogenetic dating results of Malvaceae118, was assigned to the T. cacao–G. raimondii node, a log-normal prior distribution with the mean age corresponding to 42 Ma and a standard deviation of 0.5 (offset = 34, mean = 2.08) was assigned to the Shorea–Dryobalanops node, and a log-normal distribution with the mean age corresponding to 78.2 Ma and a standard deviation of 0.3 (offset = 55.8, mean = 3.11) was assigned to the root node. The marginal prior densities for each node based on running the MCMC sampler without data are shown in Supplementary Fig. 9 for both parameter settings.
One recent study performed the most comprehensive molecular dating of Dipterocarpaceae to date23. These authors chose not to use any fossil constraints citing difficulties to assign dipterocarp fossils to particular clades, and argued that the fossil ascribed to Shorea74 that we used as a fossil constraint is likely to be of another a species within Anthoshorea. This does not affect our result as Shorea and Anthoshorea share a more recent common ancestor the node 5 that we applied the fossil constraint to. These authors instead used a log-normal distribution with a mean of 87.5 Ma as a calibration point to the most recent common ancestor of Dipterocarpoideae and Sarcolaenaceae, which is more recent than the root node in our study. This is based on a widely cited assumption among dipterocarp researchers that Sarcolaenaceae, which is endemic to Madagascar, diverged from its sister species due to the separation of India and Madagascar ~87.6 Ma119. We chose not to incorporate this age as prior information considering the results of many other plant groups suggesting an important role of dispersal over vicariance in explaining trans-oceanic distributions (see “Discussion”). We note nevertheless that this age is compatible with our results if we assume that this divergence occurred shortly after the divergence of Dipterocarpaceae and Malvaceae.
We also note that some studies have assumed that Vateriopsis, which is endemic to the Seychelles, diverged from its sister lineage containing Vatica, Upuna, and Vateria ~63 Ma by the separation of the Seychelles and India63, leading to much earlier estimates for the divergence between the lineages of Dipterocarpoideae (e.g., ∼80 Ma for node 4 and ∼55 Ma for node 5 or ∼95 Ma for node 4 and ∼70 Ma for node 5)63,120, compared with our estimates of Fig. 3a (~42–50 Ma for node 4 and ~36–40 Ma for node 5). By contrast, the aforementioned comprehensive molecular dating study of Dipterocarpacae reported mean age estimates of 54.9 Ma for node 4 and 43.3 Ma for node 5, but both with posterior density intervals of ~30 Ma23. These estimates are similar to our estimates, and these authors suggested that the divergence of Vateriopsis occurred most likely by long-distance dispersal rather than vicariance.
Characterization of duplicated genes
Ka/Ks analysis
Previous studies suggested that ancient homologs tend to show slower evolutionary rates at nonsynonymous sites49,50. We assessed whether the Ka, Ks, and Ka/Ks of the WGD-retained duplicates were significantly smaller than those of the non-retained genes between S. leprosula and T. cacao (Malvaceae), S. leprosula and H. wightiana (Dipterocarpaceae), and S. leprosula and U. borneensis (Dipterocarpaceae) using the same approach with CODEML in the PAML package explained above. The distributions of Ka, Ks, and Ka/Ks estimates between S. leprosula and T. cacao were compared between the WGD-retained duplicates and the non-retained genes. We also compared the Ka, Ks, and Ka/Ks of upregulated genes under no-irrigation treatment in section “No-irrigation treatment” below that are the WGD-retained duplicates (“WGD-retained drought-up genes”) with those of the non-retained genes. Statistical analyses were conducted using one-sided Mann–Whitney U tests considering multiple comparisons (P-value cutoff: 0.05 after Bonferroni correction).
Gene ontology (GO) enrichment test for the WGD-retained duplicates
For GO enrichment test, we used the GO information of the A. thaliana orthologs in Supplementary Table 4. The A. thaliana GO terms were downloaded from TAIR 10 (https://www.arabidopsis.org) on 21 November 2017. The GO enrichment analysis was performed using the BioConductor package topGO121 in R. For enrichment analysis, we adopted the “elim” algorithm together with Fisher’s statistic to test the functions of the retained duplicated genes. The “elim” algorithm scores P-values by considering the topology of GO graphs122. We listed the top 40 significant GO terms identified by the “elim” algorithm method and the P-values obtained (P-value cutoff: 0.05). To consider that different scoring methods may affect the result of significance, we also evaluated the significance of the enriched GO terms by Fisher’s exact test (“classic” algorithm in topGO) and multiple test corrections by false discovery rate (FDR cutoff: 0.05) using the Benjamini-Hochberg procedure.
GO enrichment test for tandemly duplicated genes
To compare with the result of GO enrichment test in the WGD-retained duplicates, we also conducted GO enrichment test for tandemly duplicated genes in the S. leprosula genome. For this purpose, we first identified the tandemly duplicated genes by using the following criteria: (i) neighboring genes on the same scaffold corresponded to the same gene in T. cacao as a result of BLASTP (see above subsection “Comparison with the proteome of Theobroma cacao”); (ii) one of the neighboring genes showed a 1:1 or 1:2 syntenic orthologous relationship with a T. cacao gene (i.e., WGD-retained duplicates or non-retained genes). In this analysis, we considered the genes that were not the tandemly duplicated genes and showed syntenic relationship with T. cacao genes as non-tandem duplicates and used them as a control of comparisons. The procedures of GO enrichment tests for the tandemly duplicated genes were the same as those described above.
No-irrigation treatment
Experimental condition of the no water treatment
To confirm the functionality of the duplicated drought-responsive genes in the WGD-retained duplicates, an experiment was conducted on six S. leprosula seedlings grown in the nursery of the Forest Research Institute Malaysia (FRIM). The seedlings were about 2 years old with an average height of 36 cm and an average collar diameter of 3.55 mm. All the seedlings were transferred to a plant growth chamber (Percival PGC-15) with the following conditions—day: 29 °C, 75% humidity; night: 26 °C, 75% humidity; day/night cycle: 12/12 h. Two types of treatment were applied: 50 mL of water daily (control) and no irrigation (artificial drought). Each treatment had three replicates and lasted for 9 days.
Sample collection, RNA extraction, and sequencing for RNA-seq data
Leaves were sampled at day 0 (before no-irrigation treatment, at 09:00) and at day 7 (during the no-irrigation treatment, at 0900). The leaves were immersed in RNAlater (Ambion) immediately after harvesting and stored at −20 °C. RNA was extracted from the leaves using the same method described above. Library preparation was carried out using an Illumina TruSeq Stranded mRNA library preparation kit in accordance with the manufacturer’s recommendations. Paired-end 125 bp sequencing using an Illumina HiSeq2500.
Analysis of RNA-seq data
All data analysis was performed using the SUSHI pipeline123. Paired-end raw sequence reads were combined and mapped onto the S. leprosula genome and the annotation file using STAR aligner87. The mapped reads (Supplementary Table 18) were then counted using the FeatureCounts function of Rsubread124. A quality control step was subsequently performed on the counted reads using CountQCApp from SUSHI. We also checked for the presence of contamination or ribosomal RNA content on our reads using FastqScreenApp from SUSHI. Finally, the genes that were differentially expressed between the two time points were detected using the BioConductor package edgeR125 in R which based on a negative binomial distribution to model the raw read counts in a gene-wise manner and followed by Trimmed Mean of M-values (TMM) method for the sequence depth normalization126.
Using the output from edgeR, we split the data into two groups of upregulated and downregulated genes in response to the dry treatment. We filtered those genes based on the significance-level false discovery rate (FDR < 0.05), obtaining 1200 upregulated genes and 914 downregulated genes. Both the upregulated and the downregulated genes underwent an enrichment analysis using the BioConductor package topGO in R with the same procedures described above.
Comparison between drought-response and duplicated genes
To test whether the drought-response genes obtained in above are enriched in the S. leprosula WGD-retained duplicates, we conducted Fisher’s exact tests using the “fisher.test” function in R. Bonferroni corrections were conducted by considering multiple comparisons.
As a comparison, we also tested whether the drought-response genes are enriched in the tandemly duplicated genes in S. leprosula, by performing Fisher’s exact tests and Bonferroni corrections as described above.
Distributions of Shorea leprosula and Shorea roxburghii, and the precipitation in their habitats
We downloaded the distribution data of S. leprosula and S. roxburghii (a closely related Shorea species to S. leprosula that grows in a more seasonal climate) from the Global Biodiversity Information Facility (GBIF) (https://www.gbif.org) using the “gbif” function in the R package, “dismo”. We further downloaded the precipitation data of the driest month (BIO14) from WorldClim (https://worldclim.org) at the resolution of 2.5 min by using the “getData” function in the R package, “raster” for every site of the two species in the GBIF data in the region ranging from −6° to 22° of latitude and from 90° to 120° of longitude. We combined these data to assess the distribution of the driest month for these two species growing in contrasting climates. We further analyzed 30-day cumulative rainfall for 13 years and 2 months from Danum Valley Field Centre in Sabah, Borneo (data downloaded from searrp.org) and Pasoh Forest Reserve, Peninsular Malaysia127 to examine the temporal rainfall and drought patterns of two sites within the distribution of S. leprosula.
Statistics and reproducibility
The data of this genome study was derived from a single diploid individual S. leprosula tree B1_19 (DNA ID 214) located at the Dipterocarp Arboretum at Forest Research Institute Malaysia (FRIM). RNA-seq reads obtained from seven organs used for genome annotation were derived from the same tree. Resequencing data were derived from 19 S. leprosula individuals obtained across its distribution range in Southeast Asia (Peninsular Malaysia, Borneo, Kalimantan, and Sumatra) and three other closely related dipterocarp species (S. platycarpa, D. aromatica, and N. heimii). RNA-seq of 10 other dipterocarp species were obtained from Dipterocarp Arboretum at FRIM for comparative genomics and molecular dating analysis. No-irrigation treatment were conducted using 2 years old S. leprosula seedlings in a plant growth chamber (Percival PGC-15). Two types of treatments were applied: 50 mL of water daily (control) and no-irrigation (artificial drought). Each treatment had three replicates. All statistical tests were conducted using publicly available programs and packages as described in sections under “Methods”. Reproducibility can be accomplished by following the sample used and methods outlined above. Statistical analysis using R were described above in each section.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Ashton, P. S. Dipterocarpaceae. In Flora Malesiana I, Vol. 9 (ed. van Steenis, C.G.G.J.) 237–552 (Springer, 1982)..
Ashton, P. S. Dipterocarp biology as a window to the understanding of tropical forest structure. Ann. Rev. Ecol. Syst. 19, 347–370 (1988).
Ghazoul, J. Dipterocarp Biology, Ecology, and Conservation (Oxford University Press, 2016).
Engelbrecht, B. M. et al. Drought sensitivity shapes species distribution patterns in tropical forests. Nature 447, 80–82 (2007).
Sakai, S. et al. Irregular droughts trigger mass flowering in aseasonal tropical forests in Asia. Am. J. Bot. 93, 1134–1139 (2006).
Kobayashi, M. J. et al. Mass flowering of the tropical tree Shorea beccariana was preceded by expression changes in flowering and drought‐responsive genes. Mol. Ecol. 22, 4767–4782 (2013).
Kobayashi, M. J. & Shimizu, K. K. Challenges in studies on flowering time: interfaces between phenological research and the molecular network of flowering genes. Ecol. Res. 28, 161–172 (2013).
Yeoh, S. H. et al. Unravelling proximate cues of mass flowering in the tropical forests of South-East Asia from gene expression analyses. Mol. Ecol. 26, 5074–5085 (2017).
Katabuchi, M., Kurokawa, H., Davies, S. J., Tan, S. & Nakashizuka, T. Soil resource availability shapes community trait structure in a species-rich dipterocarp forest. J. Ecol. 100, 643–651 (2012).
Dai, A. G. Increasing drought under global warming in observations and models. Nat. Clim. Change 3, 52–58 (2013).
Power, S., Delage, F., Chung, C., Kociuba, G. & Keay, K. Robust twenty-first-century projections of El Niño and related precipitation variability. Nature 502, 541–545 (2013).
Cai, W. et al. Increasing frequency of extreme El Niño events due to greenhouse warming. Nat. Clim. Change 5, 1–6 (2014).
O’Brien, M. J., Peréz-Aviles, D. & Powers, J. S. Resilience of seed production to a severe El Niño-induced drought across functional groups and dispersal types. Glob. Change Biol. 24, 5270–5280 (2018).
Lewis, S. L., Brando, P. M., Phillips, O. L., van der Heijden, G. M. F. & Nepstad, D. The 2010 Amazon drought. Science 331, 554 (2011).
Phillips, O. L. et al. Drought sensitivity of the Amazon rainforest. Science 323, 1344–1347 (2009).
Potts, M. D. Drought in a Bornean everwet rain forest. J. Ecol. 91, 467–474 (2003).
van der Sande, M. T. et al. Old-growth Neotropical forests are shifting in species and trait composition. Ecol. Monogr. 86, 228–243 (2016).
Motamayor, J. C. et al. The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol. 14, r53 (2013).
Tang, C. et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants 2, 16073 (2016).
Singh, R. et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 500, 335–339 (2013).
Teh, B. T. et al. The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 49, 1633–1641 (2017).
Magallon, S., Gomez-Acevedo, S., Sanchez-Reyes, L. L. & Hernandez-Hernandez, T. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207, 437–453 (2015).
Heckenhauer, J. et al. Phylogenetic analyses of plastid DNA suggest a different interpretation of morphological evolution than those used as the basis for previous classifications of Dipterocarpaceae (Malvales). Bot. J. Linn. Soc. 185, 1–26 (2017).
O’Brien, M. J., Ong, R. & Reynolds, G. Intra-annual plasticity of growth mediates drought resilience over multiple years in tropical seedling communities. Glob. Change Biol. 23, 4235–4244 (2017).
O’Brien, M. J., Reynolds, G., Ong, R. & Hector, A. Resistance of tropical seedlings to drought is mediated by neighbourhood diversity. Nat. Ecol. Evol. 1, 1643–1648 (2017).
ITTO. ITTO Biennial Review and Assessment of the World Timber Situation 2015-2016 (ITTO, 2016).
Blume, K. L. Dipterocarpaceae in Bijdragen tot de Flora van Nederlandisch indie. Batavia 1, 1–42 (1825).
Saw, L. G. & Sam, Y. Y. Conservation of Dipterocarpaceae in Peninsular Malaysia. J. Trop. Res. 12, 593–615 (1999).
Hector, A. et al. The Sabah biodiversity experiment: a long-term test of the role of tree diversity in restoring tropical forest structure and functioning. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3303–3315 (2011).
Ang, C. C. et al. Genetic diversity of two tropical trees (Dipterocarpaceae) following logging and restoration in Borneo: high genetic diversity in plots with high species diversity. Plant Ecol. Divers. 9, 459–469 (2016).
Lee, S. L., Wickneswari, R., Mahani, M. C. & Zakri, A. H. Genetic diversity of Shorea leprosula Miq. (Dipterocarpaceae) in Malaysia: implications for conservation of genetic resources and tree improvement. Biotropica 32, 213–224 (2000).
Ng, K. K. S., Lee, S. L. & Koh, C. L. Spatial structure and genetic diversity of two tropical tree species with contrasting breeding systems and different ploidy levels. Mol. Ecol. 13, 657–669 (2004).
Ng, K. K. S., Lee, S. L. & Ueno, S. Impact of selective logging on genetic diversity of two tropical tree species with contrasting breeding systems using direct comparison and simulation methods. For. Ecol. Manag. 257, 107–116 (2009).
Cao, C. P., Gailing, O., Siregar, I., Indrioko, S. & Finkeldey, R. Genetic variation at AFLPs for the Dipterocarpaceae and its relation to molecular phylogenies and taxonomic subdivisions. J. Plant Res. 119, 553–558 (2006).
Ohtani, M. et al. Nuclear and chloroplast DNA phylogeography reveals Pleistocene divergence and subsequent secondary contact of two genetic lineages of the tropical rainforest tree species Shorea leprosula (Dipterocarpaceae) in South-East Asia. Mol. Ecol. 22, 2264–2279 (2013).
Symington, C. F. Foresters’ Manual of Dipterocarps. Malayan Forest Records No. 16 (University of Malaya Press, 1943).
Pooma, R. & Newman, M. F. Shorea leprosula. The IUCN Red List of Threatened Species 2017: e.T33123A2833148. https://doi.org/10.2305/IUCN.UK.2017-3.RLTS.T33123A2833148.en (2017).
Jong, K. & Lethbridge, A. Cytological studies in the Dipterocarpaceae, I. Chromosome numbers of certain Malaysian genera. Notes Roy. Bot. Gard. Edinb. 27, 175–184 (1967).
Kaur, A. et al. Apomixis may be widespread among trees of the climax rain forest. Nature 271, 440–441 (1978).
Gnerre, S. et al. High quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011).
Ng, C. H. et al. Genome size variation and evolution in Dipterocarpaceae. Plant Ecol. Divers. 9, 437–446 (2016).
Ryan, J. F. estimate_genome_size.pl (version 0.03) [Computer software]. Bergen, Norway: Sars International Centre for Marine Molecular Biology. Retrieved from http://josephryan.github.com/estimate_genome_size.pl/ (2013). Accessed date 11 July 2019.
Mapleson, D., Accinelli, G. G., Kettleborough, G., Wright, J. & Clavijo, B. J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2016).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Ishiyama, H., Inomata, N., Yamazaki, T., Nor Aini, A. S. & Szmidt, A. E. Demographic history and interspecific hybridization of four Shorea species (Dipterocarpaceae) from Peninsular Malaysia inferred from nucleotide polymorphism in nuclear gene regions. Can. J. For. Res. 38, 996–1007 (2008).
Vanneste, K., Baele, G., Maere, S. & Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Res. 24, 1334–1347 (2014).
Fawcett, J. A., Maere, S. & Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous–Tertiary extinction event. Proc. Natl Acad. Sci. USA 106, 5737–5742 (2009).
Yang, L. & Gaut, B. S. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol. Biol. Evol. 28, 2359–2369 (2011).
Wolfe, K. H. Yesterday’s polyploids and the mystery of diploidization. Nat. Rev. Genet. 2, 333–341 (2001).
Edger, P. P. & Pires, J. C. Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 17, 699–717 (2009).
Blanc, G. & Wolfe, K. H. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16, 1679–1691 (2004).
Seoighe, C. & Gehring, C. Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 20, 461–464 (2004).
Doyle, J. J. et al. Evolutionary genetics of genomes merger and doubling in plants. Annu. Rev. Genet. 42, 443–461 (2008).
Freeling, M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453 (2009).
De Smet, R. et al. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Natl Acad. Sci. USA 110, 2898–2903 (2013).
Ton, J., Flors, V. & Mauch-Mani, B. The multifaceted role of ABA in disease resistance. Trends Plant Sci. 14, 310–317 (2009).
Bagchi, R. et al. Pathogens and insect herbivores drive rainforest plant diversity and composition. Nature 506, 85–88 (2014).
Levin, D. A. & Soltis, D. E. Factors promoting polyploid persistence and diversification and limiting diploid speciation during the K-Pg interlude. Curr. Opin. Plant Biol. 42, 1–7 (2018).
Landis, J. B. et al. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 105, 348–363 (2018).
Koenen, E. J. M. et al. The origin of the legumes is a complex paleopolyploid phylogenomic tangle closely associated with the Cretaceous–Paleogene (K-Pg) mass extinction event. Syst. Biol. 70, 508–526 (2021).
Moyersoen, B. Pakaraimaea dipterocarpacea is ectomycorrhizal, indicating an ancient Gondwanaland origin for the ectomycorrhizal habit in Dipterocarpaceae. New Phytol. 172, 753–762 (2006).
Ashton, P. S. On the Forest of Tropical Asia: Lest the Memory Fade (Royal Botanic Gardens, 2014).
Bell, C. D., Soltis, D. E. & Soltis, P. S. The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303 (2010).
Knapp, M. et al. Relaxed molecular clock provides evidence for long-distance dispersal of Nothofagus (Southern Beech). PLoS Biol. 3, e14 (2005).
Barker, N. P., Weston, P. H., Rutschmann, F. & Sauquet, H. Molecular dating of the “Gondwanan’ plant family Proteaceae is only partially congruent with the timing of the break-up of Gondwana. J. Biogeogr. 34, 2012–2027 (2007).
Beaulieu, J. M., Tank, D. C. & Donoghue, M. J. A southern hemisphere origin for campanulid angiosperms, with traces of the break-up of Gondwana. BMC Ecol. Evol. 13, 80 (2013).
Ruhfel, B. R., Bove, C. P., Philbrick, C. T. & Davis, C. C. Dispersal largely explains the Gondwanan distribution of the ancient tropical clusioid plant clade. Am. J. Bot. 103, 1117–1128 (2016).
Cardoso, D. et al. A molecular-dated phylogeny and biogeography of the monotypic legume genus Haplormosia, a missing African branch of the otherwise American-Australian Brongniartieae clade. Mol. Phyl. Evol. 107, 431–442 (2017).
Ali, J. R. & Krause, D. W. Late Cretaceous bioconnections between Indo-Madagascar and Antarctica: refutation of the Gunnerus Ridge causeway hypothesis. J. Biogeogr. 38, 1855–1872 (2011).
Paape, T. et al. Patterns of polymorphism and selection in the subgenomes of the allopolyploid Arabidopsis kamchatica. Nat. Commun. 9, 3909 (2018).
Levin, D. A. The Role of Chromosomal Change in Plant Evolution (Oxford University Press, 2002).
Soltis, D. E., Visger, C. J. & Soltis, P. S. The polyploidy revolution then…and now: Stebbins revisited. Am. J. Bot. 101, 1057–1078 (2014).
Feng, X., Tang, B., Kodrul, T. M. & Jin, J. Wing fruits and associated leaves of Shorea (Dipterocarpaceae) from the late Eocene of South China and their phytogeographic and paleoclimatic implications. Am. J. Bot. 100, 574–581 (2013).
Morley, R. J. Origin and Evolution of Tropical Rain Forests (Wiley, 2000).
Dutta, S. et al. Eocene out-of-India dispersal of Asian dipterocarps. Rev. Palaeobot. Palynol. 166, 63–68 (2011).
Hanada, K., Zou, C., Lehti-Shiu, M. D., Shinozaki, K. & Shiu, S. H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148, 993–1003 (2008).
Iwanaga, H. et al. Population structure and demographic history of a tropical lowland rainforest tree species Shorea parvifolia (Dipterocarpaceae) from Southeastern Asia. Ecol. Evol. 2, 1663–1675 (2012).
Kamiya, K. et al. Demographic history of Shorea curtisii (Dipterocarpaceae) inferred from chloroplast DNA sequence variations. Biotropica 44, 577–587 (2012).
Fernando, P. et al. DNA analysis indicates that Asian elephants are native to Borneo and are therefore a high priority for conservation. PLoS Biol. 1, 110–115 (2003).
Steiper, M. E. Population history, biogeography, and taxonomy of orangutans (Genus: Pongo) based on a population genetic meta-analysis of multiple loci. J. Hum. Evol. 50, 509–522 (2006).
Wilting, A. et al. Geographical variation in and evolutionary history of the Sunda clouded leopard (Neofelis diardi) (Mammalia: Carnivora: Felidae) with the description of a new subspecies from Borneo. Mol. Phylogenetics Evol. 58, 317–328 (2010).
Woodruff, D. S. Biogeography and conservation in Southeast Asia: how 2.7 million years of repeated environmental fluctuations affect today’s patterns and the future of the remaining refugial-phase biodiversity. Biodivers. Conserv. 19, 919–941 (2010).
Berry, N. J. et al. The high value of logged tropical forests: lessons from northern Borneo. Biodivers. Conserv. 19, 985–997 (2010).
Feeley, K. J. et al. The role of gap phase processes in the biomass dynamics of tropical forests. Proc. R. Soc. Lond. B Biol. Sci. 274, 2857–2864 (2007).
Pinard, M. A. & Putz, F. E. Retaining forest biomass by reducing logging damage. Biotropica 28, 278–295 (1996).
Malhi, Y. et al. The regional variation of aboveground live biomass in old-growth Amazonian forests. Glob. Change Biol. 12, 1107–1138 (2006).
Saatchi, S., Houghton, R. A., Avala, R., Yu, Y. & Soares, J. V. Distribution of aboveground live biomass in the Amazon basin. Glob. Change Biol. 13, 816–837 (2007).
Slik, J. W. F. et al. Environmental correlates of tree biomass, basal area, wood specific gravity and stem density gradients in Borneo’s tropical forests. Glob. Ecol. Biogeogr. 19, 50–60 (2010).
Murray, M. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325 (1980).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurences of k-mers. Bioinformatics 27, 764–770 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 21, 2987–2993 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
McGinnis, S. & Madden, T. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25 (2004).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Browning, B. L. & Browning, S. R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276 (1975).
Thorthon, K. libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics 19, 2325–2327 (2003).
Danecek, P. et al. The Variant Call Format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Xu, Y. et al. VGSC2: second generation vector graph toolkit of genome synteny and collinearity. Genomics 112, 286–288 (2020).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Proost, S. et al. PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 43, D974–D981 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Le, S. Q. & Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320 (2008).
Yang, Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 11, 367–372 (1996).
Yule, G. U. A mathematical theory of evolution, based on the conclusions of Dr. J.C. Willis, F.R.S. Philos. Trans. R. Soc. Lond. B Biol. Sci. 213, 21–87 (1925).
Carvalho, M. R., Herrera, F. A., Jaramillo, C. A., Wing, S. L. & Callejas, R. Paleocene Malvaceae from northern South America and their biogeographical implications. Am. J. Bot. 98, 1337–1355 (2011).
Smith, S. A. & Donoghue, M. J. Rates of molecular evolution are linked to life history in flowering plants. Science 322, 86–89 (2008).
Richardson, J. E., Whitlock, B. A., Meerow, A. W. & Madrinan, S. The age of chocolate: a diversification history of Theobroma and Malvaceae. Front. Ecol. Evol. 3, 120 (2015).
Ducousso, M. et al. The last common ancestor of Sarcolaenaceae and Asian dipterocarp trees was ectomycorrhizal before the India-Madagascar separation, about 88 million years ago. Mol. Ecol. 13, 231–236 (2004).
Klaus, S., Morley, R. J., Plath, M., Zhang, Y. P. & Li, J. T. Biotic interchange between the Indian subcontinent and mainland Asia through time. Nat. Commun. 7, 12132 (2016).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment analysis for gene ontology. R package version 2.30.0 (Bioconductor, 2016).
Alexa, A., Rahnenfuhrer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating go graph structure. Bioinformatics 22, 1600–1607 (2006).
Hatakeyama, M. et al. SUSHI: an exquisite recipe for fully documented, reproducible and reusable NGS data analysis. BMC Bioinformatics 17, 228 (2016).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Chen, Y. Y. et al. Species‐specific flowering cues among general flowering Shorea species at the Pasoh Research Forest, Malaysia. J. Ecol. 106, 586–598 (2018).
Ng, K.K.S. et al. The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests: Supplementary Tables (1–9 and 11–23). figshare https://doi.org/10.6084/m9.figshare.16305312 (2021).
Ng, K.K.S. et al. The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests: Supplementary Table 10. figshare https://doi.org/10.6084/m9.figshare.16306089 (2021).
Ng, K.K.S. et al. The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests: Supplementary Data 1. figshare https://doi.org/10.6084/m9.figshare.16308090 (2021).
Ng, K.K.S. et al. The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests: Supplementary Data 2. figshare https://doi.org/10.6084/m9.figshare.16306179 (2021).
Ng, K.K.S. et al. The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests: Supplementary Data 3. figshare https://doi.org/10.6084/m9.figshare.16306194 (2021).
Acknowledgements
We thank J. Ghazali, M. Yahya, P. Ramli, B. Yasri, T. Sharifah, late S. Suryani, T. Weingrill, M. Krützen, C. van Schaik, and an anonymous orangutan (who threw a twig of S. leprosula at us) for their assistance in sample collection and DNA extraction; T.L. Yao for the Pasoh rainfall data, and R. Lohaus, A. Hector, H. Iwanaga-Ishiyama, and Y. Takeuchi for discussions. We acknowledge grant support from the University of Zurich, Switzerland, under its University Research Priority Program (URPP) Global Change and Biodiversity, URPP System Biology and Functional Genomics, URPP Evolution in Action; JST CREST (number JPMJCR16O3); MEXT KAKENHI (18H04785); Beatrice Ederer-Weber Foundation; and Swiss National Foundation (SNF) grants 31003A_182318, 31003A-116376, SystemsX.ch SXPHX0-124233 to K.K.S.; a grant from the Ministry of Science, Technology and Innovation (MOSTI), Malaysia under its Science Fund program (02-03-10-SF0208) to K.K.S.N.; and a grant-in-aid for JSPS Fellows (14J11547) to M.J.K. We thank all colleagues for their support in this study in the framework of the Memorandum of Understanding (MoU) of the University of Zurich with Forest Research Institute Malaysia (FRIM), and with Bogor Agricultural University (IPB), MoU between FRIM and National Institute of Advanced Industrial Science and Technology (AIST), and MoU between FRIM and Japan International Research Center for Agricultural Sciences (JIRCAS). We thank the Malaysia Genome Institute and Functional Genomic Centre Zurich for providing computational resources and partial performance at the Research Organization of Information and Systems (ROIS), National Institute of Genetics, Japan.
Author information
Authors and Affiliations
Contributions
K.K.S., S.L.L., and K.K.S.N. conceived the project. K.K.S. and S.L.L. directed the study and supervised the research. K.K.S.N., M.J.K., S.L.L., J.A.F., C.C.A., M.J.O.B., T.N., D.C., and K.K.S. wrote the manuscript. K.K.S.N., M.J.K., and K.K.S. designed the experiment. K.K.S.N., M.J.K., S.L.L., M.H., C.C.A., and T.N. performed the experiments. C.H.N., L.H.T., C.T.L., J.S., M.J.O.B, D.C., and M.N.M.I. provided support for the experiments. K.K.S.N., M.J.K., J.A.F., and C.C.A. performed the data analysis. T.P., M.H., and Y.K. contributed to the data analysis. K.K.S.N., C.H.N., L.H.T., C.T.L., R.C.O., M.P., I.Z.S., S.I., A.I., Y.I., and K.K.S. contributed to sample collection and DNA extraction. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks Sebastien Carpentier, Arthur Zwaenepoel and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Caitlin Karniski.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ng, K.K.S., Kobayashi, M.J., Fawcett, J.A. et al. The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests. Commun Biol 4, 1166 (2021). https://doi.org/10.1038/s42003-021-02682-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02682-1
This article is cited by
-
Tree growth and survival are more sensitive to high rainfall than drought in an aseasonal forest in Malaysia
Communications Earth & Environment (2024)
-
Dipterocarpoidae genomics reveal their demography and adaptations to Asian rainforests
Nature Communications (2024)
-
Genome-wide characterization leading to simple sequence repeat (SSR) markers development in Shorea robusta
Functional & Integrative Genomics (2023)
-
Genome sequencing reveals the genetic architecture of heterostyly and domestication history of common buckwheat
Nature Plants (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
