
Abstract
Oligodendrocytes associate with axons to establish myelin and provide metabolic support to neurons. In the spinal cord of amyotrophic lateral sclerosis (ALS) mice, oligodendrocytes downregulate transporters that transfer glycolytic substrates to neurons and oligodendrocyte progenitors (NG2+ cells) exhibit enhanced proliferation and differentiation, although the cause of these changes in oligodendroglia is unknown. We found extensive degeneration of gray matter oligodendrocytes in the spinal cord of SOD1 (G93A) ALS mice prior to disease onset. Although new oligodendrocytes were formed, they failed to mature, resulting in progressive demyelination. Oligodendrocyte dysfunction was also prevalent in human ALS, as gray matter demyelination and reactive changes in NG2+ cells were observed in motor cortex and spinal cord of ALS patients. Selective removal of mutant SOD1 from oligodendroglia substantially delayed disease onset and prolonged survival in ALS mice, suggesting that ALS-linked genes enhance the vulnerability of motor neurons and accelerate disease by directly impairing the function of oligodendrocytes.
Similar content being viewed by others
Main
ALS is an adult-onset neurodegenerative disease characterized by progressive muscle weakness and eventual paralysis. Although disease culminates in the degeneration of motor neurons, non-neuronal cells such as astrocytes and microglia are critical for the pathogenic process of ALS1,2,3,4. ALS-linked genes such as Sod1 are expressed in glia and motor neurons, and glial cell dysfunction appears to exacerbate injury to motor neurons, as selective removal of mutant SOD1 from subsets of glia slows disease progression2,3. However, the vulnerability of distinct populations of glial cells to disease-induced stress and the contributions of these alterations to the pathogenesis of ALS are not well understood.
Degeneration of motor neurons in the spinal cord is associated with reactive changes in surrounding glia that include cellular hypertrophy and enhanced proliferation. In particular, recent studies have found that the behavior of NG2+ glial cells, a distinct, widely distributed class of progenitor cells that have the capacity to differentiate into oligodendrocytes, is markedly altered in the spinal cord of a mouse model of ALS (SOD1 (G93A))5,6. By end stage of disease, NG2+ cells exhibit the highest rate of proliferation of any cell type in the spinal cord of these mice, and their differentiation into oligodendrocytes is markedly enhanced5, suggesting that there may be a progressive dysfunction of oligodendrocytes in ALS.
In addition to their role in forming myelin, there is emerging evidence that oligodendrocytes provide crucial metabolic support to neurons7,8. The monocarboxylic acid transporter 1 (MCT1), a transporter that motor neurons depend on for transfer of glycolytic substrates9,10, is expressed predominantly by oligodendrocytes9,11 and is downregulated in the motor cortex of ALS patients and the spinal cord of SOD1 (G93A) mice9. Together, these abnormalities in the oligodendrocyte lineage in ALS may affect motor neuron survival; however, the cause of the enhanced proliferation and differentiation of oligodendrocyte progenitors in SOD1 (G93A) mice, and the extent of oligodendrocyte abnormalities in human ALS12,13 are uncertain.
Using in vivo genetic fate tracing of oligodendrocytes and their progenitors, we found an extensive, progressive degeneration of oligodendrocytes in the spinal cord of SOD1 (G93A) mice, with less than half of the oligodendrocytes produced in first postnatal month surviving by end stage of disease. Mobilization of oligodendrocyte progenitors occurred first in the ventral gray matter, where motor neurons are located, before behavioral manifestation of disease; however, newly formed oligodendrocytes in this region exhibited abnormal morphologies and failed to fully differentiate. Dysfunction of gray matter oligodendrocytes was also prevalent in human ALS, as reactive changes in NG2+ cells and demyelination were observed in the gray matter of the ventral spinal cord and motor cortex of ALS patients. Genetic deletion of mutant human SOD1 (G37R) from NG2+ cells and their oligodendrocyte progeny in mice substantially delayed disease onset and prolonged survival, indicating that expression of this ALS-linked gene in the oligodendrocyte lineage accelerates motor neuron degeneration. The progressive loss of gray matter oligodendrocytes, and failure to restore these crucial cells, may accelerate disease progression in ALS by depriving motor neurons of essential metabolic support.
Results
Enhanced proliferation of NG2+ cells in young ALS mice
The progressive loss of motor neurons in SOD1 (G93A) mice is accompanied by prominent changes in the behavior of NG2+ cells5. By end stage of disease, their proliferation rate is 20-fold higher than in wild-type mice5, and they comprise the majority of actively dividing cells in the spinal cord5,6. However, the cause of this enhanced proliferation in ALS is unknown. To determine when and where NG2+ cells first exhibit this altered behavior, we examined the spatio-temporal profile of NG2+ cell proliferation over the course of disease. Mice were administered BrdU for 5 d and cumulative BrdU incorporation was measured in lumbar spinal cord (Fig. 1a,b and Supplementary Fig. 1a). In wild-type mice, the number of BrdU+ NG2+ cells declined with age in all of the regions that were examined (P < 0.001, one-way ANOVA; Fig. 1c–e), following the developmental decline in the generation of oligodendrocytes from these progenitors5,14,15. However, in SOD1 (G93A) mice, NG2+ cells sustained high rates of proliferation into adulthood (Fig. 1c–e). Moreover, unlike the uniform decline in proliferation in wild-type mice, NG2+ cells in SOD1 (G93A) mice exhibited regional differences in their response. Enhanced proliferation was most prominent in ventral gray matter, where their rate of division was elevated 2.3-fold compared with wild-type mice by postnatal day 60 (P60, P < 0.001, Student's t test), before SOD1 (G93A) mice show disease symptoms16, and 12.9-fold by P95 (P < 0.0005), after they exhibit muscle weakness and tremor17. NG2+ cells in ventral white and dorsal gray matter of ALS mice eventually exhibited enhanced proliferation, although it occurred later and the magnitude of increased cell division was lower than in ventral gray matter (Fig. 1c–e). Thus, NG2+ cells in SOD1 (G93A) mice display abnormal behavior in ventral gray matter before behavioral manifestation of disease.

(a) BrdU protocol used to assess proliferation of NG2+ cells in control and SOD1 (G93A) mice. (b) Fluorescence images showing BrdU+ cells in the ventral horn of the spinal cord in control and SOD1 (G93A) mice at P60. Scale bar represents 100 μm. Right, two BrdU+ NG2+ cells (from the region highlighted by the white box) in the ventral gray matter. (c–e) Graphs showing the density of proliferating oligodendrocyte lineage cells (BrdU+ Olig2+) in different regions of spinal cord at different ages in control and SOD1 (G93A) mice. (f–h) Graphs showing the density of NG2+ cells (PDGFαR+) in different regions of the spinal cord at different stages of disease (P75 (presymptomatic), P90 (symptomatic) and P120 (end stage)), as compared with wild-type mice. GM, gray matter; WM, white matter. Data are presented as mean + s.e.m. (n = 9 sections obtained from 3 mice per group). *P < 0.05, **P < 0.001, ***P < 0.0005, unpaired Student's t test.
Despite the enhanced generation of NG2+ cells in early stages of disease, their density was not significantly altered before end stage (P > 0.05; Fig. 1f–h), suggesting that they are continually removed through death or differentiation. To determine the fate of NG2+ cells at early stages of disease, we carried out genetic lineage tracing with Pdgfra-creER; Z/EG mice5. After breeding to SOD1 (G93A) mice, we administered 4-hydroxytamoxifen (4HT) at P30 or P60 to induce EGFP expression in cohorts of NG2+ cells, and identified EGFP+ cells in lumbar spinal cord 15–60 d later (Fig. 2a and Supplementary Fig. 1b,c). Consistent with the regional differences in NG2+ cell proliferation, more EGFP+ cells were observed in ventral gray matter by P90 in SOD1 (G93A) mice (Fig. 2b and Supplementary Fig. 1d–f), whereas the number of EGFP+ cells in other areas was not significantly increased relative to controls (P > 0.05). However, by end stage, EGFP+ cell density in SOD1 (G93A) mice was higher than controls in all of the regions examined, with ventral gray matter exhibiting the greatest accumulation of NG2+ cell progeny (Supplementary Fig. 1d–f). As the number of EGFP+ cells should remain constant if they have an equal probability of dividing or dying, these results suggest that the enhanced proliferation of NG2+ cells is not induced solely by accelerated death of these progenitors.

(a) Protocol used to trace the fate of NG2+ cells in Pdgfra-creER; Z/EG; SOD1 (G93A) and Pdgfra-creER; Z/EG mice (Pdgfra-creER; Z/EG ± SOD1 (G93A)). Cohorts of NG2+ cells were labeled with EGFP by 4HT injection at P30 or P60 and then analyzed at different ages. (b) Fluorescence images showing EGFP+ NG2+ cell progeny in the ventral horn of the spinal cord in control or SOD1 (G93A) mice 60 d after 4HT administration at P30 (P30+60). Scale bar represents 100 μm. (c–e) Maps of the location of adult born oligodendrocytes generated from NG2+ cells in the spinal cord of control and SOD1 (G93A) mice from P60–75 (c), P60–90 (d) or P60–120 (or end stage for ALS mice, e). Oligodendrocytes (EGFP+ CC1+) in four randomly sampled lumbar spinal cord sections from each group (from 3–4 mice) are shown. (f–h) Graphs showing the density of newly generated EGFP+ CC1+ oligodendrocytes (OLs) in different regions of the adult spinal cord from control and SOD1 (G93A) mice. Data are presented as mean + s.e.m. (n = 9 sections from 3 mice per group). *P < 0.05, **P < 0.001, ***P < 0.0005, unpaired Student's t test. (i) Graph showing the overall density of oligodendrocytes (Olig2+ CC1+) in different regions of the spinal cord of P120 and end-stage SOD1 (G93A) mice. Data are presented as mean + s.e.m. (n = 12 sections per group from 4 mice per group) N.S., non-significant, P > 0.05, one-way ANOVA with Tukey test.
Oligodendrogenesis without oligodendrocyte accumulation
NG2+ cells in the spinal cord of end stage SOD1 (G93A) mice not only proliferate more rapidly, but also differentiate more frequently into oligodendrocytes5. To determine where and when this increase in oligodendrogenesis occurs, we followed the appearance of EGFP+ cells in Pdgfra-creER; Z/EG; SOD1 (G93A) and Pdgfra-creER; Z/EG mice that were immunoreactive to antibody to APC (CC1), a feature of mature oligodendrocytes. Consistent with the early maturation of myelinated tracts in the spinal cord18, few oligodendrocytes (EGFP+ CC1+ cells) were generated from P60–120 in controls (Fig. 2c–h). In contrast, there was a marked increase in newly generated oligodendrocytes in ventral gray matter of SOD1 (G93A) mice (P < 0.001, one-way ANOVA; Fig. 2c–f); there were 10.9-fold more EGFP+ CC1+ cells in this region relative to wild-type mice 60 d after labeling (P < 0.0005, Student's t test; Fig. 2f).
An increase in EGFP+ oligodendrocytes also was observed in ventral white matter at P90 and end stage in ALS mice, although this increase was smaller than in ventral gray matter (Fig. 2g), and there was no significant change (P > 0.05) in EGFP+ oligodendrocytes relative to control in dorsal gray matter (Fig. 2g,h). As the fate of only a small fraction (∼15%) of NG2+ cells was followed in these experiments, the total number of oligodendrocytes generated during this period is expected to be much greater (by ∼6–7-fold). Despite this marked increase in oligodendrogenesis, the overall density of oligodendrocytes in the spinal cord of SOD1 (G93A) mice at end stage was unchanged relative to wild type (Fig. 2i). These results suggest that there must be a concomitant loss of oligodendrocytes with advancing disease.
Degeneration of early-born oligodendrocytes in ALS mice
To determine the extent of oligodendrocyte survival during the course of disease, we performed genetic fate tracing of oligodendrocytes using Plp1-creER; ROSA26-EYFP; SOD1 (G93A) and Plp1-creER; ROSA26-EYFP mice (Fig. 3a,b). Administration of 4HT at P35 resulted in labeling of 15–25% of oligodendrocytes in ventral gray and white matter regions of the spinal cord. Although Cre-mediated recombination occurs in some NG2+ cells in the brains of Plp1-creER; ROSA26-EYFP mice19, EYFP was not expressed by these progenitors in the spinal cord of either control or SOD1 (G93A) mice (Supplementary Fig. 2), indicating that this approach can be used to track the survival of spinal cord oligodendrocytes in vivo. Quantitative analysis of spinal cords sampled at early time points revealed that the number of EYFP+ oligodendrocytes increased gradually for the first 2 weeks after 4HT administration in both control and SOD1 (G93A) mice19, and initial labeling of these cells was substantially higher in SOD1 (G93A) mice than in control mice in ventral gray matter (at P35+15), possibly reflecting enhanced activity of the Plp1 transgene promoter. To allow direct comparison of oligodendrocyte survival between control and SOD1 (G93A) mice, we normalized the number of EYFP-labeled oligodendrocytes in each cohort to that observed at P35+15, when oligodendrocyte labeling in SOD1 (G93A) mice was near maximum (Fig. 3c,d). In control mice, there was a small increase in EYFP+ oligodendrocytes in lumbar spinal cord (EYFP+ CC1+ Olig2+) 40 and 70 d after initial labeling (at P50) (Fig. 3c), indicating a modest, but continuous, labeling of oligodendrocytes during this period. In contrast, the number of EYFP+ oligodendrocytes in SOD1 (G93A) mice was reduced by 22% at 40 d after labeling (P < 0.05, one-way ANOVA with Tukey; Fig. 3c). This loss was progressive, as the number of labeled oligodendrocytes was reduced by 65% at end stage (P < 0.0005, one-way ANOVA; Fig. 3c,e). Moreover, as expected from the regional difference in NG2+ cell proliferation and oligodendrogenesis, loss of early-born oligodendrocytes in ALS mice was most prominent in ventral gray matter (Fig. 3c,d). These results indicate that there is already pronounced degeneration of oligodendrocytes in ventral gray matter near motor neurons when mice first exhibit overt signs of disease.

(a) Protocol used to track the fate of early-born oligodendrocytes (OLs). Plp1-creER; ROSA26-EYFP; SOD1 (G93A) and littermate control mice received 4HT (2 mg) at P35 and were analyzed at P40, P50, P90 and P120 (or end stage for ALS mice). (b) Schematic showing the rationale and possible outcomes of oligodendrocyte fate analysis. (c,d) Plot of the number of EYFP+ oligodendrocytes (CC1+ Olig2+) in ventral gray (c) or white (d) matter of Plp1-creER; ROSA26-EYFP; SOD1 (G93A) and Plp1-creER; ROSA26-EYFP mice, expressed relative to the number observed at P35+15 (P50). Note that control group had additional time point (P35+10). Data are presented as mean ± s.e.m. (n = 9 sections from 3 mice per each time point of each group). *P < 0.05, **P < 0.001, ***P < 0.0005. Unpaired Student's t test was used to compare mean values between control and SOD1 (G93A) mice at each time point. One-way ANOVA with Tukey test was used for age-dependent relative changes in EYFP+ oligodendrocytes in each group. (e) Confocal images showing the density and morphology of EYFP+ (stained with antibody to EGFP) CC1+ Olig2+ spinal cord oligodendrocytes (yellow arrows) in ventral gray matter at P120 (control) or at end stage SOD1 (G93A) mice. Scale bars represent 20 μm.
If oligodendrocytes degenerate in the spinal cord of ALS mice, other indications of cell death should be visible. Indeed, some EGFP-expressing oligodendrocytes were immunopositive for activated caspase-3 in the spinal cord of end stage Mobp-EGFP; SOD1 (G93A) mice (Fig. 4a), in which only mature oligodendrocytes express EGFP (Supplementary Fig. 3). Moreover, we observed dense cell clusters consisting of EGFP+ oligodendrocytes surrounded by activated microglial cells at P90 in ventral gray matter (Fig. 4b). Consistent with the death of early born oligodendrocytes observed through genetic fate tracing, we found similar microglia-oligodendrocyte (EYFP+) aggregates in P90 Plp1-creER; ROSA26-EYFP; SOD1 (G93A) mice, in which oligodendrocytes were labeled at P35 (P35+55) (Fig. 4c); however, we did not find microglial clustering near GFAP+ astrocytes (Fig. 4c). Given that microglia are attracted to apoptotic cells20, the presence of these cell aggregates may indicate that mature oligodendrocytes in this region are dying via apoptotic death.

(a) Confocal images from the spinal cord ventral gray matter of a Mobp-EGFP; SOD1 (G93A) mouse at end stage showing an EGFP+ oligodendrocyte (yellow arrows) that was immunopositive for activated caspase-3. Lower right panel is an orthogonal view showing colocalization of activated caspase-3 and EGFP. (b,c) Confocal images of the spinal cord ventral gray matter showing Iba1+ activated microglia surrounding oligodendrocytes labeled with EGFP (b) or EYFP (c) in Mobp-EGFP; SOD1 (G93A) (P90) or Plp1-creER; ROSA26-EYFP; SOD1 (G93A) (P30+60) mice, respectively. Yellow arrowheads highlight several labeled oligodendrocytes, and white arrows in c highlight the processes of one EYFP+ oligodendrocyte. Scale bars represent 20 μm.
Widespread axonal degeneration can lead to loss of oligodendrocytes and reactive changes in their progenitors21,22, raising the possibility that degeneration of gray matter oligodendrocytes in ALS mice is secondary to motor neuron degeneration. To address whether loss of motor neurons is sufficient to induce mobilization of NG2+ cells, we partially ablated motor neurons by injecting ricin into the sciatic nerve23. Approximately half of the motor neurons in the ventral horn degenerated on the side ipsilateral to the injection by 1 week after this manipulation (P < 0.0005, paired Student's t test; Supplementary Fig. 4a–c). However, neither the morphology nor the proliferation rate of NG2+ cells, as assessed by NG2 and Ki67 immunoreactivity, respectively, was altered by this loss of motor neurons (Supplementary Fig. 4b,d), suggesting that acute motor neuron death is not sufficient to induce activation and recruitment of these progenitors.
To determine when oligodendrocytes begin to exhibit abnormalities in ALS mice, we examined their morphology in Mobp-EGFP; SOD1 (G93A) and Mobp-EGFP mice at different stages of disease. Oligodendrocyte somata (EGFP+ Olig2+) in the spinal cord of Mobp-EGFP mice had a consistent oval shape (Fig. 5a,b and Supplementary Fig. 3). In the spinal cord of end stage Mobp-EGFP; SOD1 (G93A) mice, there was a marked increase in irregularly shaped EGFP+ structures, many of which were Olig2− and lacked nuclei (Fig. 5a,b and Supplementary Fig. 5a), reminiscent of the fragmentation that occurs during apoptotic cell death24. Despite this increase in EGFP+ structures, the density of EGFP+ Olig2+ oligodendrocytes in end stage SOD1 (G93A) mice was not different from that in control mice (control, 1,137 ± 61 cells; SOD1 (G93A), 1,233 ± 73 cells per mm2; n = 9 sections, 3 mice per group, P = 0.33, Student's t test), consistent with results obtained using CC1 immunoreactivity (Fig. 2i). We also observed irregularly shaped EGFP+ oligodendrocyte somata with reduced or absent Olig2 immunoreactivity at earlier stages of disease (Supplementary Fig. 5b,c). To evaluate the progressive nature of these changes, we measured the area of EGFP+ cell fragments following digital subtraction of EGFP+ Olig2+ somata (Supplementary Fig. 5d). EGFP+ cellular fragments were evident at P90 in SOD1 (G93A) mice, but were rarely observed in aged matched controls (volume of fragments: control, 10 ± 5.1 μm3; SOD1 (G93A), 131 ± 13 μm3; P < 7.9 × 10−6, Student's t test; Fig. 5c), indicating that pathological alterations in oligodendrocytes are widespread by the time mice begin to show behavioral manifestation of disease.

(a) Confocal images of EGFP+ structures in the ventral spinal cord gray matter from control Mobp-EGFP (P120) and Mobp-EGFP; SOD1 (G93A) mice (end stage). (b) Higher magnification of regions highlighted by white squares in a. Arrowheads indicate large EGFP+ structures that were Olig2− and CC1−. Scale bars represent 20 μm. (c) Measured volume of Olig2− EGFP+ fragments in each 250,000 μm3 imaged volume. Data are presented as mean + s.e.m. (n = 6–9 sections from 3 mice per group). **P = 1.5 × 10−5, ***P = 8 × 10−6, unpaired Student's t test.
Myelin deficits in spinal cord gray matter of ALS mice
The accelerated turnover (that is, death and subsequent regeneration) and abnormal structure of oligodendrocytes in ALS mice suggests that the state of myelination in gray matter may be altered in disease. Indeed, electron microscopic analysis of ventral gray matter revealed that the proportion of ultrastructurally normal axons with immature myelin, as evidenced by the presence of a thick layer of oligodendrocyte cytoplasm between the axon and initial myelin wraps, was 49% higher in end stage SOD1 (G93A) mice than in age-matched controls (percentage of axons with immature myelin: control, 9.3 ± 1.2%; SOD1 (G93A), 13.9 ± 1.0%; n = 3 mice per group, P < 0.01, unpaired Student's t test; Fig. 6a), suggesting that viable, uninjured axons were being remyelinated in ALS mice. Axons with disorganized microtubules, electron dense bodies and spheroids surrounded by swollen, electron dense myelin, typical of Wallerian degeneration17, were also common in the ventral gray matter of SOD1 (G93A) mice (Supplementary Fig. 6a). Moreover, myelin basic protein (MBP) immunoreactivity was more diffuse in ventral gray matter of SOD1 (G93A) mice at end stage and was often not colocalized with EGFP, in contrast with the filamentous EGFP+ processes present in Mobp-EGFP mice (Fig. 6b). Axons with mature myelin had thicker myelin sheaths and lower g ratios (ratio of axon diameter to myelin diameter) in SOD1 (G93A) mice than those in control mice (Supplementary Fig. 6b–d), possibly reflecting shrinkage of axons from metabolic stress or abnormal patterns of gene expression in oligodendrocytes.

(a) Electron micrographs of ventral gray matter spinal cord of control (P120) and SOD1 (G93A) mice at end stage. Arrows highlight partially myelinated axons. Note the presence of thick oligodendrocyte cytoplasm surrounding these axons. (b) Confocal images of MBP immunoreactivity in ventral gray matter of P120 Mobp-EGFP mice or end stage Mobp-EGFP; SOD1 (G93A) mice. Optical sections: 1.5 μm. (c) Fluorescent images of membrane-anchored EGFP in the spinal cord of Pdgfra-creER; ROSA26-mEGFP mice (P60+60). (d) Confocal images showing fine EGFP+ processes of adult-born oligodendrocytes in ventral gray matter of Pdgfra-creER; ROSA26-mEGFP; SOD1 (G93A) and Pdgfra-creER; ROSA26-mEGFP mice (P60+60). Circles indicate ChAT+ motor neuron cell bodies and white arrows indicate NG2− oligodendrocyte processes. (e) Confocal images showing colocalization of EGFP+ thin processes and MBP immunoreactivity (white arrows) in control Pdgfra-creER; ROSA26-mEGFP mice (P60+60, top). MBP immunoreactivity was more disorganized in Pdgfra-creER; ROSA26-mEGFP; SOD1 (G93A) mice (bottom) and rarely colocalized with EGFP+ processes (P60+60) (yellow arrowheads). Optical sections: 0.5 μm. (f–h) Thin section electron micrographs from Pdgfra-creER; ROSA26-mEGFP; SOD1 (G93A) and Pdgfra-creER; ROSA26-mEGFP mice at P60+60 showing silver-intensified gold labeling of EGFP+ oligodendrocyte processes. Large, non-myelinating EGFP+ structures (arrowheads) reminiscent of apoptotic bodies were observed frequently in SOD1 (G93A) mice (h). Scale bars represent 1 μm (a), 20 μm (b,d,e), 200 μm (c) and 500 nm (f–h).
To determine the structure of oligodendrocytes generated after P60, we used membrane-anchored EGFP Cre reporter mice (ROSA26-mEGFP, mT/mG) to visualize their fine cellular processes5. Pdgfra-creER; ROSA26-mEGFP; SOD1 (G93A) and Pdgfra-creER; ROSA26-mEGFP mice were injected with 4HT at P60 and analyzed 2 months later (Fig. 6c). In control mice (P60+60), numerous thin, unbranched EGFP+ processes were visible throughout the ventral gray matter, consistent with the morphology of normal internodal segments (Fig. 6d). Although oligodendrocytes generated after P60 in SOD1 (G93A) mice also expressed CC1 (Fig. 2c–e), their processes were highly branched with numerous varicosities (Fig. 6d); the processes of these newly generated oligodendrocytes often did not colocalize with MBP, in contrast with oligodendrocyte processes in control mice (Fig. 6e). Immunogold electron microscopic labeling of EGFP revealed that adult-born oligodendrocytes in SOD1 (G93A) mice were frequently associated with degenerating axons (Fig. 6f,g), suggesting that these axons had experienced at least one round of demyelination and remyelination. In addition, there were many irregularly shaped, immunogold-positive structures in SOD1 (G93A) mice that were not associated with axons (Fig. 6h), reminiscent of the oligodendrocyte fragmentation observed through confocal microscopy (Fig. 5b). Thus, oligodendrocytes generated in this disease context do not achieve the normal structure of myelinating oligodendrocytes.
Western blot analysis revealed that the protein levels of myelin-associated proteins MBP, CNPase (2′,3′-cyclic nueotide-3′-phosphodiesterase) and MOG (myelin-oligodendrocyte glycoprotein) progressively decreased with age (Supplementary Fig. 7a,b), with the mature myelin protein MOG exhibiting the earliest decline by P60; this reduction in myelin was most prominent in ventral gray matter (Supplementary Fig. 7c). The decrease in myelin protein expression was accompanied by a concomitant increase in PDGFαR expression in both symptomatic (P90) and end stage mice (Supplementary Fig. 7a,b). Although NG2+ cell density in the spinal cord of SOD1 (G93A) mice was not significantly changed (P > 0.05) at earlier stages of disease (Fig. 2f–h), the increased expression of PDGFαR from P90 is consistent with progressive mobilization of these progenitors in response to the loss of oligodendrocytes25,26,27. Together, these findings indicate that myelination is disrupted in the spinal cord ventral gray matter of ALS mice.
Reactive NG2+ cells and focal gray matter demyelination in ALS
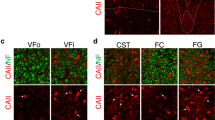
NG2+ cells are abundant in the human CNS and are likely to be major contributors to the regeneration of oligodendrocytes in demyelinating diseases such as multiple sclerosis28. To determine whether NG2+ cells also undergo reactive changes in ALS patients, we carried out immunohistochemistry on human motor cortices. We analyzed motor cortex rather than spinal cord, as this tissue is better preserved in human autopsies, which is required to preserve the NG2 proteoglycan. Sections were immunostained for the microglial antigen Iba1 to distinguish NG2+ glial cells from NG2-expressing macrophages and microglia that often appear in chronic disease and CNS injury29,30. NG2 immunoreactivity was markedly higher in the motor cortex of ALS patients than in either motor cortex from non-ALS subjects (control, 106.2 ± 5.3%; ALS, 200.2 ± 9.4%; P < 0.0001, unpaired Student's t test) or occipital cortex from the same patients, as determined by the intensity of NG2 immunoreactivity (Fig. 7a and Supplementary Fig. 8a,b). Although NG2+ Iba1+cells were observed in both control and ALS motor cortex, thicker NG2+ processes that exhibited signs of hypertrophy in ALS tissue did not express Iba1 (Supplementary Fig. 8a), indicating that NG2+ glial cells exhibit reactive changes in regions of brain in which motor neurons degenerate in ALS.

(a) Sections of motor cortex gray matter from control and ALS patients showing immunoreactivity to NG2. NG2+ cells are highlighted by arrowheads (see Supplementary Fig. 8a). Images were acquired from cortical layers IV and V. Scale bar represents 20 μm. (b) Luxol fast blue staining of motor cortices from control subjects and ALS patients. Demyelinated lesions are highlighted by arrowheads (upper right) or a dashed line (bottom). (c) MBP immunoreactivity in ALS motor cortex. Adjacent sections to those shown in the upper right and lower panels in b were used. Images show demyelinated plaques in layer III (top) and layer V (bottom). (d) Sections of lumbar spinal cord from an ALS patient stained with Luxol fast blue showing demyelination in the ventral horn gray matter and lateral corticospinal tract (arrowhead). Lower panels are higher magnification images of the boxed regions in the upper panels. Scale bars represent 1 mm (b and upper panels of d) and 200 μm (c and lower panels of d). (e,g) Western blots of oligodendrocyte lineage–specific myelin proteins in motor cortex gray matter (e) and lumbar spinal cord ventral horn (VH) gray matter (g) from controls and ALS patients. Full-length blots are presented in Supplementary Figure 11. (f,h) Graphs of MBP and CNPase protein expression levels (measured by western blot) in the motor cortex (f) and lumbar spinal cord ventral horn (h) gray matter in controls and ALS patients, normalized to average value in controls. Data are presented as mean ± s.e.m. with individual values. *P < 0.05, ***P < 0.0001, unpaired Student's t test.
To assess whether these changes in NG2+ cells also were associated with degeneration of oligodendrocytes in gray matter, we examined the integrity of myelin in motor cortex and spinal cord of sporadic and familial ALS patients. Myelin staining (Luxol fast blue) and MBP immunolabeling revealed areas of focal loss of myelin in motor cortex gray matter of ALS patients that were not observed in control patients (Fig. 7b,c and Supplementary Table 1). Moreover, in addition to the well-described reduction in myelination of corticospinal tracts, myelin was also reduced in ventral horn gray matter of the spinal cord from ALS patients (Fig. 7d). To quantify the extent of myelin loss, we determined MBP and CNPase abundance in randomly sampled regions of primary motor cortex gray matter (Fig. 7e,f and Supplementary Fig. 8c,d), as well as in spinal cord ventral gray matter (Fig. 7g,h). MBP expression was significantly lower (P < 0.001 and P < 0.05 for motor cortex and spinal cord ventral horn, respectively) in most ALS patients relative to controls, whereas CNPase levels were substantially reduced in one patient and trended lower in a second patient. Furthermore, the reduction in expression of myelin proteins was greater in motor cortices than in occipital cortices from the same ALS patients, consistent with motor cortex specific lesions (Supplementary Fig. 8c,d). Although it is not yet possible in these tissues to determine the relative contributions of primary oligodendrocyte death and secondary oligodendrocyte death because of Wallerian degeneration, these results indicate that demyelination of gray matter regions in which motor neurons are located is a common feature of human ALS.
Mutant SOD1 deletion from oligodendroglia reduces disease
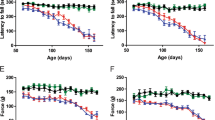
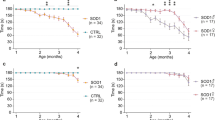
Oligodendrocytes are vulnerable to damage through cell-autonomous expression of genes linked to neurodegeneration31,32, particularly aggregation-prone proteins, raising the possibility that expression of mutant SOD1 in oligodendrocytes may impair their function and promote motor neuron degeneration. To determine whether there is an oligodendroglial contribution to disease in ALS, we selectively removed mutant human SOD1 (G37R) from NG2+ cells in Pdgfra-creER; loxSOD1(G37R) mice2. Administration of 4HT to these mice at P18 and P30 markedly delayed disease onset by 69 d (median, P = 0.0003), delayed early disease by 83 d (median, P = 0.001) and prolonged survival by more than 130 d (median, P < 0.001) (Fig. 8a,b). Consistent with this delay in disease, these mice exhibited less astrogliosis and microglial activation in the spinal cord (Supplementary Fig. 9a). The time from symptom onset to death (that is, disease duration) was not altered by removal of SOD1 (G37R) from oligodendroglia (−4HT, 180 ± 17 d; +4HT, 209 ± 15 d; mean ± s.e.m., n = 12 mice per group, P = 0.246, Mann Whitney test), indicating that the prolonged survival of these mice resulted primarily from a delay in disease onset. This manipulation decreased SOD1 (G37R) gene expression in the NG2+ cell population by ∼43%, as determined by quantitative PCR from isolated NG2+ cells (data not shown), but preserved its expression in motor neurons (Supplementary Fig. 9b). Although this approach had little effect on overall tissue levels of SOD1 (G37R) (Supplementary Fig. 9c,d), consistent with the relatively small number of NG2+ cells present (2–5% of all neural cells in the spinal cord)33, it presumably affected all oligodendrocytes generated from SOD1 (G37R)–deleted NG2+ cells.

(a) Plots of disease onset (median: −4HT, 235 d (n = 15); +4HT, 304 d (n = 20); P = 0.0003, log-rank test), early disease (median: −4HT, 330 d (n = 13); +4HT, 413 d (n = 14); P = 0.001) and survival (median: −4HT, 419 d (n = 12); +4HT, 554 d (n = 14); P = 0.0005) of Pdgfra-creER; loxSOD1(G37R) mice. (b) Comparison of mean age at disease onset (−4HT, 244 ± 10 d; +4HT, 307 ± 11 d; P = 0.0005), early disease (−4HT, 336 ± 16 d; +4HT, 415 ± 13 d; P = 0.001) and survival (−4HT, 433 ± 18 d; +4HT, 518 ± 16 d; P = 0.0025,). Data are presented as mean + s.e.m. **P < 0.01, ***P < 0.005, Mann-Whitney test. (c) Western blots of MCT1 expression from several −4HT and +4HT mice examined at disease onset. Full-length blots are presented in Supplementary Figure 11.
Previous studies have found that oligodendrocytes present at end stage of disease in SOD1 (G93A) mice express much less MCT1 (ref. 9). Given that oligodendrocytes provide metabolic support to axons via this transporter and that its expression is downregulated in SOD1 (G93A) mice9, we evaluated MCT1 expression in SOD1 (G37R)–deleted mice. Removal of SOD1 (G37R) from oligodendroglia helped preserve MCT1 expression in some mice at early stages of disease (Fig. 8c). Together, these data indicate that expression of mutant SOD1 in NG2+ cells and their oligodendrocyte progeny has a deleterious effect on motor neuron survival, and suggest that one negative consequence of mutant SOD1 expression in oligodendrocytes is to diminish their capacity to provide metabolic support to neurons.
Discussion
Oligodendrocytes form myelin sheaths around axons in the CNS that enable rapid conduction of action potentials at minimal energetic cost. However, the perception that oligodendrocytes are primarily structural elements has been challenged by recent evidence that they also provide metabolic support to neurons by transferring glycolytic intermediates34 through the monocarboxylic transporter MCT1 (ref. 9). Motor neurons in the spinal cord depend on this transporter for survival, suggesting that oligodendrocyte integrity affects motor neuron fate. We found extensive degeneration of gray matter oligodendrocytes in ALS mice that began before the onset of behavioral symptoms of disease. Although NG2+ progenitor cells are mobilized to regenerate oligodendrocytes, oligodendrocytes generated in later stages of disease exhibited aberrant morphologies and failed to restore myelin (Supplementary Fig. 10). A reduction in gray matter myelin and reactive changes in NG2+ cells were frequently observed in the motor cortex and spinal cord of ALS patients, indicating that degeneration of gray matter oligodendrocytes is prevalent in human ALS. Selective deletion of mutant SOD1 protein from oligodendroglia delayed disease onset and prolonged survival in mice, pointing to a key role for these cells in the precipitation of disease in ALS. Together, these results suggest that the marked loss of gray matter oligodendrocytes in ALS, and the inability to restore oligodendrocyte function, accelerates injury to vulnerable motor neurons.
There is accumulating evidence that ALS is not strictly a disease of motor neurons1,2,3,35,36. Selective removal of mutant SOD1 (G37R) from motor neurons slowed disease onset, but did not alter disease progression, indicating that expression of this mutant gene in other cell types is sufficient to cause motor neuron death2. In particular, astrocytes appear to be important contributors to disease, as there is a marked reduction in astrocyte glutamate transporter expression in mouse models of ALS35 and ALS patients37, astrocytes from SOD1 (G93A) mice secrete factors that are toxic to motor neurons38, and removal of mutant SOD1 from astrocytes delays microglial activation and extends animal survival3. However, expression of mutant SOD1 only in astrocytes is not sufficient to induce disease39, indicating that astrocyte dysfunction combines with other cellular alterations to accelerate the death of motor neurons.
The consequences of mutant protein expression in oligodendrocytes has been examined using chimeric mice in which oligodendrocytes and motor neurons express mutant SOD1, while all other cells expressed either wild-type or mutant SOD1 (ref. 40). Notably, most of these mice survived substantially longer than mice that expressed SOD1 (G37R) ubiquitously, and did not exhibit motor neuron degeneration at the time of death, suggesting that alterations in cells other than motor neurons and oligodendrocytes are critical for determining disease onset. Nevertheless, we found that selective removal of this mutant protein from oligodendrocyte progenitors markedly slowed disease progression. It is possible that ALS-linked gene mutations render oligodendrocytes more vulnerable to stresses created by abnormalities in other cells, and that oligodendrocyte degeneration is the critical event that triggers motor neuron degeneration. Indeed, oligodendrocyte vulnerability may be exacerbated by the presence of pro-inflammatory cytokines such as interferon-γ41, which at high levels induces apoptosis of oligodendrocytes42. Expression of mutant SOD1 in oligodendrocyte progenitors may also induce long-term changes in gene expression that increase the vulnerability of oligodendrocytes.
Mutations in genes linked to ALS, such as Sod1 (SOD1), Tardbp (TDP-43) and Fus (FUS), cause the formation of protein aggregates, often leading to endoplasmic reticulum stress that can induce apoptosis. Ubiquitinated protein aggregates containing TDP-43 have been observed in oligodendrocytes in patients with frontotemporal lobar degeneration43, and other mutant proteins, such as α-synuclein44 and tau45, accumulate in oligodendrocytes in patients with multiple system atrophy and other types of frontotemporal dementia (FTD). These observations raise the possibility that expression of aggregation-prone mutant proteins in oligodendrocytes compromises their ability to provide adequate support to neurons. Selective overexpression in oligodendrocytes of a mutant form of tau (P301L) associated with a human form of FTD (frontotemporal dementia and parkisonism linked to chromosome 17, FTDP-17) leads to formation of filamentous tau+ inclusions in these cells, eventual oligodendrocyte degeneration and reduced myelin31. These animals also exhibited impaired axonal transport, muscle atrophy and hind limb weakness, which are suggestive of motor neuron degeneration. We observed that oligodendrocyte degeneration in the spinal cord of ALS mice and demyelination in the brains of ALS patients were prominent in gray matter regions near motor neurons, indicating that oligodendrocytes in these regions are more susceptible to disease-related stresses. Notably, previous studies have shown that oligodendrocytes in the spinal cord gray matter are particularly vulnerable to endoplasmic reticulum stress when proteolipid protein, a component of myelin, is overexpressed46. Thus, the regional bias in oligodendrocyte vulnerability may depend more on metabolic demand, which is related to the type of associated neuron, than on the need for myelination.
Although some oligodendrocyte degeneration may be a secondary consequence of motor neuron death in ALS, motor axons represent only a minor portion of myelinated fibers in spinal cord gray matter. Given that each oligodendrocyte in the spinal cord forms on average ∼25 internodal segments47, each with a different axon, it seems likely that there would be sufficient remaining axons to promote oligodendrocyte survival, unless dying motor neurons produce pro-apoptotic signals. Furthermore, enhancing oligodendrogenesis without appropriate target axons to myelinate would appear maladaptive. The increase in ultrastructurally normal axons with immature myelin sheaths in ventral gray matter of end stage SOD1 (G93A) mice (Fig. 6a) suggests that oligodendrocytes attempt to remyelinate viable axons, rather than simply degenerate as a result of axon loss. Our results also indicate that selective degeneration of motor neurons is not sufficient to enhance the proliferation of NG2+ cells in the spinal cord (Supplementary Fig. 4), which is expected to occur if new oligodendrocytes are being formed17,23,24. Conversely, degeneration of oligodendrocytes and demyelination are sufficient to induce neuronal apoptosis, as seen after genetically induced oligodendrocyte ablation25,26 and in multiple sclerosis48. Thus, oligodendrocyte loss is expected to have a negative effect on the integrity and survival of motor neurons during the course of disease.
When do oligodendrocytes begin to degenerate in ALS? Although proliferation of NG2+ cells was enhanced well before behavioral onset of disease, their density did not increase, as there was a concomitant increase in the proportion of cells undergoing differentiation. If this increase in oligodendrogenesis compensates for cells that are lost or injured, as would be expected from the maintenance of oligodendrocyte number (Fig. 2), then oligodendrocyte degeneration is a very early event in the disease process. Despite the continuous regeneration of oligodendrocytes, the proportion of incompletely myelinated axons increased, myelin levels were reduced and oligodendrocytes failed to express MCT1, indicating that their maturation was impaired. In addition, adult-born oligodendrocytes exhibited morphological characteristics of apoptosis, suggesting that their turnover was accelerated. This expanding cycle of proliferation, differentiation and death in the oligodendrocyte lineage in ALS may accelerate damage to vulnerable motor neurons by consuming resources and triggering reactive changes in other glial cells (Supplementary Fig. 10).
The degeneration of oligodendrocytes and impaired maturation of oligodendrocyte progenitors in ALS share similarities to progressive forms of multiple sclerosis, where impaired remyelination following immune-mediated attack of oligodendrocytes leads to neuronal degeneration. Indeed, there is increasing appreciation of gray matter demyelination in multiple sclerosis49. Thus, therapeutic approaches that are being developed to treat multiple sclerosis, based on promotion of oligodendrocyte survival or enhancement of oligodendroglial metabolic support to neurons, may be helpful for preventing motor neuron degeneration in ALS.
Methods
Mice.
Pdgfra-creER5 and loxSOD1(G37R) (ref. 2) mice were generated and described previously. Plp1-creER50 mice were kindly provided by B. Popko (University of Chicago). SOD1 (G93A), Z/EG51, ROSA26-EYFP52 and ROSA26-mEGFP (mT/mG)53 mice were purchased from the Jackson Laboratory. Mobp-EGFP mice were generated by GENSAT54 and purchased from the Mutant Mouse Regional Resource Center. All experiments were carried out in strict accordance with protocols approved by the Animal Care and Use Committee at Johns Hopkins University.
BrdU labeling and Cre activity induction.
For continuous exposure to BrdU (Sigma), mice were provided with BrdU-containing drinking water (1 mg ml−1 supplemented with 1% sucrose), and received additional BrdU injections twice a day (50 mg per kg of body weight, intraperitoneal, at least 8 h apart) for 5 consecutive days. 4HT (Sigma H7904) was administered, 2 mg (to Plp1-creER; ROSA26-EYFP) or 4–5 mg (to Pdgfra-creER; Z/EG or Pdgfra-creER; mTmG) by intraperitoneal injection at either P30 or P60. For the excision of SOD1 (G37R), 4HT was injected to Pdgfra-creER; loxSOD1(G37R) mice at P18 (1 mg) and at P30∼31 (total 4 mg). Each mouse received up to two injections per day (1 mg per injection), at least 8 h apart.
Ricin injection.
After anesthesia with ketamine (120 mg per kg) and xylazine (8 mg per kg, intraperitoneal), the sciatic nerves of P20 mice were exposed and crushed with a fine forcep for 20 s, followed by 1 μl of 1 mg ml−1 ricin (Sigma) injection into the nerve in the proximal side of the crush, and 1 μl of phosphate-buffered saline (PBS) in the other side. The mice were killed 1 week later.
Immunohistochemistry.
Mice were deeply anesthetized with sodium pentobarbital (100 mg per kg) and perfused transcardially with 4% paraformaldehyde (PFA in 0.1 M phosphate buffer, pH 7.4). Spinal cords were further post-fixed overnight at 4 °C. For immunofluorescence, lumbar spinal cords were stored at 4 °C for more than 36 h in 30% sucrose solution (in PBS, pH 7.4), and then sectioned with a cryostat (35 or 25 μm thick). Immunofluorescence was performed on free-floating sections as described previously5. We used primary antibodies to APC (mouse, clone CC1, Calbiochem, OP80,1:50), BrdU (rat, clone BU1/75, Accurate, 1:500), cleaved caspase-3 (rabbit, Cell Signaling, Asp175, 1:500), ChAT (goat, Millipore, AB144P, 1:300), EGFP (goat, Frontier Institute, GFP-Go-Af1480-1, 1:500), EGFP (rabbit, a gift from R. Huganir, Johns Hopkins University, 1:500), GFAP (mouse, clone N206A/8, NeuroMab, 1:500), GFAP (rabbit, DAKO, Z0334,1:1,000), Iba1 (rabbit, Wako, 019-19741, 1:1,000), Ki67 (rabbit, Abcam, ab15580, 1:500), NG2 (guinea pig, Bergles' Lab, 1:4,000), Olig2 (guinea pig, 1:20,000, a gift from B. Novitch, University of California, Los Angeles), Olig2 (rabbit, Millipore, AB9610, 1:500), PDGFαR (rabbit, a gift from W. Stallcup, Burnham Institute, 1:500), SMI32 (mouse, Covance, SMI-32P, 1:1,000) and human SOD1 (rabbit, Rothstein Lab, 1:70). Secondary antibodies were Alexa Fluor 488– (Invitrogen, A-21206 and A-11055, 1:500), Cy3–, or DyLight 647–conjugated donkey F(ab')2 fragments to rabbit (711-166-152, and 712-606-152), goat (705-166-147), mouse (715-166-151 and 715-606-151), rat (712-166-150 and 712-606-153) or guinea pig (706-166-148 and 706-516-148) (Jackson ImmunoResearch). For Olig2, ALDH or APC (CC1) immunostaining, tissue sections were incubated in LAB solution (Polysciences) for 10 min before blocking. For BrdU staining, sections were pre-incubated in 2 N HCl at 37 °C for 30 min, followed by neutralization with 0.1 M sodium borate buffer (pH 8.5) before immunolabeling. DAPI was added during the secondary antibody incubation for most cases. Sections were mounted on slides with ProLong antifade reagent (Invitrogen).
For human tissue histopathological analysis, paraffin-embedded and fresh frozen human primary motor cortex and lumbar spinal cord sections (all 5 μm thick) were used. Sections were deparaffinized in a series of xylene, 100% alcohol, 95% alcohol, 70% alcohol and water, followed by antigen retrieval (boiling in 10 mM citrate buffer at pH 6.0 for 10 min). Sections were incubated with 3% H2O2 (vol/vol), blocked with 3% BSA in PBS, and incubated with the primary antibody for 60 min at 20–25 °C or at 4 °C overnight. We used primary antibodies to Iba1 (rabbit, Wako, 019-19741, 1:1,000), MBP (rabbit, Millipore, AB980,1:200) and NG2 (mouse, BD Science, 554275, 1:1,000). Sections were incubated with biotinylated secondary antibodies (Vector Lab), followed by VECTASTAIN Elite ABC reagent according to the manufacture's instruction (Vector Lab) and colorimetrically developed using the 3,3′-diaminobenzidine (DAB) substrate kit (Vector Lab). For double labeling of NG2 and Iba1, mounted fresh frozen sections (5 μm) were fixed with 4% PFA at 20–25 °C for 15 min, incubated with 3% H2O2, blocked with 3% BSA (wt/vol), and then incubated with antibody to NG2 at 4 °C overnight. Sections were incubated with HRP-labeled secondary antibody and Renaissance TSA Biotin system (PerkinElmer) according to manufacturer's instructions, and developed with DAB substrate. For Iba1 staining, sections was blocked with the BLOXALL solution (Vector Lab), followed by alkaline phosphatase–conjugated secondary antibody (Promega) and developed with NBT/BCIP substrate (Vector Lab).
Luxol fast blue myelin staining.
Tissue sections were stained with 0.1% luxol fast blue solution (vol/vol, Sigma) at 60 °C overnight. Sections were rinsed with 95% ethanol and water, reacted with 1% lithium carbonate solution (wt/vol) and 70% ethanol, followed by rinsing in water. Counter staining with hematoxylin and eiosin was performed as necessary.
Microscopy and cell counting.
Mounted slides were imaged using an epifluorescence microscope (Zeiss Axio-imager M1), and Axiovision software (Zeiss), or a confocal laser-scanning microscope (Zeiss LSM 510 Meta) using appropriate excitation and emission filters. A total of 3–5 sections were examined per mouse, and 3–4 mice were analyzed per each data point. Confocal images represent projected stacks of 15–45 images collected at 0.5–1.5-μm steps.
Volume measurement of oligodendrocyte cellular fragments.
EGFP+ elements of Mobp-EGFP mice that are not associated with Olig2 were quantified using a commercially available three-dimensional analysis package (Imaris, Bitplane). EGFP+ structures were defined by applying an absolute intensity threshold, followed by thresholding for structure size to remove small objects. Identical thresholds were applied to all images. EGFP+ structures that were also associated with Olig2 staining were deleted, as were structures which were positioned at the edge of the imaged field. The volume of Olig2− EGFP+ was scaled according to total imaged volume.
Electron microscopy.
Mice were perfused transcardially with 4% PFA/2.5% glutaraldehyde (vol/vol) in 0.1 M phosphate buffer under deep anesthesia, and brain tissue was isolated and post-fixed for 4 h at 4 °C. Brains were treated with 2% OsO4 (wt/vol) for 1 h, and washed in water. Samples were incubated in 2% uranyl acetate (wt/vol) for 30 min and dehydrated using 50%, 70%, 90%, 100% ethanol and propylene oxide. Samples were embedded in Epon 812 resin (Ted Pella). Ultrathin sections were obtained using Ultracut UCT (Leica) and stained with 2% uranyl acetate and lead citrate. Electron micrographs were taken with an H-7600 electron microscope (Hitachi). ImageJ was used to measure the diameter of axons and myelin sheath.
For pre-embedding immunoelectron microscopy, 4HT-injected Pdgfra-creER; ROSA26-mEGFP; SOD1 (G93A) and Pdgfra-creER; ROSA26-mEGFP mice (P60+60) were perfused transcardially with 4% PFA/0.1% glutaraldehyde in 0.1 M phosphate buffer under deep anesthesia. After blocking with 5% normal donkey serum (vol/vol) in PBS, spinal cord sections (50 μm thick) were incubated overnight with rabbit antibody (IgG) to EGFP and then with antibody (IgG) to rabbit conjugated to 1.4-nm gold particles (Nanoprobes). Following silver enhancement (HQ silver, Nanoprobes), sections were osmificated, dehydrated and embedded in Epon 812 resin. Ultrathin sections were prepared with an ultramicrotome (Leica) and stained with 2% uranyl acetate.
Clinical materials.
Human autopsy materials, including frozen and paraffin-embedded control and ALS patient cortex and spinal cord tissues, were obtained from the Johns Hopkins University ALS Clinic, Brain Resource Center/Department of Pathology, VA Biorepository Brain Bank. All experiments were performed using motor cortex, cervical and/or lumbar spinal cord tissues. All available demographic information for the ALS and control cases analyzed in this study is presented in Supplementary Table 1.
Western blotting.
Lumbar spinal cords were collected from control and SOD1 (G93A) mice, snap frozen in liquid nitrogen and then stored at −80 °C until use. For human cortex protein analysis, the gray matter and white matter were separated grossly. The tissues were homogenized in RIPA buffer (Thermo) supplemented with proteinase inhibitor (Roche) and phosphatase inhibitor (Calbiochem) on ice. 30 μg of protein was separated on 4–12% gradient Bis-Tris gels (BioRad) or NuPage and transferred to nitrocellulose membranes (BioRad). After blocking with 5% nonfat milk (wt/vol), membranes were probed with primary antibodies at 4 °C overnight, following by HRP-conjugated secondary antibodies, Supersignal Pico detection reagent (Pierce), and then exposed to HyBlot CL autoradiography film (Denville). The intensity for each band was quantified with ImageJ software. We used antibodies to CNPase (mouse, Millipore, MAB326, 1:1,000), MBP (mouse, Millipore, AB980, 1:2,000), PDGFαR (rabbit, Santa Cruz, sc-338, 1:200), GAPDH (rabbit, Cell Signaling, 2118, 1:1,000) and MCT1 (rabbit, Santa Cruz, sc-50325, 1:100).
Analysis of disease progression.
For Pdgfra-creER; loxSOD1(G37R) mice, the time of disease onset, early disease and end stage were defined as the time when mouse body weight reached the peak, body weight declined to 10% of the maximum weight and paralyzed mice could not right themselves within 20 s when placed on their side respectively, as previously described2. The total disease duration was defined as the duration between disease onset and end stage. Mice were weighted once a week when they were 150 d old.
Quantitative PCR determination of transgene excision in NG2+ cells.
The brain neural cells from 2-month-old mice (with and without 4HT injection, n = 3 each group) were dissociated using Dissociation Kit (P) (Milteny) by following the manufacturer's instructions. Myelin was removed by using Percoll generated gradient. Live cell fraction was collected and the cells were submitted to staining for PDGFαR (1:200, Santa Cruz) followed by Alexa 488–conjugated antibody to rabbit before fluorescence-activated cell sorting (FACS). Dead cells were excluded by propidium iodide staining during sorting using MoFlo MLS high-speed cell sorter (Beckman Coulter) at Johns Hopkins School of Public Health FACS core. Only PDGFαR+ and propidium iodide− cells were sorted for transgene evaluation. Genomic DNA was extracted by using QIAmp DNA micro kit (Qiagen) following the manufacturer's instructions. qPCR for SOD1 (G37R) transgene was performed using the primers and cycler parameters on ABI Plusone cycler as described previously2.
Statistical analysis.
All data are presented as mean ± s.e.m. For multiple groups, data were evaluated by one-way ANOVA and further evaluated with Tukey post hoc comparisons. Otherwise, unpaired Student's t test was applied. P < 0.05 was considered to be statistically significant. The paired Student's t test was used to evaluate contralateral and ipsilateral side of cell counts in ricin-injected mouse experiments. Log-rank test was used for survival curve analysis. Mann Whitney test was used for disease duration.
References
Clement, A.M. et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117 (2003).
Boillée, S. et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 (2006).
Yamanaka, K. et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 (2008).
Ilieva, H., Polymenidou, M. & Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772 (2009).
Kang, S.H., Fukaya, M., Yang, J.K., Rothstein, J.D. & Bergles, D.E. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681 (2010).
Magnus, T. et al. Adult glial precursor proliferation in mutant SOD1G93A mice. Glia 56, 200–208 (2008).
Nave, K.A. Myelination and support of axonal integrity by glia. Nature 468, 244–252 (2010).
Nave, K.A. & Trapp, B.D. Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 31, 535–561 (2008).
Lee, Y. et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448 (2012).
Suzuki, A. et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823 (2011).
Rinholm, J.E. et al. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci. 31, 538–548 (2011).
Niebroj-Dobosz, I., Rafalowska, J., Fidzianska, A., Gadamski, R. & Grieb, P. Myelin composition of spinal cord in a model of amyotrophic lateral sclerosis (ALS) in SOD1G93A transgenic rats. Folia Neuropathol. 45, 236–241 (2007).
Cosottini, M. et al. Magnetization transfer imaging demonstrates a distributed pattern of microstructural changes of the cerebral cortex in amyotrophic lateral sclerosis. AJNR Am. J. Neuroradiol. 32, 704–708 (2011).
Rivers, L.E. et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 11, 1392–1401 (2008).
Zhu, X. et al. Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–753 (2011).
Gurney, M.E. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994).
Dal Canto, M.C. & Gurney, M.E. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 676, 25–40 (1995).
Woodruff, R.H., Tekki-Kessaris, N., Stiles, C.D., Rowitch, D.H. & Richardson, W.D. Oligodendrocyte development in the spinal cord and telencephalon: common themes and new perspectives. Int. J. Dev. Neurosci. 19, 379–385 (2001).
Guo, F. et al. Pyramidal neurons are generated from oligodendroglial progenitor cells in adult piriform cortex. J. Neurosci. 30, 12036–12049 (2010).
Davalos, D. et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 (2005).
Jamin, N., Junier, M.P., Grannec, G. & Cadusseau, J. Two temporal stages of oligodendroglial response to excitotoxic lesion in the gray matter of the adult rat brain. Exp. Neurol. 172, 17–28 (2001).
Wu, Y.J., Tang, Y.F., Xiao, Z.C., Bao, Z.M. & He, B.P. NG2 cells response to axonal alteration in the spinal cord white matter in mice with genetic disruption of neurofilament light subunit expression. Mol. Neurodegener. 3, 18 (2008).
Coutts, M., Kong, L.X. & Keirstead, H.S. A model of motor neuron loss: selective deficits after ricin injection. J. Neurotrauma 27, 1333–1342 (2010).
Henson, P.M., Bratton, D.L. & Fadok, V.A. Apoptotic cell removal. Curr. Biol. 11, R795–R805 (2001).
Locatelli, G. et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat. Neurosci. 15, 543–550 (2012).
Pohl, H.B. et al. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J. Neurosci. 31, 1069–1080 (2011).
Woodruff, R.H., Fruttiger, M., Richardson, W.D. & Franklin, R.J. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol. Cell Neurosci. 25, 252–262 (2004).
Chang, A., Nishiyama, A., Peterson, J., Prineas, J. & Trapp, B.D. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. 20, 6404–6412 (2000).
Pouly, S., Becher, B., Blain, M. & Antel, J.P. Expression of a homologue of rat NG2 on human microglia. Glia 27, 259–268 (1999).
Bu, J., Akhtar, N. & Nishiyama, A. Transient expression of the NG2 proteoglycan by a subpopulation of activated macrophages in an excitotoxic hippocampal lesion. Glia 34, 296–310 (2001).
Higuchi, M. et al. Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J. Neurosci. 25, 9434–9443 (2005).
Yazawa, I. et al. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45, 847–859 (2005).
Horner, P.J., Thallmair, M. & Gage, F.H. Defining the NG2-expressing cell of the adult CNS. J. Neurocytol. 31, 469–480 (2002).
Fünfschilling, U. et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521 (2012).
Bruijn, L.I. et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 (1997).
Pramatarova, A., Laganiere, J., Roussel, J., Brisebois, K. & Rouleau, G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21, 3369–3374 (2001).
Rothstein, J.D., Van Kammen, M., Levey, A.I., Martin, L.J. & Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84 (1995).
Nagai, M. et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622 (2007).
Gong, Y.H., Parsadanian, A.S., Andreeva, A., Snider, W.D. & Elliott, J.L. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis, but does not cause motoneuron degeneration. J. Neurosci. 20, 660–665 (2000).
Yamanaka, K. et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 105, 7594–7599 (2008).
Aebischer, J. et al. Elevated levels of IFNgamma and LIGHT in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Eur. J. Neurol. 19, 752–759 (2012).
Buntinx, M. et al. Cytokine-induced cell death in human oligodendroglial cell lines. I. Synergistic effects of IFN-gamma and TNF-alpha on apoptosis. J. Neurosci. Res. 76, 834–845 (2004).
Neumann, M. et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 66, 177–183 (2007).
Tu, P.H. et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 44, 415–422 (1998).
Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Brain Pathol. 9, 663–679 (1999).
Bauer, J. et al. Endoplasmic reticulum stress in PLP-overexpressing transgenic rats: gray matter oligodendrocytes are more vulnerable than white matter oligodendrocytes. J. Neuropathol. Exp. Neurol. 61, 12–22 (2002).
Chong, S.Y. et al. Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc. Natl. Acad. Sci. USA 109, 1299–1304 (2012).
Dutta, R. & Trapp, B.D. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology 68, S22–S31 (2007).
Rudick, R.A. & Trapp, B.D. Gray-matter injury in multiple sclerosis. N. Engl. J. Med. 361, 1505–1506 (2009).
Doerflinger, N.H., Macklin, W.B. & Popko, B. Inducible site-specific recombination in myelinating cells. Genesis 35, 63–72 (2003).
Novak, A., Guo, C., Yang, W., Nagy, A. & Lobe, C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28, 147–155 (2000).
Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001).
Muzumdar, M.D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Gong, S. et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925 (2003).
Acknowledgements
We thank N. Ye, I. Srivastava, S. Singh and T. Le for their excellent technical support, M. Pucak for help with Imaris software operation and quantitative analysis, J. Carmen for her contributions to the Cre/lox animal study, and H. Zhang at the Johns Hopkins University School of Public Health FACS core for assistance with NG2+ cell isolation. We thank B. Trapp (Cleveland Clinic Lerner Research Institute) for advice regarding human NG2+ cell staining, R. Dutta (Cleveland Clinic Lerner Research Institute) for advice regarding protein extraction from human tissues and B. Popko (University of Chicago) for providing Plp1-creER mice. Human samples were provided by R. Bowser (Barrow Neurological Institute), K. Trevor (SACTL-VA Biorepository Trust), T. Hyde (Lieber Institute for Brain Development and the Johns Hopkins School of Medicine), J. Glass (Emory Alzheimer's Disease Research Center, 5P50AG025688-07), and the Johns Hopkins School of Medicine Department of Neuropathology. We thank E. Mosmiller for helping with patient demographic information. We also thank A. Agarwal and A. Langseth for critical discussions. This work was supported by grants from P2ALS (D.E.B. and J.D.R.), the US National Institutes of Health (NS27036 to D.W.C., NS33958 to J.D.R. and NS051509 to D.E.B.), the ALS Association (4ZMUDE to Y.L.), the Robert Packard Center for ALS Research at Johns Hopkins, and the Brain Science Institute.
Author information
Authors and Affiliations
Contributions
S.H.K., Y.L., J.D.R. and D.E.B. designed the experiments. S.H.K. carried out the experiments involving NG2+ cell proliferation, cell fate analysis of NG2+ cells and oligodendrocytes, and immunohistochemical analysis of oligodendrocyte structure (Figs. 1,2,3,4,5,6 and Supplementary Figs. 1–3 and 5). Y.L. performed western blot analysis from SOD1 (G93A) mice (Supplementary Fig. 7) and ALS patients (Supplementary Fig. 8), histological analysis of human tissue (Fig. 7), analysis of ricin-injected mice (Supplementary Fig. 4), and analysis of the SOD1 (G37R)–deleted mice (Fig. 8 and Supplementary Fig. 9). M.F. performed the electron microscopic and immunogold analysis (Fig. 6 and Supplementary Fig. 6). I.L. assisted with the ricin injections. D.W.C. provided the loxSOD1 (G37R) mice. L.W.O. provided assistance with analysis of the human ALS samples. S.H.K., Y.L., J.D.R. and D.E.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–11 and Supplementary Table 1 (PDF 3864 kb)
Rights and permissions
About this article
Cite this article
Kang, S., Li, Y., Fukaya, M. et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16, 571–579 (2013). https://doi.org/10.1038/nn.3357
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.3357
This article is cited by
-
Disruption of MAM integrity in mutant FUS oligodendroglial progenitors from hiPSCs
Acta Neuropathologica (2024)
-
Axonal energy metabolism, and the effects in aging and neurodegenerative diseases
Molecular Neurodegeneration (2023)
-
Regulation of cortical hyperexcitability in amyotrophic lateral sclerosis: focusing on glial mechanisms
Molecular Neurodegeneration (2023)
-
Elevated peripheral levels of receptor-interacting protein kinase 1 (RIPK1) and IL-8 as biomarkers of human amyotrophic lateral sclerosis
Signal Transduction and Targeted Therapy (2023)
-
Integrative transcriptomic analysis of the amyotrophic lateral sclerosis spinal cord implicates glial activation and suggests new risk genes
Nature Neuroscience (2023)
