
Abstract
AUTS2 syndrome is characterized by low birth weight, feeding difficulties, intellectual disability, microcephaly and mild dysmorphic features. All affected individuals thus far were caused by chromosomal rearrangements, variants at the base pair level disrupting AUTS2 have not yet been described. Here we present the full clinical description of two affected men with intragenic AUTS2 variants (one two-base pair deletion in exon 7 and one deletion of exon 6). Both variants are de novo and are predicted to cause a frameshift of the full-length transcript but are unlikely to affect the shorter 3′ transcript starting in exon 9. The similarities between the phenotypes of both men are striking and further support that AUTS2 syndrome is a single gene disorder.
Similar content being viewed by others
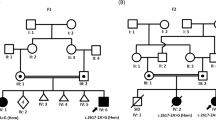


Introduction
Disruption of AUTS2 by translocations, inversions or deletions causes a syndromic form of intellectual disability.1 Forty-four pathogenic disruptions of AUTS2 have been described: 6 translocations and 2 inversions with one breakpoint in AUTS2 and 36 deletions (with a size of 50 kb to 4.5 Mb) containing at least one exon and a maximum of two downstream genes. In most cases parental DNA was available. De novo occurrence could be confirmed in the majority of the patients, but in five families the deletions were inherited from an affected parent (twice), an unaffected father (once) or a parent of whom no clinical data were available (twice).1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 A detailed study of 17 affected individuals with a disruption of AUTS2 revealed a distinct AUTS2 syndrome characterized by intellectual disability, microcephaly, mild short stature, feeding problems, hypertonia or hypotonia and facial features, including ptosis, highly arched eyebrows, narrow mouth and micro/retrognathia.1 The AUTS2 syndrome severity score, expressed as the sum of all features seen more than once in unrelated affected individuals, is a measure of the severity and specificity of the phenotype. The median AUTS2 syndrome severity score of individuals with a genomic rearrangement/deletion involving the 3′ region of AUTS2 was significantly higher than that of individuals with 5′ deletions. The 3′ end of the gene harbors an alternative transcript that is (like the full-length transcript) expressed in human brain and starts in exon 9. The short transcript is able to rescue the phenotype of AUTS2 zebrafish morphants. These two observations indicate that the 3′ region of AUTS2 contains important functional domains.1
Here we report a deletion of two nucleotides, the first pathogenic variant at the base pair level, in a young adult with a syndromic form of intellectual disability. His phenotype is compared with another new individual with AUTS2 syndrome of similar age and gender with an intragenic deletion of exon 6 (NG_034133.1). Both variants result in a frameshift and only disrupt the full-length transcript. Both adult men have a very similar phenotype and fit the AUTS2 syndrome, confirming it to be a single gene disorder that can be caused by disrupting the full-length transcript only and reemphasizing the importance of AUTS2 in neurodevelopment.
Subjects and methods
Clinical reports
Proband 1 is the second child of healthy nonconsanguineous parents. He has a healthy older sister and younger brother. He was born after 40 weeks of gestation. The pregnancy was complicated by growth restriction from 6 months of gestation. Apgar scores were 9 and 10 after 1 and 5 minutes, respectively. Birth weight was 2745 g (<2nd percentile) and length was 49 cm (10th percentile). In the first year, feeding difficulties and mild delay in development and eye contact were observed. At the age of 1 year he got a tympanostomy, after which his social interaction improved. Intellectual development remained delayed. He started walking at 20 months of age with a tendency to walk on his toes and started talking at 2.5 years of age. He has been operated for a misalignment of his feet and for cryptorchidism.
Physical examination at the age of 3 years showed a height of 92 cm (p10), a weight of 12.2 kg (<p10) and a head circumference (OFC) of 46 cm (<p10), mild ptosis, narrow os frontale and down-slanting palpebral fissures. Neurological examination at that time revealed an immature motor function with normal muscle tone, no paresis, no extrapyramidal movement disorder, no ataxia and normal to high deep tendon reflexes, and a normal sensibility. Psychological evaluation revealed a global developmental delay with a severe delay in speech and language development and a pervasive developmental disorder not further classified. Hyperactive behavior and sound sensitivity were also observed. CT of the brain was performed and showed no anomalies. A nerve conduction study was normal. The Brainstem Auditory Evoked Potential (BAEP) was normal and electroencephalogram (EEG) was diffusely abnormal with too many slow waves and diffuse beta-waves but no signs of epilepsy. Chromosomal analysis showed a normal male karyotype (46,XY). At the age of 13 years he was evaluated by a clinical geneticist. Fragile-X testing was negative and no other causal diagnosis could be made. Reevaluation at the age of 24 years showed a length of 177 cm (−1 SD) and a head circumference of 52.5 cm (−3 SD) and mild dysmorphisms, thick eyebrows, a mild down slant of the palpebral fissures, a deep nasal bridge and a prominent nasal tip with a broad nasal base, a wide mouth with thick lips and a short mildly upturned philtrum. He has a slender build, a long neck, long fingers and contractures of the proximal interphalangeal (PIP) joints of digit V of both hands (see Figure 1 and Table 1). He has a moderate intellectual disability with a severe language delay and an autism spectrum disorder. His IQ was tested to be 45. Array analysis, metabolic screening and sequencing of the PQBP1 gene to exclude Renpenning syndrome were normal (data not shown).

(a) Proband 1 at the age of 24 years. (b) Proband 2 at the age of 20 years. These photographs show the slender build, the mild ptosis, small ears, deep nasal bridge, prominent nasal tip and broad nasal base, the thick lips and short philtrum in both men. The mild down-slanting palpebral fissures and the camptodactyly of the thumbs without other abnormalities of the hands were seen in proband 1, and shallow palmar creases of the PIP joints on digit V in proband 2. Parental consent was obtained.
The second proband is a 20-year-old male. He is the second child of healthy nonconsanguineous parents and has a healthy older brother. The pregnancy and delivery were uneventful. His birth weight was 3200 g (15th percentile), length was 49 cm (10th percentile), and head circumference was 34 cm (25th percentile) at a gestational age of 40 weeks. Apgar scores were 2/5/10 after 1, 5 and 10 minutes respectively. He had feeding difficulties and a poor weight gain. Evaluation for failure to thrive that occurred in the first month of his life did not reveal an underlying cause. His social and motor development was delayed. He started walking at the age of 2 years. He was hypertonic, had joint contractures and frequent infections as an infant. His joint contractures improved significantly with physiotherapy as did his hypertonia. Extensive evaluation by a pediatric neurologist at the age of 2 years and 10 months did not reveal a cause for the motor delay. Hypertonia was not objectified anymore, but he still had a camptodactyly of his thumbs and immature, stiff movements. EEG, BAEP, electromyography, visual evoked potential, CT and MRI brain, metabolic screening, karyotyping and DNA analysis for fragile-X were all normal (data not shown). Eye examination revealed strabismus. He visited a special school where he was tested to have an IQ of 74 at the age of 5 years with a disharmonic profile, verbal IQ of 87 and performance IQ of 64. He had signs of autism, namely poor interaction with peers, obsessions, stereotypic behavior and movements. His personality was described as kind and calm.
Now at the age of 20 years, he still has stiff and immature movements. He has some degree of independence (he can dress and feed himself and is able to use public transport), but needs too much assistance in his daily care to live independently. A recent IQ test is not available, but based on his educational and independence level his IQ is estimated to be between 60 and 70. Physical examination reveals a slender build and a height of 175 cm (−1 SD), a weight of 52.5 kg (−2 SD) and a head circumference of 52 cm (−3.2 SD). He has a mild brachycephaly, low frontal hairline, thick hair, highly arched eyebrows, mild ptosis and low-set small but normal-formed ears. He has a prominent nasal tip, a low nasal bridge and a broad nasal base. He has a short philtrum, thick lips, normal palate and normal teeth (see Figure 1 and Table 1). Limb proportions are normal. His hands are long and narrow with absent/shallow dermatoglyphics of the distal interphalangeal joints – joint movement is normal. He has highly arched feet, hammer toes and a valgus deformity of his left foot. Neurologic examination showed mild peripheral hypertonia with normal muscle strength of hands and feet, biceps and quadriceps. He has stiff movements and a poor coordination. Deep tendon reflexes are normal except for the high reflexes at his Achilles tendon and a clonus of the right ankle.
Written consent was obtained from the parents of both men for using the clinical and genetic information in this study and for publishing their photographs.
Genomic DNA was isolated from blood samples using standard procedures.
Exome sequencing (Proband 1)
Exome enrichment of DNA samples of proband 1 and both parents was performed with the SureSelectXT human All Exon 50 Mb kit (Agilent, Santa Clara, CA, USA), followed by sequencing on a SOLiD 5500XL machine (Life Technologies, Leusden, the Netherlands). For data analysis LifeScope software (Life Technologies) was used. The mean coverage is >20%.12 We selected candidate de novo variants by excluding common variants and variants inherited from either parent as described before.13
Candidate de novo variants were validated by conventional Sanger sequencing methods in DNA samples obtained from the proband and his parents. Primer sequences and PCR conditions are available upon request.
Array analysis (Proband 2)
SNP array analysis was performed using a HumanCytoSNP-12 Chip following standard protocols as provided by the manufacturer on an iScan System (Illumina, San Diego, CA, USA). CNV analysis was performed using CNV-WebStore (http://sourceforge.net/projects/cnv-webstore/).14
Results
Standard diagnostic exome sequencing of proband 1 and his healthy parents revealed two de novo variants that could be confirmed with Sanger sequencing. These two variants were submitted to the LOVD gene variant database at www.LOVD.nl/AUTS2 and www.LOVD.nl/ABI2 (patient ID 0016365). The first variant was predicted to cause a frameshift in exon 7 of AUTS2, NM_015570.2: c.857_858delAA (p.(Lys286fs)) (hg 19, build 37). Exon numbering is according to NG_034133.1. This variant is predicted to cause haploinsufficiency of the longest AUTS2 transcript. The variant lies outside a shorter transcript that starts in exon 9.1 The other de novo variant was found in Abl-interactor 2 (ABI2), NM_005759.4: c.1223C>T, p.(Pro408Leu) (hg 19, build 37). This regards a highly conserved amino acid of the ABI2. This protein is a fusion partner of MLL in acute myeloid leukemia cells. No germ-line mutations in this gene have been described in human so far.15 Missense mutations in the ABI2 gene are not described to be pathogenic. In contrast, knock-down experiments in mouse neurons showed that loss of function of the ABI2 protein causes dysfunction of the WAVE2–ABI2 complex that seems to be involved in neuronal migration.16 It is very likely that the AUTS2 variant causes the phenotype in this individual because of its resemblance to individuals with AUTS2 deletions. It can however not be excluded that the de novo variant in ABI2 has an additional effect on the phenotype.
Array analysis of proband 2 revealed a small intragenic deletion encompassing exon 6 of AUTS2 (NG_034133.1), chr7.hg 19:g.(69985843_69991859)_(70,221,259_70,228,020)del (ISCN nomenclature: arr 7q11.22(69,985,843 × 2,69,991,859-70,221,259 × 1,70,228,020 × 2)dn (hg 19, build 37)) (submitted to www.LOVD.nl/AUTS2 (patient ID 0016366)). This deletion is predicted to cause a frameshift of the full-length transcript and does not affect the shorter 3′ transcript.1 No other rare CNVs were found. The deletion was not detected in the parents.
Clinical evaluation of both probands resulted in an AUTS2 syndrome severity score of 12 in proband 1 and 16 in proband 2, see also Table 1.1
Discussion
We describe two new adult individuals with AUTS2 syndrome, one with a two-nucleotide deletion and one with a single exon deletion. To our knowledge this is the first report of a person with a pathogenic variant in AUTS2 at the base pair level. Both probands have comparable age and sex and a genomic defect that causes an early frameshift in the full-length AUTS2 transcript, likely to cause haploinsufficiency. Both men have intellectual disability, an autism spectrum disorder, feeding difficulties after birth, mild distal joint contractures and mild dysmorphic features. These clinical features are consistent with the phenotypic spectrum reported in the AUTS2 syndrome. As with other individuals with AUTS2 syndrome, the facial dysmorphisms are subtle, but show clear similarities to each other and other adult individual with AUTS2 syndrome (see Figures 1 and 2 of Beunders et al1). Especially the morphology of the eyes (mild ptosis, short palpebral fissures and arched eyebrows), nose (prominent nasal tip and a broad nasal base) and the prominent midface is characteristic.1, 6, 7 However, both men have a rather wide mouth with thick lips, whereas almost 60% of the individuals with AUTS2 syndrome in literature have a narrow mouth (see Table 1).1 The misalignment of the feet and the mild camptodactyly, with absent or shallow palmar creases that have been described as infrequent features of the AUTS2 syndrome, are present in both men. Detailed clinical studies of more affected individuals will be necessary to delineate the frequency of these features as they may be very subtle and can easily be missed. Cryptorchidism has not been described before, but as it is a frequent birth defect, this might be a coincidental finding. The AUTS2 syndrome severity scores of 12 and 16 are similar to the average score of 15 other persons with a genomic rearrangement that affects the C-terminal domain of AUTS2 (average of 12.8).1

(a) Schematic representation of the AUTS2 gene and the variants of probands 1 and 2. (b) Exome sequencing result of proband 1. The two-nucleotide deletion at the genomic position g.70227971 is shown in purple. The blue and green lines indicate the reads of the parents (only part of the reads are shown in this overview). The Sanger sequence result of the forward strand shows the frameshift (upper) and is compared with the normal Sanger sequence (lower). (c) The array results of proband 2 show the deletion of exon 6 (NG_034133.1) as indicated by the log R ratio of −0,5 and the absence of heterozygosity.
Both deletions in AUTS2 described here are likely to cause haploinsufficiency. The variants cause a frameshift and the messenger RNA formed is expected to be subject to nonsense-mediated decay in both cases. A dominant-negative effect of the variants is unlikely because heterozygous deletions of the entire AUTS2 gene also cause AUTS2 syndrome.1 Furthermore, several variants have been reported that affect only the full-length AUTS2 transcript and not the shorter transcript starting in exon 9. Comparison of the AUTS2 syndrome severity score (ASSS, determined as described in Beunders et al1) of patients with variants that disrupt the C terminus of the long isoform only (the patients described here and patients 5–8 and 16–17, Beunders et al1) and patients with variants that affect both the long and the short isoform of AUTS2 (patients 9–15, Beunders et al1) shows comparable scores for both groups. The mean ASSS is 11.5 in the first group and 13.6 in the last, which is not significantly different (Kolmogorov–Smirnov test). (The exons mentioned here are numbered like in NG_034133.1). Although the number of patients is small, the typical presentation of AUTS2 syndrome in these affected individuals, where normal levels of the shorter transcript are expected, indicates that the shorter transcript can not compensate for loss of the normal full-length transcript in humans. Nevertheless, this shorter transcript can rescue the microcephaly phenotype of AUTS2 zebrafish morphants, strongly suggesting that it contains important information for proper protein function.
In conclusion, our findings confirm the significance of AUTS2 in neurodevelopment and show that a small frameshift mutation in AUTS2 can cause AUTS2 syndrome.
References
Beunders G, Voorhoeve E, Golzio C et al: Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet 2013; 92: 210–220.
Amarillo IE, Li WL, Li X, Vilain E, Kantarci S : De novo single exon deletion of AUTS2 in a patient with speech and language disorder: A review of disrupted AUTS2 and further evidence for its role in neurodevelopmental disorders. Am J Med Genet 2014; 164: 958–965.
Bakkaloglu B, O'Roak BJ, Louvi A et al: Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet 2008; 82: 165–173.
Girirajan S, Brkanac Z, Coe BP et al: Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 2011; 7: e1002334.
Huang XL, Zou YS, Maher TA, Newton S, Milunsky JM : A de novo balanced translocation breakpoint truncating the autism susceptibility candidate 2 (AUTS2) gene in a patient with autism. Am J Med Genet A 2010; 152A: 2112–2114.
Jolley A, Corbett M, McGregor L et al: De novo intragenic deletion of the autism susceptibility candidate 2 (AUTS2) gene in a patient with developmental delay: a case report and literature review. Am J Med Genet A 2013; 161: 1508–1512.
Kalscheuer VM, FitzPatrick D, Tommerup N et al: Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum Genet 2007; 121: 501–509.
Nagamani SC, Erez A, Ben-Zeev B et al: Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur J Hum Genet 2013; 21: 343–346.
Sultana R, Yu CE, Yu J et al: Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 2002; 80: 129–134.
Talkowski ME, Rosenfeld JA, Blumenthal I et al: Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012; 149: 525–537.
Tropeano M, Ahn JW, Dobson RJ et al: Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS One 2013; 8: e61365.
Neveling K, Feenstra I, Gilissen C et al: A Post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 2013; 34: 1721–1726.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG : Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012; 367: 1921–1929.
Vandeweyer G, Reyniers E, Wuyts W, Rooms L, Kooy RF : CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinformatics 2011; 12: 4.
Coenen EA, Zwaan CM, Meyer C et al: Abl-interactor 2 (ABI2): a novel MLL translocation partner in acute myeloid leukemia. Leuk Res 2012; 36: e113–e115.
Xie MJ, Yagi H, Kuroda K et al: Wave2-ABi2 complex controls growth cone activity and regulates the multipolar-bipolar transition as well as the initiation of glia-guided migration. Cereb Cortex 2013; 23: 1410–1423.
Acknowledgements
We are grateful to the families for their cooperation. We thank the Genome Diagnostics laboratories of the VU University Medical Center, Amsterdam, The Netherlands; the Radboud University Medical Centre, Nijmegen, The Netherlands; and Antwerp University Hospital, Antwerp, Belgium, for their assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Beunders, G., de Munnik, S., Van der Aa, N. et al. Two male adults with pathogenic AUTS2 variants, including a two-base pair deletion, further delineate the AUTS2 syndrome. Eur J Hum Genet 23, 803–807 (2015). https://doi.org/10.1038/ejhg.2014.173
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.173
