
Abstract
An accumulation of misfolded proteins in the endoplasmic reticulum (ER) triggers the unfolded protein response (UPR) mediated via the activation of three transmembrane proteins IRE1, PERK and ATF6. Signalling through these proteins is aimed at enhancing the ER folding capacity and reducing the folding load. If these processes fail to re-establish protein homeostasis within the ER, then cell death prevails via apoptosis. How the shift from pro-survival to pro-apoptotic signalling is regulated remains unclear with both IRE1 and PERK signalling associated with pro-survival as well as pro-apoptotic signalling. To investigate the temporal activation of IRE1 and PERK in live cells and their relationship to cellular fate, we devised single cell reporters for both ER stress signalling branches. SH-SY5Y neural cells stably expressing these fluorescent protein reporter constructs to monitor IRE1-splicing activity and PERK-mediated ATF4-translation were imaged using single cell and high content time lapse live cell microscopy. We could correlate an early onset and attenuation of XBP1 splicing in the IRE1-reporter cells as cytoprotective. Indeed, silencing of IRE1 expression using shRNA inhibited splicing of XBP1 resulting in an early onset of cell death. In contrast, in the PERK-reporter cells, we observed that a slow rate of ATF4-translation and late re-initiation of general translation coincided with cells which were resistant to ER stress-induced cell death. Interestingly, whereas silencing of PERK did not affect overall levels of cell death in response to ER stress, it did increase sensitivity to ER stressors at early time points following treatment. Our results suggest that apoptosis activation in response to ER stress is not caused by a preferential activation of a single UPR branch, or by a switch from one branch to the other. Rather, our data indicated that the relative timing of IRE1 and PERK signalling determines the shift from cell survival to apoptosis.
Similar content being viewed by others


Main
The endoplasmic reticulum (ER) provides an environment for the folding and posttranslational modification of proteins in eukaryotic cells. Stresses that lead to a build-up of unfolded proteins in the ER activate a signalling network called the unfolded protein response (UPR), which activates the IRE1, PERK and ATF6 signalling pathways resulting in the re-establishment of cell homeostasis or, if the stress remains unresolved, result in apoptosis. Activation of the endonuclease activity of IRE1 leads to unconventional splicing of Xbp1 mRNA, resulting in the translation of the active transcription factor XBP1s.1 The functions of genes upregulated by XBP1s are aimed at clearing the ER of unfolded proteins,2, 3 thus splicing of XBP1 is generally thought to enhance pro-survival functions. However, IRE1 endonuclease activity has been shown to splice additional mRNAs and microRNAs, which has been interpreted both as a pro-survival mechanism when occurring early during ER stress and as contributing to apoptosis when occurring late during ER stress.4, 5, 6, 7 Additionally, it has been shown that IRE1-mediated splicing is eventually attenuated despite continuous ER stress. This has been proposed to be a switch into cell death facilitated by the pro-apoptotic outputs of the PERK signalling pathway.8 However, in the light of potential late pro-apoptotic IRE1 signalling outputs, it might be the case that attenuation of IRE1-activity is protective.
Similiarly, activation of the PERK signalling cascade leads to pro-survival as well as pro-apoptotic outputs. ER stress-activated PERK phosphorylates eIF2α resulting in general translational attenuation, which is considered cytoprotective as it reduces the load of newly synthesised proteins in the ER.9 Phosphorylation of eIF2α also allows for specific initation of translation from the of 5’UTR of transcription factor ATF4.10, 11 Among the transcriptional targets of ATF4 is CHOP, which has been associated with pro-apoptic signalling.12 ATF4 and CHOP can interact in the induction of their targets, which include genes involved in protein synthesis and amino acid synthesis and transport.13 Furthermore, GADD34, a regulatory subunit of the protein phosphatase 1 complex, which dephosphorylates eIF2α, is induced by ATF4.14 Dephosphorylation of eIF2α allows re-initiation of general translation, which has been proposed to lead to cell death through increase in protein synthesis and load in the ER leading to an increase in ROS.13
To address the dual roles of IRE1 and PERK signalling in triggering ER stress-dependent cell death, we chose a single cell time lapse imaging approach. This technique allowed monitoring the activation patterns of the IRE1 and PERK signalling pathways in real time on a single cell level. Thus, we were able to resolve the heterogeneity of responses within the population and link particular activation patterns of IRE1-mediated splicing of Xbp1 or PERK-dependent translation of ATF4 to cell survival or cell death. High content time lapse imaging of the effect of silencing IRE1 and PERK signalling further complemented our findings. We found that the specific timing of IRE1 and PERK signalling, rather than a switch between these UPR signalling branches was crucial for individual cell survival or cell death.
Results
Generating a reporter cell system to monitor IRE1 and PERK activity at single cell level
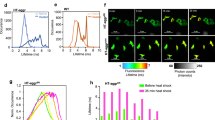
To investigate activation of the UPR on a single cell level, we created fluorescent protein reporters for the IRE1 and the PERK signalling pathways. IRE1 activity was monitored by constructing a vector encoding the full length spliced XBP1 protein in frame with yellow fluorescent protein (YFP). Under ER stress, a 26 bp intron is spliced from the Xbp1-Yfp mRNA by the endonuclease activity of IRE1, leading to a frame shift and translation of the XBP1s-YFP fusion protein (Figure 1a). The reporter construct was stably transfected into human SH-SY5Y neuroblastoma cells. To test the responsiveness of the IRE1-reporter, cells were treated with 3 μM Thapsigargin (Tg). After 24 h, YFP fluorescence was detected in the treated cells (Figure 1c). Co-staining with Hoechst showed that the spliced XBP1s-YFP fusion protein was mainly localised in the nucleus (Supplementary Figure 1a).

Generation and characterisation of the IRE1- and PERK-reporter cell lines. (a) Schematic depicting the IRE1α reporter construct. Under conditions of ER stress, activation of IRE1α splices Xbp1 mRNA, which enables translation of the in-frame XBP1-YFP reporter fusion protein. In the absence of IRE1α activation, a premature stop codon prevents the translation of the fluorescent protein. (b) Schematic of the PERK-reporter construct. Under ER stress, activated PERK phosphorylates translation initiation factor eIF2α, which initiates translation from the 5′UTR of the YFP-reporter. (c) SH-SY5Y cells stably expressing the IRE1-reporter or (d) the PERK-reporter. Cells were treated with 3 μM Tg (right column) or DMSO for 24 h (left column) and YFP fluorescence imaged (scale bar=20 μM). (e) Expression of ER stress-induced proteins in the IRE1-reporter cell line. Cells were treated with 3 μM Tg and harvested at times indicated. Protein levels were analysed by western blotting and probing with antibodies against XBP1s, which detected the spliced endogenous and reporter XBP1, IRE1 and IRE1-p [Ser724]. β-actin served as loading control. (f) Expression of ER stress-induced proteins in lysates of PERK-reporter cells treated with 3 μM Tm. Protein levels were analysed by western blotting and probing with antibodies against GFP, detecting ATF4(1-28)-YFP, eIF2α, eIF2α-p [Ser51] and CHOP. β-actin served as loading control for both. (g, h) Real-time qPCR analysis of levels of spliced Xbp1 mRNA in IRE1-reporter and parental cells in response to ER stress (g) and Chop mRNA in PERK-reporter and parental cells (h). Cells were treated with 3 μM Tm and harvested at the indicated times. Results were normalized to β-actin levels and expressed relative to cultures harvested at 0 h (mean of n=3, error bars indicate S.E.M.). ANOVA was carried out to compare all groups. Post hoc Tukey pairwise comparison found no significant differences between parental and reporter cells at the different time points
Next, we tested whether expression of the spliced reporter XBP1-YFP fusion protein correlated with expression of the endogenous spliced XBP1s over time. Western blot analysis showed that levels of spliced endogenous XBP1s and spliced reporter XBP1s-YFP started to increase 4 h after treatment up until 24 h (Figure 1e). This increase correlated with increased levels of phosphorylated IRE1, which is associated with the activation of the endonuclease domain.15 We also compared the mRNA levels of spliced XBP1s between the IRE1 reporter and parental SH-SY5Y cells. qRT-PCR employing primers binding specifically to spliced XBP1 showed that both cell lines displayed similar levels of spliced XBP1 over time (Figure 1g). Indeed, the mRNA levels of transcriptional targets of spliced XBP1, Herp and ERdj42 were similar between the reporter and parental cells (Supplementary Figure 1b and c), suggesting that the IRE1 reporter construct did not disrupt the endogenous IRE1-XBP1 signalling pathway.
To construct the PERK reporter plasmid, a fragment of the 5’UTR comprising three regulatory upstream ORFs of the human ATF4 gene were cloned in frame with YFP under the control of a CMV promoter (Figure 1b). This was sufficient for translation initiation under ER stress conditions following phosphorylation of eIF2α. In SH-SY5Y cells stably transfected with the PERK reporter construct, a low basal fluorescence could be detected, which increased after treatment (Figure 1d). To characterise the expression kinetics of the ATF4(1-28)-YFP fusion protein in the PERK-reporter cell line, levels of key components of the PERK signalling pathway were analysed by western blotting (Figure 1f). An increase in the PERK reporter could be detected at 4 h, downstream of the increase in phosphorylation of eIF2α and prior to an increase in the levels of CHOP, a transcriptional target of ATF4, from 8 h on. To further insure that the construct did not obstruct PERK-ATF4 signalling, we compared the expression levels of Chop mRNA between the PERK reporter cell line and parental cells. Kinetics of mRNA levels in response to ER stress were similar in both cell lines (Figure 1h). Taken together, these data confirm that the IRE1 and the PERK reporter cell lines represented adequate tools to monitor ER stress responses.
IRE1 and PERK reporter cell lines respond to ER stress in a concentration-dependent manner
Next, we characterised the rate of fluorescent reporter activation in response to treatment with varying concentrations of ER stress inducers using a high content live cell imaging approach. IRE1 or PERK reporter cells were treated with a range of different concentrations of Tunicamycin (Tm) or Tg, incubated on stage and imaged at 1 h intervals for 48 h (Figure 2). YFP mean intensities per cell were pooled for all cells of the same treatment group and plotted over time (Figures 2a and c). In IRE1 reporter cells, we observed a concentration-dependent increase in YFP intensity in response to both stimuli, which was absent in DMSO-treated cells (Figure 2a). After 25 h, the YFP intensity in both the Tm- and Tg-treated IRE1 reporter cells decreased, suggesting an attenuation of IRE1 endonuclease activity (Figure 2b).

The IRE1 and the PERK reporters are activated in response to ER stress in a concentration-dependent manner. IRE1 reporter cells were stained with Hoechst and PI and treated with varying concentrations of Tm (0.03–1 μM) or Tg (0.03–1 μM) or 0.1% DMSO. Images were taken at 1 h intervals starting immediately after treatment for 48 h. (a) Mean YFP fluorescence intensity per cell over time following treatment with different concentrations of Tm (left panel) or Tg (right panel). Error bars indicate S.E.M. of all cells per time point and treatment. Experiment representative for at least two experiments. (b) Mean YFP fluorescence intensity 16 h after treatment with Tm (left panel) or Tg (right panel). Error bars indicate S.E.M. of at least n=10 wells from at least two independent experiments. ANOVA found significant differences between all treatment groups (P<0.01). Post hoc Tukey pairwise comparison found all Tm/Tg-treated groups to be significantly different from DMSO control (* indicates P<0.01). (c) PERK reporter cells were stained with Hoechst and PI and treated with varying concentrations of Tm (0.03–1 μM) or Tg (0.03–1 μM) or 0.1% DMSO. Images were taken at 1 h intervals starting immediately after treatment for 48 h. Mean YFP fluorescence intensity per cell over time following treatment with Tm (left panel) or Tg (right panel). Error bars indicate S.E.M. of all cells per time point and treatment. Experiment representative for at least two experiments. (d) Mean YFP fluorescence intensity 30 h after treatment with Tm (left panel) or Tg (right panel). Error bars indicate S.E.M. of n=8 wells from two independent experiments. ANOVA found significant differences between all treatment groups (P<0.05). Post hoc Tukey pairwise comparison found all Tm/Tg-treated groups to be significantly different from DMSO control (* indicates P<0.05)
The PERK reporter cell line was similarly activated in a concentration-dependent manner with the fluorescence intensity in vehicle-treated control cells remaining at baseline level throughout the experiment (Figure 2c). The highest level of fluorescence intensity was reached at 30 h (Figure 2d); interestingly a decrease in the fluorescence intensity levels was noted in the Tm-treated cells after 30 h suggesting a decrease in the translation of ATF4 and decreased phosphorylation of eIF2α.
Silencing of IRE1 results in early onset of cell death
To further elucidate the role of IRE1 and PERK signalling in ER stress-induced cell death, we silenced IRE1 and PERK and monitored the effect on cell death. We employed a lentiviral-delivered shRNA construct to effectively suppress the expression of IRE1 (Supplementary Figure 2a). Transduction of the construct into IRE1 reporter cells followed by treatment with Tg resulted in the loss of YFP fluorescence intensity in comparison to the IRE1 reporter cells transduced with scrambled control (Figure 3a). YFP fluorescence intensity in the IRE1 reporter cells silenced for IRE1 and treated with Tg was similar to the intensity values measured in vehicle-treated cells, suggesting little or no splicing of the XBP1 occurred in these cells and indicating loss of functional IRE1 expression.

IRE1 silencing resulted in increased sensitivity to ER stress and early onset of cell death whereas PERK gene silencing did not cause higher amounts of cell death. (a) IRE1 reporter cells were transduced with shRNA against IRE1 or scrambled control vector. Ninety-six hours after transduction, the cells were stained with Hoechst and PI and treated with 0.1, 0.3 or 1 μM Tg or 0.1% DMSO. Images were taken at 1 h intervals starting immediately after treatment for 48 h using high content time lapse live cell imaging. Left panel: YFP mean fluorescence intensity in IRE1 reporter cells over time following IRE1-kd or scrambled control group treated with 0.3 μM Tg or 0.1% DMSO, respectively. Error bars indicate S.E.M. of all cells per time point and treatment. Data shown are representative of two experiments. Right panel: YFP mean fluorescence intensity 16 h after treatment with 0.1, 0.3 or 1 μM Tg or 0.1% DMSO. Error bars indicate S.E.M. of n=5 wells from two independent experiments. ANOVA found significant differences between all treatment groups (P<0.05). Post hoc Tukey pairwise comparison between Tg-treated groups and the corresponding DMSO ctrl was significant as indicated (*P<0.05) or IRE1-kd and scrambled control groups treated with the same concentration of Tg (#P<0.05). (b) Left panel: Percentage of PI-positive cells in IRE1 reporter cells over time in IRE1-kd or scrambled control group treated with 0.3 μM Tg or 0.1% DMSO, respectively. Error bars indicate S.E.M. of n=3 wells. Data shown are representative for two experiments. Right panel: Percentage of PI-positive cells 30 h after treatment with 0.1, 0.3 or 1 μM Tg or 0.1% DMSO. Error bars indicate S.E.M. of n=5 wells from two independent experiments. ANOVA found significant differences between all treatment groups (P<0.05). Post hoc Tukey pairwise comparison between Tg-treated groups and the corresponding DMSO ctrl was significant as indicated (*P<0.05) or IRE1-kd and scrambled control groups treated with the same concentration of Tg (#P<0.05). (c) PERK reporter cells were transduced with shRNA against PERK or scrambled control vector. Treatment and imaging was carried out as above. Left panel: YFP mean fluorescence intensity over time in PERK-kd or scrambled control group treated with 0.3 μM Tg or 0.1% DMSO, respectively. Error bars indicate S.E.M. of all cells per time point and treatment. Data shown are representative of four experiments. Right panel: YFP mean fluorescence intensity 30 h after treatment with 0.1, 0.3 or 1 μM Tg or 0.1% DMSO. Error bars indicate S.E.M. of n=9 wells from four independent experiments. ANOVA found significant differences between all treatment groups (P<0.05). Post hoc Tukey pairwise comparison between Tg-treated groups and the corresponding DMSO ctrl was significant as indicated (*P<0.05) or PERK-kd and scrambled control groups treated with with the same concentration of Tg (#P<0.05). (d) Left panel: Percentage of PI-positive cells over time in PERK-kd or scrambled control group treated with 0.3 μM Tg or 0.1% DMSO, respectively. Error bars indicate S.E.M. of n=2 wells. Data shown are representative for four experiments. Right panel: Percentage of PI-positive cells 45 h after treatment with 0.1, 0.3 or 1 μM Tg or 0.1% DMSO. Error bars indicate S.E.M. of n=9 wells from four independent experiments. ANOVA found significant differences between all treatment groups (P<0.05). Post hoc Tukey pairwise comparison between Tg-treated groups and the corresponding DMSO ctrl was significant as indicated (*P<0.05) or PERK-kd and scrambled control groups treated with the same concentration of Tg (#P<0.05)
To assess cell death throughout the experiment, the cells were stained with propidium iodide (PI). In response to treatment with Tg, the increase in the percentage of PI-positive cells occurred earlier (10 h) in the cells silenced for IRE1 expression compared with scrambled control (20 h) (Figure 3b). As a result of the earlier onset of cell death in the IRE1-silenced cells, the percentage of PI-positive cells was higher at 30 h in these cells compared with scrambled control cells (Figure 3b). To ensure that the effect was not limited to the IRE1 reporter cell line, we also transduced parental SH-SY5Y cells with the shRNA construct. Here the effect was similar, cells transduced with the IRE1 silencer construct started to die earlier in response to ER stress in comparison with scrambled control cells, and the overall levels of PI-positive cells were higher in the IRE1-silenced cells compared with the control cells (Supplementary Figure 2c and d).
Silencing of PERK does not affect overall levels of cell death in response to ER stress
To investigate the effect of silencing PERK on ER stress-induced cell death levels, PERK expression was also reduced with a lentiviral-delivered shRNA (Supplementary Figure 2b). Monitoring the fluorescence intensity of the PERK reporter cells transduced with PERK silencing construct or scrambled control, we found that although some increase in YFP-fluorescence intensity could be detected in the PERK silenced cells treated with Tg, it was significantly lower than levels measured in the scrambled control cells, suggesting that PERK expression was efficiently silenced (Figure 3c). Levels of cell death in the PERK-silenced cells were found to be higher in the early time points after treatment with Tg (10–25 h) compared with scrambled control-transduced cells. However, after 48 h, there was no significant difference in the amount of cell death between the PERK-silenced and the scrambled cells (Figure 3d).
Early onset and early attenuation of XBP1 splicing are cytoprotective
To relate the kinetics of the fluorescent reporter in the IRE1 or PERK reporter cells to survival or cell death on a single cell level, it was necessary to track individual cells throughout the experiment. For this, we employed single cell time lapse live cell imaging which allowed for higher magnification and temporal resolution (10 min intervals), thus allowing individual cells to be tracked throughout the course of the experiment (48 h). Imaging conditions were optimised so that no phototoxicity could be detected.
To test whether the reporter cells responded in a similar manner as in the high content experiments, IRE1 reporter cells were exposed to ER stress and the mean YFP intensity was plotted over time along with cell death monitored as PI-positive cells (Figure 4a). In response to the Tm treatment, up to 30% PI-positive cells were observed at the end of the experiment (48 h), whereas little or no cell death occurred in the DMSO-treated cells (Figure 4b). Next, individual cells were randomly chosen and tracked throughout the experiment with the YFP and PI fluorescence intensities measured for each time point (Figures 4c–e). Traces of dying and resistant cells both showed an increase in YFP fluorescence intensity until a plateau of YFP fluorescence was reached. In either case, this was followed by a decrease in fluorescence intensity until no YFP signal could be detected for the cells, suggesting attenuation of XBP1 splicing and degradation of XBP1-YFP.

Single cell time lapse imaging of IRE1 reporter cells. Cells were stained with Hoechst and PI and treated with 3 μM Tm or DMSO and imaged live on stage. Images were acquired every 10 min for 48 h using the Axiovert 200M epi-fluorescence microscope. (a) Mean YFP fluorescence intensity of all cells imaged per time point is plotted in response to Tm or DMSO. (b) The number of PI-positive cells after treatment with 3 μM Tm or DMSO is plotted over time. Average number of imaged cells was n=281 in the Tm treatment and n=434 in the DMSO ctrl-treated experiment. Both Tm- and DMSO-treated experiments were repeated at least three times each with similar results. (c) YFP and PI intensities of a single dying cell measured over time. Black arrows and times correspond to images in panel e below. (d) YFP and PI intensities of a single resistant cell measured over time. (e) Phase, Hoechst, YFP and PI images for selected time points: before increase of fluorescence intensity (5 h), during increase in YFP fluorescence intensity (10 h), time of maximum YFP fluorescence intensity (13 h), decreasing YFP fluorescence intensity (25 h), no YFP fluorescence intensity detectable (36 h), cell death marked by increase in Hoechst intensity caused by condensation of the nucleus and increase in PI intensity marking cell membrane permeabilisation (42 and 42.6 h). Yellow arrows indicate the cell of interest which was tracked. (Scale bar=20 μM). Experiment was repeated twice with similar results
To examine the potential differences in IRE1 activity in surviving versus dying cells in more detail, the tracked cells were grouped according to cellular outcome within the time frame of the experiment (48 h), and key parameters of the YFP traces were compared between groups (Figure 5a). We noted that in resistant cells, the fluorescence intensity increased earlier compared with dying cells (Figure 5b), suggesting that an early onset of XBP1 splicing was protective. Furthermore, the fluorescence intensity plateau was reached earlier in the resistant compared with the dying cells (Figure 5c). Additionally, we observed a trend towards a faster rate of fluorescence intensity increase in the surviving compared with the dying cells (Figure 5d). This suggested that in surviving cells, XBP1 splicing occurred at a faster rate with IRE1 activity attenuated earlier in the surviving compared with the dying cells.

IRE1 reporter cells resistant to ER stress showed earlier onset of fluorescence increase and reached a fluorescence intensity plateau earlier than dying cells. Parameters of IRE1 reporter activation kinetics in response to ER stress measured in individual cells in relation to cellular outcome. IRE1 reporter were cells stained with Hoechst and PI were treated with 3 μM Tm and images acquired every 10 min for 48 h. PI-positive or PI-negative cells were randomly chosen and tracked. (a) Median YFP fluorescence intensity of the traces of surviving (green diamonds) or dying cells (red diamonds) were plotted over time. Resistant cells n=30, dying cells n=31, from three independent experiments. (b-e) YFP-traces were analysed and the following curve parameters as indicated in the schematics above the box plots were compared between dying and resistant cells: (b) Time point of onset of increase in fluorescence intensity (resistant cells n=27, dying cells n=31). (c) Time point the fluorescence intensity stops to increase and a plateau is reached (resistant cells n=30, dying cells n=31). (d) Rate of fluorescence intensity increase (resistant cells n=27, dying cells n=31). (e) Mean fluorescence intensity reached at plateau (resistant cells n=30, dying cells n=31). Mean is indicated as red and median as blue line. Top and bottom of boxes represent 25th and 75th percentile, respectively. Whiskers refer to 10th and 90th percentile and outliers are shown as dots. Mann-Whitney Rank Sum tests were performed to compare the median of resistant and dying groups, #P<0.05, n.s. P >0.05
We also compared the mean YFP fluorescence intensity, indicative of the amount of spliced XBP1-YFP but found no significant difference between surviving and dying cells (Figure 5e), which would suggest that the overall amount of spliced XBP1 was not decisive for cellular outcome.
Other trace parameters that were compared between dying and surviving cells were the ‘time point fluorescence starts to decrease’, ‘duration of intensity plateau’, ‘rate of fluorescence intensity decrease’ and the ‘time point fluorescence intensity reaches baseline’ (Supplementary Figure 3a and d). No significant differences between the groups for any of these parameters were identified, suggesting that these did not play a critical role in determining cellular outcome.
Slow rate of ATF4 translation and late re-initiation of general translation are cytoprotective
Next, we investigated how the activation of PERK and subsequent downstream induction of ATF4 translation related to cell death using single cell time lapse imaging and tracking individual cells. Plotting the YFP fluorescence of Tm-treated PERK reporter cells imaged in a single cell experiment showed an increase in YFP fluorescence intensity in the cells exposed to ER stress, whereas in the DMSO-treated cells, the fluorescence intensity remained at baseline (Figure 6a). In the treated cells, we observed an increase in cell death up to 30% PI-positive cells at 48 h, whereas there was little cell death in vehicle-treated cells (Figure 6b).

Single cell time lapse imaging of PERK-reporter cells. Cells were stained with Hoechst and PI and treated with 3 μM Tm or 0.1% DMSO. Images were taken every 10 min for 48 h using the Axiovert 200M epi-fluorescence microscope. (a) Mean YFP fluorescence intensity of all cells imaged per time point in response to Tm or DMSO. (b) Amount of PI-positive cells after treatment with 3 μM Tm or 0.1% DMSO. Average number of imaged cells was n=271 in the Tm treatment and n=205 in the DMSO ctrl-treated experiment. Both Tm- and DMSO-treated experiments were repeated at least three times each with similar results. (c) YFP and PI intensities of a dying single cell measured over time. Black arrows and times correspond to images in panel e. (d) YFP and PI intensities of a resistant single cell measured over time. (e) Phase contrast, Hoechst, YFP and PI images for selected time points: baseline YFP fluorescence intensity before onset of increase (0.6 h), during increase in YFP fluorescence intensity (10 h), time of YFP fluorescence intensity plateau (32.6 h), cell death marked by increase in Hoechst and PI intensity (47.5 and 47.7 h). Yellow arrows indicate the cell of interest which was tracked. (Scale bar=20 μM). Experiment was repeated twice with similar results
Tracking of randomly chosen single cells and measurement of their YFP fluorescence intensity over time yielded a number of traces. Traces of PERK reporter cells showed a detectable YFP fluorescence baseline at the beginning of the experiment and an increase in fluorescence intensity until a plateau was reached (Figures 6c–e). Comparing parameters of the traces between the resistant and the dying cells showed that there was no significant difference in the time point of the onset of the YFP fluorescence intensity increase, the fluorescence intensity at baseline or the mean fluorescence intensity reached at the plateau (Figures 7a–d). This indicated that the time at which ATF4 translation was initiated was not a decisive parameter for cellular fate, nor was the overall amount of ATF4. However, we found that in resistant cells, the rate of fluorescence intensity increase was lower than in dying cells and the fluorescence plateau was reached later (Figures 7e and f), suggesting that a slow translation rate and a late attenuation of ATF4 translation were protective.

PERK reporter cells resistant to ER stress showed slower rate of fluorescence intensity increase and reached a fluorescence intensity plateau later than dying cells. Parameters of PERK reporter activation kinetics in response to ER stress measured in individual cells in relation to cellular outcome. PERK reporter were cells stained with Hoechst and PI and treated with 3 μM Tm. Images were taken every 10 min for 48 h. PI-positive or PI-negative cells were randomly chosen and tracked. (a) Median YFP fluorescence intensity of the traces of surviving (green triangles) or dying cells (red diamonds) were plotted over time. Resistant cells n=30, dying cells n=32, from three independent experiments. (b-f) YFP traces were analysed and the following curve parameters as indicated in the schematics above the box plots were compared between dying and resistant cells: (b) Time point of onset of increase in fluorescence intensity (resistant cells n=30, dying cells n=28). (c) Mean fluorescence intensity at baseline (resistant cells n=29, dying cells n=28). (d) Mean fluorescence intensity at plateau (resistant cells n=29, dying cells n=28). (e) Rate of fluorescence intensity increase (resistant cells n=32, dying cells n=30). (f) Time point fluorescence intensity plateau is reached (resistant cells n=30, dying cells n=30). Mean is indicated as red and median as blue line. Top and bottom of boxes represent 25th and 75th percentile, respectively. Whiskers refer to 10th and 90th percentile and outliers are shown as dots. Mann-Whitney rank sum tests were performed to compare the median of resistant and dying groups, #P<0.05, n.s. P>0.05
Discussion
Here, we set out to investigate the temporal activation patterns of the UPR signal transducers IRE1 and PERK and the relative contribution of these signalling events to cell death decisions. To this end, we constructed cell lines stably expressing fluorescent reporters for the IRE1 and the PERK signalling pathway. To link the activation of the UPR reporters to cellular outcome, we developed a single cell time lapse live cell imaging assay allowing for resolution of the heterogeneity of responses and resulting cell fates within the cell population.
Early onset of XBP1 splicing is protective
In response to accumulation of unfolded proteins, Xbp1 mRNA is spliced through the activation of IRE1 leading to the translation of active XBP1. Applying for the first time single cell time lapse imaging to observe XBP1 splicing in live cells, we found that surviving cells had an earlier onset of XBP1 splicing and splicing was attenuated earlier compared with cells that succumbed to ER stress. Earlier onset and faster rate of XBP1 splicing would suggest a faster accumulation of downstream transcriptional targets, many of which are chaperones and ERAD components,2, 16 resulting in a more effective clearance of folding intermediates from the ER. Although the protective output of the IRE1-XBP1 branch of the UPR has been proposed in previous studies,8, 17 our experiments emphasise the critical importance of the early onset of these signalling processes. Interestingly, the amount of spliced XBP1 was not significantly different between resistant and surviving cells pointing to a fast activation of IRE1 signalling and initiation of XBP1 splicing but not overall amounts of spliced XBP1 being crucial for cell survival.
The requirement for protective IRE1-XBP1 signalling outputs to ensure cell survival early after induction of ER stress is further supported by the results of the high content time lapse imaging of IRE1 reporter cells silenced for IRE1 activity. Here, the onset of cell death in response to ER stress occurred earlier in cells with reduced IRE1 activity in comparison with control cells, suggesting that the loss of spliced XBP1s results in cells not being able to cope effectively with the unfolded protein load and thus succumb to death earlier.
Our single cell imaging further showed a decrease in YFP fluorescence indicating an attenuation in IRE1 endonuclease activity which occurred in both dying and resistant cells. Previous studies have interpreted the attenuation of IRE1 activity as a switch from pro-survival to pro-apoptotic signalling.8 Indeed, in our experiments on a populational level, the time of decrease of XBP1-YFP fluorescence intensity coincides with an increase in cell death. However, our single cell analysis shows that the reduction of IRE1 endonuclease activity occurs in both dying and resistant cells, which suggests a scenario whereby IRE1 activity is attenuated independently of cell fate.
In addition to Xbp1, the RNase activity of IRE1 can cleave a range of mRNA substrates targeting them for degradation.4, 5, 7 Pro-survival as well as pro-apoptotic roles have been suggested for IRE1-dependent decay of mRNAs (RIDD).5, 6, 18 Our system does not allow direct monitoring of RIDD, however, the data available to date would indicate that RIDD cannot occur independent of XBP1 splicing.5, 7, 18 Thus, we assume that the observed attenuation of XBP1-YFP splicing is indicative of a decrease in all IRE1 endonuclease activities including mRNA decay. In fact, if RIDD contributes to pro-apoptotic cell decision in the later stages of the UPR, this could explain why in our single cell experiments, the time point the fluorescence intensity of the IRE1 reporter reached a plateau, indicating an attenuation in IRE1 activity, was observed earlier in resistant compared with dying cells. Early attenuation might protect the cells from detrimental signalling outputs of hyperactivated IRE1.
Slow translation of ATF4 is protective
We also applied single cell imaging to investigate signalling events in the PERK-ATF4 branch of the UPR. Here, the PERK reporter cells resistant to ER stress showed a slower increase rate in YFP fluorescence intensity, which would indicate that ATF4 translation was slower in the resistant compared with the dying cells. Slow translation of ATF4 would in turn lead to a slower increase in downstream transcriptional targets of ATF4, such as CHOP.
Pro-apoptotic outputs of CHOP signalling have been suggested by a number of studies.19, 20, 21 CHOP can trigger the downregulation of pro-survival BCL2 and upregulation of pro-apoptotic BH3-only proteins BIM22, 23 or PUMA.20 Additionally, a recent study proposed that increased protein synthesis driven by ATF4/CHOP after re-initiation of general translation increases the amount of reactive oxygen species, thus leading to apoptosis.13 Therefore, a slower increase in ATF4 levels could be protective. Indeed, in our experiments, we found that the rate of ATF4 translation was slower in surviving cells compared with cells that succumbed to ER stress.
We also observed that the plateau in fluorescence intensity, indicating a decrease in ATF4 translation and possibly a switch to general translation was reached later in surviving compared with dying cells. This could be explained by a slower accumulation of the phosphatase subunit GADD34 in the surviving cells, which exhibit slower ATF4 translation rates, as GADD34 is also a downstream target of ATF4. GADD34 dephosphorylates eIF2α, leading to re-initiation of general translation.24 Thus, our experiments would suggest that surviving cells might retain eIF2α phosphorylation longer and switch to general translation later than dying cells. This is in line with reports, which showed that inhibition of eIF2α dephosphorylation protects cells from ER stress.25 Taken together, with our single cell analysis of activation of IRE1 signalling this would suggest a scenario where the length of time between activation of IRE1 and re-initiation of general translation, rather than a switch between signalling branches is critical for cellular outcome, with a longer window between these events being potentially advantageous for cell survival (Figure 8).

Assumed kinetics of XBP1 splicing and ATF4 translation in response to Tm-induced ER stress in a resistant and a dying cell. In a hypothetically resistant cell, IRE1 is activated early and spliced XBP1 accumulates quickly, until the IRE1 endonuclease activity is attenuated at a relatively early time point, whereas ATF4 translation would occur at a slow rate and dephosphorylation of eIF2α occurs late (Top panel). In a dying cell, XBP1 splicing starts late and occurs at a slow rate, whereas ATF4 accumulates fast and general translation is re-initated early (bottom panel)
Our high content image analysis of the effect of PERK silencing further underlined the protective role of eIF2α phosphorylation and arrest of general translation. PERK deficiency resulted in higher sensitivity of the cells to ER stress at early time points, probably due to the failure to attenuate general protein synthesis in the PERK-silenced cells. At the end of the experiment (48 h after treatment), the percentage of cell death was slightly higher in the scrambled control, compared with the PERK deficient cells. Thus, loosing PERK did not seem to have a strong effect on cellular outcome.
Conclusion
Taken together, our ER stress reporter systems in combination with single cell time lapse imaging allowed here for the first time to investigate activation kinetics of ER pathway components and correlate these findings with cell fate. Our study highlights that rather than a switch of activity from one UPR branch to another, the timing of IRE1 and PERK signalling events is critical to determining cellular outcome.
Materials and Methods
Cell culture, transfection and treatments
SH-SY5Y neuroblastoma cell lines were cultured in DMEM/F12 (Sigma Aldrich, Wicklow, Ireland) supplemented with 15% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin/streptomycin. SH-SY5Y cells were transfected using Metafectene (Biontex, Martinsried, Germany) following the manufacturer’s protocol. HEK293TN cells were grown in DMEM (5 g/l Glucose) (Lonza, Verviers, Belgium) supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 100 U/ml penicillin/streptomycin. Stable cell lines were selected in medium containing 0.5 mg/ml G418 (Sigma, Arklow, Ireland) for up to 2 weeks. Monoclonal cell lines were isolated by seeding the cells sparsely into a 6 well plate. After ~5 days, colonies were manually picked by carefully scraping off and taking up the cells using a pipette. Monoclonal cell lines were cultivated in the presence of 0.1 mg/ml G418. ER stress was induced by treating the cells with Tm (Sigma Aldrich) or Tg (Sigma Aldrich) at indicated concentrations. Control cultures received an equivalent volume of solvent (DMSO).
Construction of plasmids
For the IRE1 reporter plasmid, full length Xbp1 (NM_001079539) containing the 26 bp intron was amplified from human cDNA using forward primer 5′-CGTAGATCTCGAGATGGTGGTGGTGG-3′ and reverse primer 5′-CTCGCACAAGCTTGACACTAATCAGC-3′ and cloned into vector pEYFP-N1 (Clontech, Saint Germain-en-Laye, France) using the XhoI and HindIII restriction sites. For the PERK reporter, a 360 bp long sequence comprising 280 bp of the 5′UTR of the human ATF4 gene (NM_182810.1) immediately upstream of the coding region and 84 bp coding for the first 28 amino acids was amplified from human cDNA using primer pair 5′-CTCGCACCTCGAGTTTCTACTTTGCC-3′/5′-ACTTACTAAGCTTTTCAGCCCCCAAACC-3′ which contained XhoI and HindIII restriction sites, respectively, for cloning into vector pEYFP-N1.
Western blotting
Cells were lysed and western blotting performed as described previously.26 Blots were probed with the following primary antibodies: rabbit polyclonal anti-IRE1α (1 : 1000) (Cell Signaling Technology, Inc., Danvers, MA, USA); rabbit polyclonal anti-IRE1-p [Ser724] (1 : 500) (Novus Biologicals, Littleton, CO, USA); rabbit monoclonal anti-GFP (1 : 1000) (Epitomics, Burlingame, CA, USA), rabbit polyclonal anti-EIF2α (1 : 1000) (Cell Signaling), rabbit polyclonal anti-EIF2α-P [Ser51] (1 : 500) (Cell Signaling), mouse monoclonal anti-XBP1 (1 : 500) (BioLegend, San Diego, CA, USA), mouse monoclonal anti-CHOP (1 : 100) (Santa Cruz Biotechnology Inc., Heidelberg, Germany), mouse monoclonal anti-β-actin (1 : 10 000) (Sigma Aldrich). Secondary antibodies conjugated to horseradish peroxidase (Thermo Fisher Scientific, Dublin, Ireland) were detected using Immobilion chemoluminescent HRP substrate (Millipore, Cork, Ireland). Blots were imaged using LAS-4000 imaging system (Fujifilm, Sheffield, UK).
Real time qPCR
RNA was extracted using RNeasy Mini Kit (Qiagen, Manchester, UK). cDNA was generated from 500 ng total RNA using random hexamer primers (Fisher Scientific, Dublin, Ireland) and Superscript II Reverse Transcriptase (Invitrogen, Life Technologies Products, Dun Laoghaire, Ireland). Real-time quantitative PCR was carried out using SYBR Green PCR Master Mix. The following primer pairs were used: spliced Xbp1 5′-TGAGTCCGCAGCAGGTGCA-3′, 5′-CTGGGTCCTTCTGGGTAGACCTC-3′; Erdj4 5′-TGGCCATGAAGTACCACCCTGACAA-3′, 5′-TCCACTACCTCTTTGTCCTTTACCACT-3′, Herp 5′-GAGCCTGCTGGTTCTAATCG-3′ 5′-TCAGCTGGAACTGGTTGTG-3′, Chop 5′-GGTCCTGTCTTCAGATGAAAATG-3′ 5′-CTTGGTGCAGATTCACCATTC-3′ and Actin 5′-TCACCC;ACACTGTGCCCATCTACGA-3′ 5′-CAGCGGAACCGCTCATTGCCAATGG-3′. Reactions were performed using the LightCycler (Roche Diagnostics, West Sussex, UK). The data were analysed using the LightCycler Software 4.0. All samples were normalized to β-actin. Statistical significance was tested employing ANOVA and post hoc Tukey tests, carried out using SPSS (IBM Ireland Ltd., Dublin, Ireland) statistical analysis software.
shRNA and lentivirus production
MISSION shRNA vectors for gene silencing of IRE1 (clone number TRCN0000000530) or PERK (clone number TRCN0000001399) in mammalian cells and scrambled control containing plasmids were obtained from Sigma. Lentiviral particles were produced by co-transfecting HEK293TN cells seeded at a density of 2 × 105 cells/well in 3 ml medium per well in a 6 well plate with viral 50 ng/well envelope protein encoding vector pMD2.G (Addgene, Cambridge, MA, USA) and 450 ng/well packaging protein coding vector psPAX2 (Addgene) together with 500 ng/well of the MISSION shRNA containing pLKO.1-puro vector and 3 μl/well Metafectene. The medium containing virus particles was harvested 48 and 72 h after transduction. Both batches were pooled, centrifuged at 3000 × g for 10 min, filtered through a 40 μm syringe filter, aliquoted and stored at −80 °C.
Transduction with lentiviral particles
For transduction with shRNA, SH-SY5Y cells were seeded at a density of 3 × 104 cells/well in a 12 well plate and incubated with 250 μl virus suspension in 1 ml medium and 5 μg/ml polybrene per well. The cells were centrifuged at 1000 × g at 20 °C for 90 min. Medium was changed after 24 h. Transduced cells were cultivated for 72 h before being moved to imaging plates or 96 h subjected to ER stress treatment.
Fluorescence microscopy
Images of cultured cells in Figures 1c and d were taken using the Nikon TE 300 fluorescence microscope (Nikon, Dusseldorf, Germany), equipped with a SPOT RT SE 6 CCD camera. The cells were imaged using × 20 magnification combined with × 0.6 sideport magnification. Images were analysed using ImageJ software.27 For time lapse imaging, cells were seeded at a density of 4 × 104 cells in 2 ml phenolfree DMEM medium (Sigma) onto a 12 mm glass bottom dish (Willco BV, Amsterdam, The Netherlands). Two hours prior to the experiment, cells were stained with 50 ng/ml Hoechst 33588 and 2 μg/ml PI (Sigma). The dish was covered with cling film to prevent evaporation of medium during the experiment and placed in a covered heating element (37 °C) mounted on the microscope which was supplied with CO2 (5%) throughout the experiment. Fluorescence was observed using an Axiovert 200 m inverted microscope equipped with a 20 × 0.5 numerical aperture (NA) objective (Carl Zeiss, Jena, Germany), polychroic mirror and filter wheels in the excitation and emission light path containing the appropriate filter sets for Hoechst (excitation 350–409 nm, emission 415–480 nm); PI (excitation 510–555 nm, emission 580–618 nm) and YFP (excitation 488–513 nm, emission 526–556 nm) (Semrock, Laser 2000, Cambridge UK) and a 6% Neutral Density (ND) filter. For excitation in the light path of the Hoechst channel, an additional 25% ND filter was used to minimize phototoxicity. Images were acquired using a back-illuminated cooled EM CCD camera (Andor Ixon BV 887-DCS, Andor Technologies, Belfast, Northern Ireland). Automated image acquisition and stage positioning protocols were generated and executed using MetaMorph Microscopy Automation and Image Analysis software (Molecular Devices, LLC, Sunnyvale, CA, USA). Images were taken every 10 min for 48 h of nine overlapping fields of view. One hour after starting the experiment, 3 μM Tm or the same volume DMSO was added to the cells on stage.
High content time lapse imaging
SH-SY5Y reporter cells were seeded at a density of 8000 cell/well in 150 μl medium in a Nunc Micro Well 96 well optical bottom plate. Two hours prior to the experiment, the medium was exchanged for 150 μl imaging medium containing 100 ng/ml Hoechst 33588 and 2 μg/ml PI. Immediately before the experiment, 50 μl imaging medium containing different concentrations of Tm, Tg or the same volume DMSO were added to each well. Each experimental condition corresponded to at least two wells on the plate.
The plate was imaged using a Cellomics ArrayScan VTi instrument (Fisher Scientific) equipped with a × 10 PlanApo objective lens (NA 0.45), a 120 W Hg arc illumination source (EXFO, Chandlers Ford, UK) and a monochrome CCD camera (Orca Hamamatsu Photonics, Hertfordshire, UK). The following filters sets were used: Hoechst excitation 387±11 nm, emission open; PI excitation 504±12 nm, emission 645±75 nm; YFP excitation 504±12 nm, emission 542±27 nm (Semrock, AHF, Analysentechnik, Tubingen, Germany). A 25% ND filter was used in the excitation light path of all channels. Images were taken of 9 FOV per well in 1 h intervals for 48 h, with 1024 × 1024 pixel resolution (6.45 μM pixel size).
Image analysis for single cell tracking
To obtain image sequences in which single cells could be tracked over the length of the experiments the imaging data were restructured using Fiji image processing software.28, 29
All raw images for each channel (Hoechst, YFP or PI) were opened as image sequence and brightness and contrast were adjusted. The nine adjacent fields of views (512 × 512 pixel) which were imaged with a 13% overlap at each time point were combined together into a 1432 × 1432 pixel image using the ‘Stitch Sequence of Grids of Images’ plugin.28, 29 CellProfiler 2.0 image analysis software30 was used to analyse mean YFP intensity and percentage of PI positive cells of the whole cell population in these image sequences (see below). For single cell traces, cells were randomly chosen and tracked manually starting at the latest frame by defining a region of interest around the object, which was adjusted for each frame to accommodate cell movement. The integrated fluorescence intensity of the cell within the region of interest was measured for each frame in the YFP channel. The values were imported into Microsoft Excel and the resulting traces analysed manually.
Depending on outcome, the traces were grouped into dying and surviving cells. SigmaPlot software (Systat Software Inc, London, UK) was used to calculate mean and median and to generate box plots. The significance of the results was tested using the Mann-Whitney rank sum test.
Analysis and data processing of high content time lapse imaging data
CellProfiler 2.0 cell image analysis software was used to identify individual objects and measure their fluorescence intensity in stitched images collected with the Axiovert 200 m and images collected using the Cellomics ArrayScan (Fisher Scientific).30 Intensities of all images of the same channel were scaled between 0 and 1 upon loading into CellProfiler. To identify objects, images were corrected for shading and background by subtracting a strongly smoothed image (Gaussian Filter, pixel size 25). YFP images were additionally corrected for cross-talk from the PI channel.
Objects were identified in the corrected images employing an intensity threshold calculated using an Otsu Adaptive algorithm. The mean intensity per object of all objects identified in the Hoechst channel was measured in the input PI images and the cross-talk-corrected YFP images.
A purpose built Matlab script (Matlab; MathWorks Inc., Cambridge, UK) was used to calculate and plot the fraction of dying cells and the mean of the mean YFP fluorescence intensity values of all cells per treatment group and time point as well as standard errors of these values. Statistical significance of the imaging data shown in bar graphs was tested employing ANOVA and post hoc Tukey tests, carried out using SPSS statistical analysis software.
Abbreviations
- ATF4:
-
activating transcription factor 4
- a.u.:
-
arbitrary units
- ER:
-
endoplasmic reticulum
- ERAD:
-
endoplasmic reticulum associated degradation
- fluo.int.:
-
fluorescence intensity
- IRE1:
-
inositol requiring enzyme
- ORF:
-
open reading frame
- PERK:
-
PKR-like ER-kinase
- PI:
-
propidium iodide
- RIDD:
-
regulated IRE1-dependent decay
- Tg:
-
Thapsigargin
- Tm:
-
Tunicamycin
- UTR:
-
untranslated region
- XBP1:
-
x-box binding factor 1
- YFP:
-
yellow fluorescent protein
References
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002; 415: 92–96.
Lee AH, Iwakoshi NN, Glimcher LH . XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003; 23: 7448–7459.
Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 2009; 323: 1693–1697.
Hollien J, Weissman JS . Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006; 313: 104–107.
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS . Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 2009; 186: 323–331.
Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012; 338: 818–822.
Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009; 138: 562–575.
Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318: 944–949.
Harding HP, Zhang Y, Ron D . Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397: 271–274.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6: 1099–1108.
Lu PD, Harding HP, Ron D . Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 2004; 167: 27–33.
Ma Y, Brewer JW, Diehl JA, Hendershot LM . Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 2002; 318: 1351–1365.
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nature Cell Biol 2013; 15: 481–490.
Novoa I, Zeng H, Harding HP, Ron D . Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 2001; 153: 1011–1022.
Li H, Korennykh AV, Behrman SL, Walter P . Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci USA 2010; 107: 16113–16118.
Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 2013; 3: 1279–1292.
Lin JH, Li H, Zhang Y, Ron D, Walter P . Divergent effects of PERK and IRE1 signaling on cell viability. PloS One 2009; 4: e4170.
Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S et al. Allosteric Inhibition of the IRE1alpha RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell 2014; 158: 534–548.
Ghosh AP, Klocke BJ, Ballestas ME, Roth KA . CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS One 2012; 7: e39586.
Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP . Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci 2010; 30: 16938–16948.
Yamaguchi H, Wang HG . CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 2004; 279: 45495–45502.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ . Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21: 1249–1259.
Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007; 129: 1337–1349.
Ma Y, Hendershot LM . Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem 2003; 278: 34864–34873.
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005; 307: 935–939.
Ramapathiran L, Bernas T, Walter F, Williams L, Dussmann H, Concannon CG et al. Single-cell imaging of the heat-shock response in colon cancer cells suggests that magnitude and length rather than time of onset determines resistance to apoptosis. J Cell Sci 2014; 127: 609–619.
Schneider CA, Rasband WS, Eliceiri KW . NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9: 671–675.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682.
Preibisch S, Saalfeld S, Tomancak P . Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009; 25: 1463–1465.
Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O et al. Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006; 7: R100.
Acknowledgements
This research was supported by grants from Science Foundation Ireland (13/IA/1881 and 08/IN1/B1949) to JHMP.
Author Contributions
FW, HD, CGC and JHMP conceived and designed experiments; FW and HD performed experiments; FW, JS, HD and CGC analysed the data; FW, CGC and JHMP wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H Ichijo
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Walter, F., Schmid, J., Düssmann, H. et al. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ 22, 1502–1516 (2015). https://doi.org/10.1038/cdd.2014.241
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.241
This article is cited by
-
Endoplasmic reticulum stress mediates the myeloid-derived immune suppression associated with cancer and infectious disease
Journal of Translational Medicine (2023)
-
Toxoplasma gondii dense granule protein 3 promotes endoplasmic reticulum stress-induced apoptosis by activating the PERK pathway
Parasites & Vectors (2022)
-
ATF4 leads to glaucoma by promoting protein synthesis and ER client protein load
Nature Communications (2020)
-
Decomposition profile data analysis of multiple drug effects identifies endoplasmic reticulum stress-inducing ability as an unrecognized factor
Scientific Reports (2020)
-
ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling
Cell Communication and Signaling (2019)
