
Abstract
Cells depend for their survival on stimulation by trophic factors and other prosurvival signals, the withdrawal of which induces apoptosis, both via the loss of antiapoptotic signaling and the activation of proapoptotic signaling via specific receptors. These receptors, dubbed dependence receptors, activate apoptotic pathways following the withdrawal of trophic factors and other supportive stimuli. Such receptors may feature in developmental cell death, carcinogenesis (including metastasis), neurodegeneration, and possibly subapoptotic events such as neurite retraction and somal atrophy. Mechanistic studies of dependence receptors suggest that these receptors form ligand-dependent complexes that include specific caspases. Complex formation in the absence of ligand leads to caspase activation by a mechanism that is typically dependent on caspase cleavage of the receptor itself, releasing proapoptotic peptides. Cellular dependence receptors, considered in the aggregate, may thus form a system of molecular integration, analogous to the electrical integration system provided by dendritic arbors in the nervous system.
Similar content being viewed by others


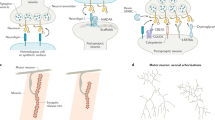
Introduction
Cells depend for their survival on stimulation mediated by various receptors and sensors. For example, some cells respond to the loss of extracellular matrix adhesion by undergoing anoikis (‘homelessness’),1 that is, programmed cell death (PCD) initiated by a loss of adhesion. Other cells may undergo PCD following the withdrawal of trophic factors (e.g., the neurotrophins), cytokines, hormonal support, electrical activity, or other stimuli.2
Depending on the cell type and its state of differentiation, cells require different supportive stimuli for survival. For example, prostate epithelial cells may require testosterone for survival, and for such cells the withdrawal of testosterone leads to apoptosis. Therefore, prostate neoplasms are often treated by withdrawing testosterone because this induces apoptosis, and thus tumor shrinkage; unfortunately, the few remaining cells that are androgen independent typically repopulate the tumors, and therefore alternative therapy is required.
For any given required stimulus, withdrawal leads to PCD; that is, the loss of trophic support somehow triggers an active process of cell suicide. It is of both theoretical and practical importance to understand how cells ‘recognize’ a lack of supportive stimuli and activate PCD in response to the withdrawal of such stimuli. For years it was generally assumed that cells dying as a result of the withdrawal of required stimuli do so because of the loss of a positive survival signal. For example, nerve growth factor (NGF) is a trophic factor for certain sympathetic neurons and sensory neurons (as well as some other populations of neurons). Its trophic effect is mediated by dimerization of the receptor tyrosine kinase (RTK) TrkA, autophosphorylation, and downstream signaling, including PI3 kinase and Akt phosphorylation.3 Although such positive survival signals are clearly extremely important, data obtained over the past 10 years argue for a complementary and novel form of signal transduction that is proapoptotic, and is activated or propagated by stimulus withdrawal.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 Moreover, whereas positive survival signals – such as those mediated by RTKs that bind trophic factors – involve classical signal transduction (i.e., ligand–receptor interaction initiates the signal), negative survival signals (i.e., ligand withdrawal initiates the signal), such as those mediated by the netrin-1 receptors, deleted in colorectal cancer (DCC) and uncoordinated gene 5 (Unc5)H1–3 (see below for a more detailed description), involve nonclassical signal transduction, in which typically the unbound receptor (or possibly the receptor bound by a hypothetical ‘antitrophin’) is activated to induce cell death by proteolytic processing, generating proapoptotic fragments (Figure 1). In this latter case, ligand binding blocks the proapoptotic effect of the fragments, at least in some cases by inhibiting the proteolytic processing of the receptor.2, 10 This form of signal transduction has been dubbed negative signal transduction.2

Schematic representation of some dependence receptors. Modified from Bredesen et al.15
Cellular dependence on trophic influences for their antiapoptotic effect is mediated, at least in part, by specific ‘dependence receptors’ or ‘addiction receptors.’ These receptors induce apoptosis in the absence of the required stimulus (when unoccupied by a trophic ligand, or possibly when bound by a competing ‘antitrophin’), but block apoptosis following binding of their respective ligands.2, 4, 5 Expression of these dependence receptors creates cellular states of dependence on the associated trophic ligands. These states are not absolute, since they can be blocked downstream in some cases by the expression of antiapoptotic genes such as bcl-2 or p35;2, 13, 17 however, they result in a shift of the apostat18, 19 – that is, the probability that a given cell will undergo apoptosis – toward an increased likelihood of undergoing apoptosis.
It should be pointed out that the terms ‘dependence receptor’ and ‘addiction receptor,’ although used interchangeably here (since it is not yet clear whether these receptors serve both functions, or whether specific ones mediate cellular dependent states and others mediate cellular addictive states), are not identical: by definition, addiction requires previous exposure, whereas dependence does not. In vivo, it is likely that at least some of these receptors are expressed following initial cellular exposure to their associated ligands, which would make them addiction receptors; however, in cell culture experiments, the expression of these receptors in cells not previously exposed to the ligands in question may lead to apoptosis induction, and therefore create cellular states of dependence.2, 4, 9, 10 Functionally, therefore, these receptors may be dependence receptors, but physiologically they may be either addiction receptors or dependence receptors or both; hence, here the terms are used interchangeably.
Discussion
DCC: Initiating a third class of tumor-associated genes?
Vogelstein et al. have shown that the development of colorectal carcinoma from normal colorectal epithelium is associated with the mutation of a specific set of genes.20 Over 70% of primary colorectal neoplasms display allelic deletions, resulting in a loss of heterozygosity (LOH) on chromosome 18q.20 This observation prompted a search for a tumor suppressor gene located in this region, which resulted in the cloning of a putative cell-surface receptor, DCC.20 DCC expression is absent or markedly reduced in over 50% of colorectal tumors, and in multiple other tumor types, such as gastric carcinoma, pancreatic carcinoma, esophageal carcinoma, prostatic carcinoma, carcinoma of the bladder, carcinoma of the breast, male germ cell tumors, neuroblastomas, gliomas, and some leukemias.21, 22 Since the initial description, however, DCC has been an ‘odd’ tumor suppressor candidate: although in vitro studies have supported DCC's potential role as a tumor suppressor gene,23, 24, 25, 26, 27 point mutations have rarely been identified in DCC coding sequences;28 mice heterozygous for DCC-inactivating mutations29 do not display the expected tumor predisposition phenotype; and other known and candidate tumor suppressor genes30 have been identified in the same region of chromosome 18q as DCC. Reconciliation of these apparently paradoxical data has been suggested by recent work from Mazelin et al.,31 who found that a netrin receptor (or receptors) functions not as a classical tumor suppressor but rather as a conditional tumor suppressor. In other words, unlike a classical tumor suppressor such as Rb, the suppressive effect of the netrin receptor is reversed by the presence of high concentrations of netrin.
The DCC gene encodes a 1447 amino-acid type I transmembrane protein with a relative molecular mass of approximately 200 kDa. It displays homology in its extracellular domain with cell adhesion molecules,21 suggesting a role in cell–cell or cell–matrix interactions.32 However, DCC-mediated cell aggregation has not been clearly proven.33
In addition to its putative role in neoplasia, DCC also mediates axon guidance and cell survival during neural development. As the nervous system develops, migrating axons and cells receive guidance cues that originate from several sources, such as members of the netrin, semaphorin, ephrin, and slit protein families.34, 35 Tessier-Lavigne and his collaborators have demonstrated that DCC functions as a component of a receptor complex that mediates the effects of the axonal guidance molecule netrin-1.36, 37, 38 The role of DCC in mediating growth cone extension has been supported by the analysis of DCC-null mice, which display abnormal development of the central nervous system.29
The relationship between the initially proposed role of DCC as a tumor suppressor and the ability of DCC to bind netrin-1 and mediate axon guidance was initially unclear. This dual role in neural development and neoplasia has turned out to be a common feature of dependence receptors (see below). Indeed, DCC was shown to act as a dependence receptor, that is, its expression creates a state of cellular dependence on its trophic ligand, netrin-1: its expression was shown to induce apoptosis in the absence of netrin-1,36, 37, 38 but to manifest an antiapoptotic effect when bound by netrin-1.9, 39 Site-directed mutagenesis identified residues 1243–1264 in the intracytoplasmic region of DCC as required for apoptosis induction. Expression of this dependence domain was sufficient for apoptosis induction. It is noteworthy that the dependence domain of DCC displays no similarity to the dependence domains of other dependence receptors at the level of primary structure or predicted secondary structure.
During studies of the proapoptotic effect of DCC, it was noted that DCC is a caspase substrate, with cleavage occurring primarily at Asp1290, just distal to the dependence domain.9 Point mutation of this caspase site completely suppressed the proapoptotic effect of DCC, resulting in a modestly antiapoptotic effect of the mutant.9, 13
Functionally, therefore, DCC may amplify cellular caspase activity in the absence of ligand, via exposure of a proapoptotic domain lying in the amino-terminal region of the intracellular domain, proximal to Asp1290. Whether DCC may also initiate apoptosis – as opposed to functioning simply as an amplifier – is a subject of current studies. DCC was shown to induce apoptosis by a mechanism independent of Apaf-1 and cytochrome c, yet dependent on caspase-9 (and not on caspase-8), suggesting that DCC may induce or amplify an apoptotic signal via a novel pathway.13 The requirements for caspase activation by DCC and other dependence receptors are being defined (see below), but temporally involve caspase recruitment followed by activation, followed by receptor cleavage.
A search for interactors of DCC revealed caspase-3 and -9, as well as DCC-interacting protein 13α (DIP13α), to interact (directly or indirectly) with DCC.39 The caspase interactions suggested a conformational dependence, in that caspase-9 interacted with DCC (or a DCC-containing complex) in the absence of ligand, whereas no such interaction was detected in the presence of ligand, or when DCC was mutated at Asp1290. In a complementary fashion, caspase-3 was found to interact with DCC in the presence of ligand (and also with the Asp1290 → Asn mutant), but not in the absence of ligand.13 These results suggest that DCC may assume more than one conformation, depending on the presence of ligand binding. The interaction of DCC with both caspase-3 and -9 (albeit under different conditions) is reminiscent of the apoptosome complex that catalyzes caspase-3 activation.40 In vitro studies supported the hypothesis that DCC also initiates a caspase-activating complex; however, the precise mechanism involved, and the identification of the molecular components of the complex, are as yet undefined.
In addition to caspase-3 and -9, DCC was also found to interact with a protein dubbed DIP13α.39 Based on several observations, DIP13α appears to be an excellent candidate as a mediator of DCC-induced apoptosis. First, DIP13α was shown to interact with the dependence domain of DCC, that is, the domain required for apoptosis induction following ligand withdrawal (residues 1243–1264). Second, DCC–DIP13α co-expression was found to increase apoptosis induction over that observed with DCC alone. Third, small interfering RNA (siRNA) directed against DIP13α inhibited DCC-induced apoptosis.39 The mechanism by which DIP13α mediates the DCC-induced proapoptotic effect is unknown.
The A2b purinoceptor, which is a G-protein-coupled receptor,11 has been found to serve as a co-receptor for netrin-1 with DCC. These co-receptors mediate neurite outgrowth and cAMP production induced by netrin-1.41, 42 DCC also interacts with other guidance-related receptors such as Unc5H (see below)43 and Robo,38 but whether or not these interactions mediate or modulate any of the proapoptotic effects of DCC is not yet known.
DCC thus functions as a dependence receptor – inducing apoptosis following ligand withdrawal, but suppressing this effect (and actually supporting survival) in the presence of appropriate concentrations of its ligand, netrin-1. As has been noted for other dependence receptors, DCC exerts effects on both neoplasia and neural development. With respect to neural development, DCC may mediate developmental neuronal apoptosis and neurite outgrowth. Results to date suggest that netrin-1 not only mediates chemoattractive and chemorepulsive axonal effects, but also functions as a survival factor during the process of neuronal guidance. Supporting this notion, DCC-expressing olivary neurons that normally migrate through a netrin-1-dependent mechanism undergo apoptosis in the netrin-1 null mice at the time when migration would otherwise be initiated.12 Similarly, developing commissural neurons require netrin-1 for their survival in ex vivo experiments.13
On the other hand, with respect to neoplasia, DCC may potentially function as a ‘metastasis suppressor’ by inducing apoptosis when ligand is limited by metastasis or growth beyond local ligand availability. However, in vivo experiments will be required to validate this hypothesis.
Finally, a receptor closely related to DCC is neogenin, which demonstrates 50% amino-acid identity with DCC. Perhaps somewhat surprisingly, given this similarity, neogenin appears to prefer a different ligand than DCC – rather than netrin-1, neogenin binds repulsive guidance molecule (RGM) with high affinity.44 The RGM–neogenin pair mediates axonal retraction, but RGM–neogenin also appear to function in cellular dependence. In the developing neural tube, downregulation of RGM or overexpression of neogenin led to apoptosis.44 Furthermore, just as for DCC, neogenin displays a caspase cleavage site in its intracytoplasmic domain. However, unlike for DCC, LOH in the neogenin genomic region (15q22) does not appear to be a frequent phenomenon in malignancies.45
Unc5H2: Conditioning the response to the guardian, p53
Netrin-1 not only binds DCC but also binds another family of receptors – the Unc5 family. These family members include the Caenorhabditis elegans protein Unc5 and four mammalian orthologues, Unc5H1–4 (Unc5 A–D in humans). Based on genetic screens, Unc6 (the worm ortholog for netrin-1) was linked to both Unc-40 – the ortholog of DCC – and Unc5. It was therefore proposed that the mammalian Unc5H1–3 may function as netrin-1 receptors.46, 47 Subsequent studies suggested that DCC/Unc-40 mediates the chemoattractive effect of netrin-1, whereas Unc5H is involved with the chemorepulsive effect of netrin-1, as an interactor of DCC within the intracytoplasmic region.43 This notion of attraction/repulsion, however, did not explain settings in which netrin-1 appears to serve as a chemoattractant for axons that do not express DCC, or in which neurons that express both DCC and Unc5H nonetheless undergo attraction to netrin-1 rather than repulsion by it.
Unc5H1–3 have also been shown to function as dependence receptors.12 Just as for DCC, these proteins display a caspase cleavage site in their intracytoplasmic domains, and mutation of this site prevents apoptosis induction. Furthermore, also as for DCC, netrin-1 null mice display an increase in developmental neural apoptosis in cells expressing Unc5H.12 Unlike DCC, however, Unc5H proteins display a classical death domain in the carboxyterminal region. Also contrasting with DCC is the caspase site, which is typical in Unc5H (DXXD), but atypical in DCC (LSVD, with R in the P1′ position, which is also atypical).
The Unc5H death domain is somewhat similar to the p75NTR death domain, and, although not clearly classified as a type I or type II death domain, it is more similar to the type II death domain of p75NTR than to the type I death domains of Fas and TNFR I.12 The Unc5H death domain is required for apoptosis induction – thus it functions as a dependence domain – and this effect requires membrane targeting. The caspase cleavage requirement for the proapoptotic effect of Unc5H suggests the possibility of a conformational change following cleavage.
The mediation of cell death by Unc5H1–3 appears to involve multiple mechanisms, potentially varying among family members. Unc5H1 interacts, through its ZU5 domain, with the NRAGE protein, and mediates apoptosis via NRAGE.48 This finding is reminiscent of what was observed for the common neurotrophin receptor, p75NTR.49 On the other hand, Unc5H2 and 3 do not interact with NRAGE, and appear to require their death domains to induce apoptosis.
The findings that DCC and Unc5H1–3 all induce apoptosis that is suppressed by netrin-1 lends further support to the notion that netrin-1 serves, at least in part, as a survival factor during neural development. Beyond neural development, however, since both netrin-1 and Unc5H are also expressed in the adult, it was hypothesized that, like DCC, Unc5H1–3 may also play roles as conditional tumor suppressors that would induce apoptosis in locations of limiting netrin-1 concentration, such as may occur with metastasis or growth beyond local ligand support. Compatible with this hypothesis, it was observed that, just as for DCC, the expression of Unc5A–C is decreased in multiple neoplasms, including colorectal tumors and those of the breast, ovary, uterus, stomach, lung, and kidney. In colorectal tumors, this downregulation is associated with LOH occurring within Unc5H genes, but may also be partially related to epigenetic processes such as methylation.50 Such a decrease in expression of Unc5H may be one mechanism for neoplastic growth or metastasis, since the expression of Unc5H1, 2, or 3 inhibits tumor cell anchorage-independent growth and invasion in vitro. Furthermore, the abrogation of these hallmarks of malignant transformation by Unc5H1–3 can be reversed by netrin-1 addition or by apoptosis inhibition, providing further support for the notion that Unc5H1–3 may function as dependence receptors and conditional tumor suppressors.50
An interesting extension of the notion of conditional tumor suppression was provided by the finding that p53-induced apoptosis is also dependent on extracellular signaling, specifically on netrin-1.34 In that study, Tanikawa et al.51 reported that Unc5B is a direct transcriptional target of p53. Surprisingly, netrin-1 was indeed found to block p53-induced apoptosis; moreover, the inhibition of Unc5B blocked p53-induced apoptosis. Thus, Unc5B expression was required for induction of apoptosis by p53.51 This finding suggests that p53 is, itself, a conditional tumor suppressor. The interplay between p53, Unc5B, and netrin-1 suggests that the output signal of p53 activity (e.g., cell death induction or cell cycle inhibition) may depend on the presence or absence of the netrin-1–Unc5B interaction. Thus, p53 expression may create a cellular state of dependence on netrin-1 (and, by extrapolation, perhaps on other trophic factors or supportive stimuli): expressing cells would be forced into a choice, in which trophic support would spare the cells PCD, but a lack of trophic support (which may be tolerated in cells with minimal or no dependence receptor expression (here, Unc5B, but potentially also other such receptors)) would prove lethal following p53-induced upregulation of dependence receptors such as Unc5B.
Rearranged during transfection (RET): one receptor, two diseases
The RET proto-oncogene is a type I transmembrane protein that displays an extracellular ligand-binding domain, a transmembrane domain, and an intracytoplasmic tyrosine kinase domain similar to other RTKs.52 In addition, RET displays a cadherin-like domain extracellularly, suggesting that it may function in cell–cell interaction.53
Just as for the other dependence receptors, RET has been shown to play a role in both neural development and neoplasia: RET forms part of the receptor complex for glial-derived neurotrophic factor (GDNF) and its related trophic factors neurturin, artemin, and persephin.54 These four trophic factors are similar to members of the transforming growth factor-β (TGF-β) family. The receptor complexes include RET and various glycosylphosphatidylinositol (GPI)-anchored proteins that are required for RET dimerization; these GPI-anchored co-receptors include GFRα-1, 2, 3, and 4.55 RET and GFRα-1 transduce a GDNF signal that plays a role in the development of the enteric nervous system and the kidney; thus, null mutations in GDNF, RET, and GFRα-1 display similar phenotypes.56, 57, 58
Ligand binding to the RET complex triggers RET autophosphorylation, followed by interaction with effectors that include phospholipase Cγ, Shc, Enigma, Grb2, Grb7/Grb10, Src kinase, and Ras-GAP, and resultant downstream signaling.59, 60, 61
Mutations in the RET proto-oncogene may be associated with neoplastic disease or with a neural developmental disease. Multiple endocrine neoplasia type 2 (MEN-2) occurs in association with one set of RET mutations,62 whereas Hirschsprung syndrome occurs in association with a different set of RET mutations.63 MEN-2 is a neoplastic syndrome that includes the development of tumors associated with endocrine organs: tumors of the parathyroid (parathyroid adenomas), adrenal gland (pheochromocytomas), and medullary thyroid carcinomas. Hirschsprung syndrome is a relatively common (one in 5000 live births) neural developmental syndrome in which neural crest-derived parasympathetic neurons of the hindgut are congenitally absent, resulting in a loss of peristalsis and associated intestinal dilation.
This profile – a combination of neoplastic and developmental neural diseases in association with RET mutations – led Bordeaux et al.11 to evaluate the possibility that RET may function as a dependence receptor. They reported that the expression of RET in the absence of GDNF induced apoptosis, whereas GDNF blocked this effect. Furthermore, RET was found to be a caspase substrate, with caspase cleavage sites at residues 708 and 1017. Cleavage at both sites was found to be required for apoptosis induction by RET, and the dependence domain was shown to lie between these two sites. This domain did not, however, display similarity to the dependence domains of other dependence receptors.
Interestingly, the disease-associated RET mutants could be divided into two clearly distinct groups: those associated with neoplasia displayed an antiapoptotic effect irrespective of ligand presence, whereas those associated with Hirschsprung syndrome demonstrated constitutive proapoptotic activity, again irrespective of ligand.11 Thus, both the proliferative and the developmental syndromes appear to result from mutations that result in a loss of ligand response to the receptor signaling: the ligand-dependent on–off apoptosis ‘switch’ of this dependence receptor is ‘stuck’ in the ‘on’ position for the Hirschsprung-associated mutants, and in the ‘off’ position for the MEN-2-associated mutants. Such findings suggest that the cell culture phenotypes may turn out to be predictive of those in vivo by indicating what apoptosis-related effects the receptors will have on their expressing cells, and what modifying effects (if any) the ligands will exert. The results suggest further that Hirschsprung syndrome may result, at least in part, from apoptosis induction during development, resulting in the observed lack of ganglionic neurons in the hindgut.
Patched: Hedging bets on neural tube development
During development, the neural tube is a site of neuronal proliferation, differentiation, and migration, as well as neuronal cell death. These events are regulated in part by ventral structures, the floor plate, and notochord. Among the cues that determine the fates of cells within the developing neural tube is the classic morphogen, sonic hedgehog (SHH), a diffusible molecule whose secretion sets up a ventral-to-dorsal concentration gradient. Recently, Thibert et al. proposed that the 12-transmembrane receptor for SHH, Patched (Ptc), functions as a dependence receptor.64 This hypothesis is compatible with earlier observations made by LeDouarin and colleagues, in which the experimental withdrawal of SHH in chick embryos led to massive cell death of the neuroepithelial cells in the developing neural tube, suggesting that SHH is indeed a survival factor.65, 66 Thibert et al. found that, both in vitro and in ovo, expression of Ptc in settings of SHH absence induces apoptosis. Moreover, as for DCC, RET, UNC5H and the androgen receptor (see below), Ptc is cleaved at a caspase site – for Ptc, this site is Asp1392 – and, just as for these other dependence receptors, such cleavage is required for cell death induction. In the case of Ptc, however, there is no clear evidence yet of the nature of the dependence domain, which appears to lie proximal to the caspase cleavage site.64
The common neurotrophin receptor p75NTR: death receptor and dependence receptor
Although p75NTR was the first dependence receptor to be described,6, 67 it has remained the most enigmatic, at least in part because it appears to function both as a death receptor and a dependence receptor.15 Classic experiments by Levi-Montalcini68 and Hamburger demonstrated the phenomenon of trophic factor dependence: developing neurons pass through a critical phase, at which time typically approximately half of the neurons from various subsets fail to survive. Death of developing neurons may occur in three different morphological patterns: the majority undergoes apoptosis (which has, in the past, also been referred to as type I or nuclear PCD). However, other developing neurons die by one of two alternative morphological patterns: type II, also referred to as autophagic, cell death; or type III, also referred to as cytoplasmic PCD. Most developmental neuronal cell death is apoptotic,69 and is critically dependent on the availability of neurotrophic factors: supraphysiological concentrations of neurotrophic factors block developmental neuronal apoptosis, whereas neurotrophin withdrawal leads to enhanced apoptosis.
NGF, the first trophic factor identified, binds to two distinct receptors, p75NTR70, 71 and TrkA.72, 73, 74, 75 TrkA was initially shown to be capable of mediating the described responses to NGF, such as neurite outgrowth and neuronal survival,73 leaving the role of p75NTR unexplained. Subsequent studies demonstrated that p75NTR and TrkA collaborate to produce high-affinity sites for NGF binding, and that p75NTR expression enhances the selectivity of neurotrophin binding for specific Trks (TrkA, B, and C;76).
However, p75NTR also displays Trk-independent effects. p75NTR is a member of the tumor necrosis factor receptor superfamily.77 The relationship between ‘death factor’ (proapoptotic factor) receptors such as Fas, and p75NTR, which binds a trophic factor (which by definition displays antiapoptotic, rather than proapoptotic, effects), was unclear until 1993, when reports began to appear implicating p75NTR in neural cell death.4, 5, 6, 78, 79, 80 Consistent with the notion that p75NTR binds a trophic factor rather than a death factor, these reports suggested a novel phenomenon: that the expression of p75NTR induced apoptosis when p75NTR was unoccupied by NGF, whereas binding of NGF blocked apoptosis.4, 5, 6 This is the reverse of the effect of binding of death factors to death receptors, such as Fas ligand binding to the Fas receptor.81 One report suggested that a similar phenomenon may occur with another member of the TNFR superfamily, CD40.82
These findings suggested that p75NTR expression creates a state of cellular dependence on NGF and other neurotrophins (such as brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and neurotrophin-4/5 (NT-4/5)). Follow-up studies provided further evidence for this notion: for example, p75-deficient mice were observed to have an increase in cholinergic neurons in the medial septal and diagonal band regions, and hyperinnervation in some areas of the hippocampus.80, 83 NGF hemizygous mice, with reductions in cholinergic cell number and size within the medial septal region, were rescued by crossing with p75NTR null mice,56 restoring somal dimensions and cell number to supranormal (indistinguishable from p75-deficient controls). Thus in the presence of a reduced concentration of NGF, p75NTR mediates a reduction in cholinergic neuronal number and size.
The effect of p75NTR on cellular neurotrophin dependence has potential implications for both neural and extraneural cellular behaviors: for example, prostate epithelial cells express p75NTR, which may tie them to a source of neurotrophin,84 supplied by the stromal cells. Pflug et al.85 documented a progressive decrease in p75NTR expression associated with the development of prostate neoplasia. The re-expression of p75NTR in PC3 prostate carcinoma cells restored a state of neurotrophin dependence, resulting in apoptosis if NGF was not supplied.67 Thus, p75NTR may mediate neurotrophin dependence in both neural and extraneural cellular paradigms.
It is not yet clear whether the proapoptotic form of the dependence receptors requires monomerization following trophic factor withdrawal, or if in fact there are competing nontrophic ligands (‘antitrophins’). However, studies utilizing FK binding protein chimeras86 support the notion that receptor monomerization is indeed associated with the proapoptotic state, with dimerization and higher-order multimerization completely suppressing this effect.67, 87 Moreover, Longo et al.88 found that cyclic dimeric peptides that bound p75NTR inhibited apoptosis, whereas monomers were completely ineffective. These studies demonstrated that p75NTR homomultimerization does not induce apoptosis (at least under the conditions of the reported studies), but did not exclude the possibility that monomeric p75NTR leads to apoptosis by a mechanism requiring heteromultimerization with another receptor, or, alternatively, that monomeric p75NTR may undergo an activating modification (e.g., cleavage, phosphorylation, etc.) that leads to multimerization and cell death induction.
Two regions appear to be crucial for the proapoptotic effect of p75NTR: the juxtamembrane intracytoplasmic region, dubbed chopper,89 and a region that lies in the fourth and fifth helices of the six-helical death domain. The latter, a 30-amino-acid region, was dubbed a dependence domain because of its requirement for apoptosis induction following the withdrawal of serum or trophic factors.67 Reversal of the death-inducing effect of p75NTR by ligand binding, on the other hand, was shown to require a region at the carboxyterminus, dubbed the neurotrophin response domain.90, 91
Following the descriptions of apoptosis induction by p75NTR in the absence of NGF, studies appeared showing that p75NTR may also induce apoptosis following NGF binding, that is, p75NTR may also function as a death receptor.75, 78, 79 However, despite this apparent functional similarity to Fas and TNFR I, and the similarities in structure of these three proteins, Fas-p75NTR chimeras consisting of the extracellular domain of Fas and the intracellular domain of p75NTR failed to induce apoptosis.92 Thus, apoptosis induction following NGF binding to p75NTR is somehow different from following the binding of death factors to Fas and TNFR I. This may be a Trk-dependent phenomenon, since it has been described almost exclusively in systems in which mismatched Trk members (e.g., Trk B with NGF or Trk A with BDNF) have been expressed.2, 8 It is also possible that the decision between ligand-induced apoptosis and ligand-inhibited apoptosis mediated by p75NTR may depend on specific downstream mediators: for example, the interaction of p75NTR with NADE induces apoptosis following ligand binding;93 in contrast, TRAF2 has been shown to interact preferentially with monomeric p75NTR and to induce apoptosis in the absence of NGF.94 Other potential death-mediating p75NTR interactors include NRAGE49 and NRIF.95 Another proapoptotic transmembrane protein has been identified that has a death domain very similar to that of p75NTR, and this protein interacts with p75NTR.96 The protein, dubbed p75NTR-like apoptosis-inducing death domain protein (PLAIDD) and also referred to as neurotrophin receptor homologue 2 (NRH2),97 displays a type II death domain (which is structurally slightly different from the type I death domains displayed by death receptors such as Fas and TNFR I) that is closely related to the type II death domain of p75NTR, with 42% identity.96
Thus, p75NTR appears to function both as a dependence receptor and a death receptor, apparently depending on associated Trk expression, ligand presentation, expression of p75NTR-interacting molecules, patent downstream signaling pathways, and probably additional factors. One potential implication of this dual role in cell death is that p75NTR may mediate the cellular response to neurotrophin withdrawal through its function as a dependence receptor, whereas it may mediate the cellular response to a mismatched neurotrophin (e.g., exposure of a neuron-expressing TrkB and p75NTR to NGF, which binds TrkA and p75NTR) via an alternative mechanism, through its function as a death receptor.2
Since p75NTR may mediate PCD both in response to ligand withdrawal and in response to ligand binding, a model has been suggested in which p75NTR serves a ‘quality control’ function,15 mediating apoptosis when cells experience a decline in trophic support, the inappropriate trophic support (mismatched for the expressed Trk), unprocessed trophic factor (pro-NGF, which binds p75NTR and induces apoptosis via sortilin and p75NTR),98 or the binding of neurotrophin to an inappropriately immature cell.99 As noted above, p75NTR also mediates neurotrophin binding specificity by reducing the promiscuity of neurotrophin–Trk interactions, which is also compatible with the ‘quality control’ hypothesis.
Subapoptotic events such as neurite retraction and somal atrophy may also be mediated by p75NTR, based on transgenic results.80, 84 p75NTR has also been shown to be the transducing receptor for Nogo, which binds to a GPI-anchored receptor, the Nogo receptor.100 In this case, p75NTR may be a mediator of neurite retraction. Although the effect of neurotrophin binding to p75 on Nogo signal transduction is not yet clear, the involvement of p75 in neurite retraction suggests the possibility that at least part of the neurotrophic effect on process outgrowth occurs via a block of Nogo-mediated neurite retraction.
The androgen receptor as a dependence receptor
The androgen receptor is a nuclear/cytosolic steroid receptor that includes an aminoterminal-region polyglutamine stretch, a DNA-binding domain, and a carboxyterminal-region ligand-binding domain. Binding of androgens such as testosterone by the androgen receptor leads to nuclear translocation and transcriptional activity. Gene regulation by the androgen receptor affects widespread processes such as male gonadal development, prostate cellular survival, motor neuron survival, and muscular development, among other effects.
As was noted for RET, mutations in the androgen receptor are associated both with neoplasia and neural cell loss. However, in contrast to the RET-associated developmental loss of neurons in Hirschsprung syndrome, some androgen receptor mutations are associated with a neurodegenerative syndrome, that is, the neurons apparently undergo normal development but degenerate at some point following differentiation (typically during adulthood). This clinical syndrome is referred to as Kennedy's disease, or spinobulbar muscular atrophy (SBMA), and involves the degeneration of motor neurons in the brainstem and spinal cord, resulting in weakness and muscular atrophy. The associated mutations are not point mutants of the androgen receptor, but rather polyglutamine expansion mutations similar to those associated with Huntington's disease, dentatorubropallidoluysian atrophy (DRPLA), and other diseases such as spinocerebellar degenerations. Disease-associated polyglutamine tracts are typically longer than 30 glutamines, whereas those with fewer than 30 glutamines in the polyglutamine tract of the androgen receptor do not develop Kennedy's disease.
The androgen receptor displays a profile similar to that of other dependence receptors: expression induces apoptosis in the absence of ligand, whereas the addition of ligand inhibits the receptor-induced cell death.10 The androgen receptor is also a caspase substrate, with a cleavage site at Asp146, and, as for other dependence receptors, mutation of this site results in a marked reduction in the proapoptotic effect of the receptor.10
The dependence domain of the androgen receptor spans the polyglutamine region.10 An expanded polyglutamine domain is associated with an increase in cell death, whereas a polyglutamine-deleted androgen receptor is associated with a decrease in cell death, as compared to the wild-type androgen receptor. However, deletion of the polyglutamine tract did not completely suppress the proapoptotic activity of the androgen receptor, arguing that the proapoptotic dependence domain extends beyond the polyglutamine tract.
These findings suggest that the length of the polyglutamine tract in the androgen receptor correlates with its proapoptotic effect, with short polyglutamine tracts having a lesser proapoptotic effect than the wild type androgen receptor. This may turn out to have important clinical implications: epidemiological studies have shown that men with short polyglutamine stretches (⩽15 glutamines) in their androgen receptors have a statistically significantly higher likelihood of developing prostate cancer, especially metastatic prostatic cancer.86, 101 Although clearly not by an identical mechanism, this association of neoplasia with a reduction in the proapoptotic effect of a dependence receptor is reminiscent of the finding noted above for RET.
In contrast, an increase in the polyglutamine stretch is associated with the neurodegeneration of Kennedy's disease. Again, analogous to the situation for RET, a neoplastic syndrome is associated with a decrease in dependence, and a syndrome of neuronal loss is associated with an increase in dependence. One dissimilarity between the observations of patients with Hirschsprung syndrome due to RET mutations, and those with Kennedy's disease due to polyglutamine expansion of the androgen receptor: whereas the former is associated with a developmental neuronal loss, the latter is associated with post-developmental neurodegeneration; similarly, there is a distinction in the results of the mutations on the dependence receptor effects. Specifically, Hirschsprung-associated mutants fail to respond to ligand inhibition but have a proapoptotic effect similar to that of the wild type; in contrast, Kennedy's-associated mutants do respond to ligand inhibition but demonstrate an increased overall proapoptotic effect.10
Other dependence receptors
Giancotti and Ruoslahti102 found that the overexpression of the α5β1 integrin led to apoptosis and reduced tumor growth in vitro and in vivo. More recently, Stupack et al.14 proposed that specific integrins, such as the αvβ3 integrin, function as dependence receptors. Expression of unligated integrins, the β-subunit cytoplasmic domain, or the juxtamembrane sequence KLLITIHDRKEF, led to apoptosis induction, associated with the recruitment of a proapoptotic complex that included caspase-8. However, unlike DCC, neogenin, Unc5H1–3, RET, Ptc, and the androgen receptor, no required caspase cleavage site has yet been identified in the intracytoplasmic domain.
The β-amyloid precursor protein (APP) may also function as a dependence receptor:103 it is cleaved intracytoplasmically at Asp664, releasing a cytotoxic peptide, APP-C31. The possibility that such an effect may play a role in the pathogenesis of Alzheimer's disease is discussed below.
Dependence receptors and neurodegeneration: is Alzheimer's disease a state of altered dependence?
A number of proteins associated with neurodegeneration – such as huntingtin, the androgen receptor, atrophin-1, ataxin-3, and APP – have been shown to be caspase substrates. As noted above, the androgen receptor functions as a dependence receptor, with its proapoptotic activity being inhibited by androgen binding and by mutation of the caspase site, Asp146.10 Furthermore, the proapoptotic effect requires a region that includes the polyglutamine region that is expanded in Kennedy's syndrome (SBMA), a neurodegenerative syndrome that involves motor neurons. These findings raise the question of whether some neurodegeneration-associated proteins demonstrate features of dependence receptors; in other words, are neurodegenerative syndromes states of altered dependence within the nervous system?
Some of the recent findings in Alzheimer's disease are indeed compatible with such a notion: APP is cleaved in its intracytoplasmic region by caspases at Asp664,104 and this cleavage releases a cytotoxic carboxyterminal fragment, APP-C31.103 If this caspase cleavage event plays an important role in the pathogenesis of Alzheimer's disease, then the prevention of this cleavage should mitigate the behavioral and pathological alterations associated with the disease. Galvan et al.105 created transgenic mice carrying APP mutations characteristic of familial Alzheimer's disease, added a D664A mutation to prevent C31 formation, and then compared these mice to similar mice lacking the additional D664A mutation. No effect was observed on APP expression, amyloid Aβ production, or the development of senile plaques characteristic of Alzheimer's disease. However, the hippocampal synaptic loss, dentate gyral atrophy, electrophysiological abnormalities, and neural precursor dysregulation that characterize the Alzheimer's disease phenotype were all prevented by the D664A mutation. To carry the notion of Alzheimer's disease as a state of altered dependence, and APP as a dependence receptor, a step further, would require that the pathological process be inhibited not only by the D664A mutation of APP but also by supplying an APP ligand. APP has been shown to bind laminin,106 as well as type IV collagen,107 and it is possible that other ligands will also be identified. It is not yet known whether any APP ligand does indeed mimic the effect of the D664A mutation on Alzheimer's disease pathogenesis. However, one potentially interesting implication of this view of Alzheimer's disease is that it implies that the Aβ peptide that binds to APP and activates cell death via C31 formation108 functions as an ‘antitrophin’ – that is, it may potentially interfere with trophic effects of a ligand or ligands such as laminin. This view would be compatible with previous reports that Aβ induces neurite retraction, and with the dramatic effect that Alzheimer's disease has on synapse loss in affected areas.109, 110 As a broad generalization, then, one might suggest that Alzheimer's disease may result from an imbalance reminiscent of that between oncogene function and tumor suppressor gene function that is associated with neoplasia; in this case, however, it would be between the neurite-retractive (and antitrophic) effects of Aβ and the neurite-extending (and trophic) effects of laminin or other trophic APP ligands.
Is there a role for dependence receptors in tumor invasion and metastasis?
As the expression of proapoptotic receptors creates a state of cellular dependence on their respective ligands, it has been proposed9, 84 that such receptors tie cells to specific contexts in which the dependent ligands are available. This might conceivably represent a strategy used by the organism to prevent tumor growth beyond specific ligand fields, and thus block metastasis. Indeed, a number of observations support this notion.
For example, the dependence domain of the androgen receptor includes the polyglutamine tract that lies near the receptor's aminoterminus.10 Longer stretches of glutamines led to a greater proapoptotic effect, and shorter stretches were less effective at apoptosis induction; however, in all cases, testosterone binding inhibited the proapoptotic effect. Deletion of the polyglutamine region destroyed most, but not all, of the proapoptotic effect. These findings suggest that the degree of dependence is a function of the polyglutamine length.
The length of this polyglutamine tract varies from individual to individual. As noted above, individuals with abnormally long polyglutamine tracts in the androgen receptor (typically, >30 glutamines) develop the motor neuron degenerative Kennedy's syndrome.111 Conversely, individuals with short androgen receptor polyglutamine tracts (⩽16 glutamines) are at significantly increased risk for the development of metastatic prostate cancer.86, 101
Similarly, p75NTR is expressed by normal prostate epithelial cells, with the ligand source being the prostate stromal cells. Pflug et al.85 demonstrated a progressive decline in p75NTR expression accompanying prostate neoplasms, from benign prostatic hypertrophy to carcinoma in situ, with cell lines derived from metastatic prostatic carcinomas failing to express p75NTR. Re-expression of p75NTR in PC3 prostate carcinoma cells returned apoptosis induction in the absence of NGF.67
The loss of expression of DCC is associated with invasive and metastatic colorectal carcinomas.20, 28 Ectopic expression of DCC in a tumorigenic keratinocyte cell line lacking endogenous DCC expression was shown to suppress tumorigenic growth of the cells in nude mice.25 Interestingly, in that study, tumorigenic reversion was associated with loss of DCC expression and loss or rearrangement of the transfected DCC expression vector.25 Several more recent studies also indicate that restoration of DCC expression can suppress tumorigenic growth properties in vitro or in nude mice,26, 27 supporting a role for DCC in tumor suppression. Therefore, one of the roles of DCC may be to induce apoptosis in colorectal cancer cells that grow outside the ligand field.9 This hypothesis is supported by the recent observation that the Unc5H family of netrin-1 dependence receptors, may also show similar features to those of DCC – loss of Unc5H expression in cancer, LOH in Unc5H genes, and suppression of the hallmarks of cell transformation following re-expression of Unc5H in a colorectal cancer cell line.50 Complementing these results is the recent finding that transgenic mice overexpressing netrin-1 show an increased propensity to tumor formation, and an increased likelihood of malignant progression when combined with a second ‘hit’ provided by a common APC mutation.31
These results suggest that dependence receptors may play a novel role in tumorigenesis, as conditional tumor suppressors; in this role, they may function to suppress or support neoplasia, depending on their local environment.
A role for dependence receptors in nervous system development
As originally pointed out by Levi-Montalcini68 for the first trophic factor described – NGF – trophic influences play major roles in the developing nervous system, and the inhibition of PCD is one of their many important effects. One mechanism by which trophic factors inhibit PCD in the developing nervous system is by binding to dependence receptors, blocking their induction of apoptosis. As described above, ligand–receptor pairs that play a role in neural development include SHH-Ptc, netrin-DCC, netrin-Unc5H1–3, GDNF-RET, NGF/BDNF/NT-3/NT4/5-p75NTR, and dependence receptors may potentially exist for many other trophic systems.
DCC, Unc5H, and netrin are involved in axon outgrowth and turning. Netrin and DCC-null mice both support a role for this pair in the development of numerous commissural projections.29, 37 However, in addition to pathfinding, netrin-1 mediates survival: pontine cells and olivary neurons in the netrin-1 knockout mutant mice were not simply incorrectly targeted, but were lacking,112, 113 probably due to premature developmental neuronal death.12 Indeed, Llambi et al.12 demonstrated increased cell death at the level of olivary neuron precursors that express DCC and Unc5H. These data support the view that netrin-1 acts both as a survival factor and a guidance factor.
One of the predictions that stems from the dependence receptor hypothesis is that, whereas transgenic mice null for a given trophic ligand should demonstrate an increase in PCD in cells expressing the associated dependence receptor, mice null for the receptor may display a decrease in cell death (if ligand is normally limiting), an increase in cell death (if ligand is abundant), or neither, depending on whether the dominant effect of the receptor during development is proapoptotic or antiapoptotic. On the other hand, when crossed with the ligand-hemizygous mice, which are more likely to express subphysiological concentrations of the ligand in question, deleting the dependence receptor should result in a restoration of at least some of the cell survival lost by the decrease in trophic factor. This appears to be the case for the netrin–DCC pair, in which the DCC nulls display a much less pronounced olivary neuronal cell death than the netrin nulls (E Bloch-Gallego, personal communication). Thus, the presence of netrin-1 along the pathway of migration may be important not only to attract or repel axons or neurons but also as a key to support survival of these neurons. Beyond neuronal survival, netrin-1 availability may also contribute to physiological cell turnover, since in the intestine it is expressed in the crypts but not at the tips of the villi, where cells turn over following migration from the crypts.50
RET is expressed in the migratory neural crest cells that colonize the enteric nervous system, and RET plays a critical role in this migration, since the loss of function of RET or its ligand GDNF by gene inactivation in mice results in aganglionosis.114 However, a similar pathological state in humans – Hirschsprung disease – is not simply due to the loss of a trophic function mediated by RET, since some RET mutations associated with Hirschsprung disease do not interfere with trophic function (at least with the trophic function mediated by tyrosine kinase-related signaling).115 Therefore, it has been hypothesized that Hirschsprung disease is not simply due to an aberrant or reduced migration of neural crest cells but also due to their death.11 This observation is supported by work from Bordeaux et al. (noted above), who observed that five Hirschsprung-associated mutations convert RET's cell death-inducing profile from that of a typical dependence receptor to that of a constitutive proapoptotic molecule – that is, cell death is induced irrespective of ligand (here, GDNF).11 Thus, neural crest cell migration appears to be dictated by both positive signaling and by an inhibition of cell death induction by RET, the latter of which may serve to restrict cell migration to the region of adequate ligand availability.
Ptc, and its ligand, SHH, are crucial for determining cell fate during development, and in particular during early development of the neural tube.116 A putative ventro-dorsal gradient of SHH is produced by the floor plate and the notochord, allowing cell fate determination/differentiation of the ventral neurons of the neural tube. LeDouarin and colleagues have demonstrated that SHH specifies not only the differentiation of these cells, but also their survival, since the withdrawal of SHH leads to widespread PCD in the neural tube. Thibert et al. found that blocking Ptc-induced cell death during chick neural development inhibits the PCD occurring in a neural tube from which SHH has been withdrawn. Surprisingly, the introduction of a dominant-negative Ptc actually revealed the development of a neural tube-like structure despite the lack of SHH. Clearly, a more detailed study of the differentiation profile of the neuroepithelial cells in this rescued neural tube would be of interest; nonetheless, this initial result argues for a role of cell death not simply as a consequence of aberrant or inadequate differentiation but also as an active process that shapes the neural tube. It also supports a role for the dependence receptor Ptc in regulating cell fate during neural tube development. Interestingly, a recent study by Tessier-Lavigne and colleagues suggested that SHH – like DCC, UNC5H, and RET – may function as a guidance cue for specific (here, commissural) neurons via Ptc.117
Other dependence receptors may also turn out to be involved with neuronal migration or axonal guidance. Indeed, p75NTR was found to be a co-receptor for Nogo (along with the Nogo receptor), and as such p75NTR may be involved in the retraction of axons.100, 118 However, whether the proapoptotic activity of p75NTR is modulated by Nogo (or the Nogo receptor) is unknown. Similarly, integrins and their ligands (e.g., laminin) are likely to be involved in axonal guidance.119, 120 However, the proapoptotic role of the β-integrin in the absence of ligand has not been explored during neuronal migration or axonal guidance.
Conclusions
Potential mechanisms of environment-dependent apoptotic switching by dependence receptors
The receptors that create cellular dependence, when considered as a group, are relatively dissimilar at the level of amino-acid sequence, three-dimensional structure (in cases in which structure has been determined), and functional domains. Some, such as UNC5H1–3, display death domains, while most do not; most are type I transmembrane proteins, but Ptc and the androgen receptor are not. Most (and potentially all) display intracytoplasmic caspase cleavage sites, but this feature does not distinguish the group from many other proteins. One region of similarity between all of the dependence receptors is a region christened the DART domain (del Rio et al., unpublished data), but the function of this stretch is as yet unknown. On the other hand, all seem to share the functional similarity of apoptosis induction that is blocked by ligand binding. How is this achieved by a group of such disparate proteins?
One proposed mode features (at least) two distinct pathways mediated by these receptors: a classical signal transduction pathway that leads to signals that include an antiapoptotic response, and a nonclassical signal transduction pathway that is initiated by ligand withdrawal and results in a proapoptotic signal. In the absence of ligand, most if not all dependence receptors undergo proteolytic processing, generating stable peptides.9, 10 In at least some cases (e.g., the androgen receptor), processing is blocked when the receptors are ligand bound.
In the model proposed, the presence or absence of ligand determines the signal transduction pathway that will be utilized, and feedback loops create two stable states – full apoptosis induction or complete suppression – rather than supporting intermediate states.
In the absence of ligand, it is proposed that caspase cleavage leads to the production of contingency peptides, at least one of which includes a dependence domain. It is possible that the cleavage event may also lead to a change in secondary structure that allows presentation of the dependence domain.13
The proapoptotic peptides that are produced then, by definition of their proapoptotic activity, lead directly or indirectly to the processing of caspase zymogens to active caspases. For DCC, caspase-3 and -9 were found to interact with the intracellular domain of DCC. Similarly, caspase-8 was found to interact with β-integrin. However, in vitro, the presence of DCC, caspase-9, and -3 was not sufficient to induce caspase activation unless a cell lysate was added. This would suggest that intermediates are probably necessary to allow caspase activation.13 The pathway invoked by DCC required caspase-9 but not cytochrome c.13 For APP, it has been proposed that cleavage at D664 reveals a Smac/DIABLO-like activity requiring the amino-terminus of the APP-C31 peptide.121 Whether or not this model will prove to extrapolate to other dependence receptors is yet unknown, but most do not display a Smac/DIABLO-like region at the aminotermini of the dependence peptides.
Thus, the activation of apoptosis by dependence receptors in the absence of ligand binding (or potentially in the presence of a nontrophic ligand such as Aβ peptide) appears to involve caspase interaction, caspase cleavage of the receptor's intracytoplasmic domain, and potentially a Smac/DIABLO-like effect on iaps; however, none of these effects explains the initiation of the proapoptotic signal. If induced proximity is proposed to occur due to the caspase interactions, then it is unclear why multimerization, at least in some cases, inhibits the death induction.90 Furthermore, any potential Smac/DIABLO-like effect requires an initial caspase cleavage, and therefore does not explain the initiating event. A recent report from Llambi et al.,122 however, describes one potential initiating event: an interaction of UNC5H2 with DAP kinase via their death domains was observed. In the absence of netrin, DAP kinase autophosphorylation was reduced on Ser308 in the presence of UNC5H2, leading to an activation of the catalytic activity of DAP kinase and resultant cell death. The cell death was partially but not completely blocked by dominant-negative DAP kinase and in DAP kinase mutant cells. The addition of netrin restored DAP kinase autophosphorylation, reducing the catalytic activity of DAP kinase. Thus, for netrin-UNC5H2, netrin-1 withdrawal may initiate apoptotic signaling by mediating reduced DAP kinase autophosphorylation via UNC5H2. Nonetheless, this finding does not offer a mechanism by which other dependence receptors, especially those lacking death domains, may go beyond caspase amplification and function as apoptotic initiators.
Abbreviations
- DCC:
-
deleted in colorectal cancer
- LOH:
-
loss of heterozygosity
- NGF:
-
nerve growth factor
- PCD:
-
programmed cell death
- RTK:
-
receptor tyrosine kinase
- TrkA:
-
tyrosine receptor kinase A
- Unc5:
-
uncoordinated gene 5
References
Frisch SM and Francis H (1994) Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell. Biol. 124: 619–626
Bredesen DE, Ye X, Tasinato A, Sperandio S, Wang JJ, Assa-Munt N and Rabizadeh S (1998) p75NTR and the concept of cellular dependence: seeing how the other half die (see comments). Cell Death Differ. 5: 365–371
Yao R and Cooper GM (1995) Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 267: 2003–2006
Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL and Bredesen DE (1993) Induction of apoptosis by the low-affinity NGF receptor. Science 261: 345–348
Rabizadeh S and Bredesen DE (1994) Is p75NGFR involved in developmental neural cell death? Dev. Neurosci. 16: 207–211
Barrett GL and Bartlett PF (1994) The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc. Natl. Acad. Sci. USA 91: 6501–6505
Barrett GL and Georgiou A (1996) The low-affinity nerve growth factor receptor p75NGFR mediates death of PC12 cells after nerve growth factor withdrawal. J. Neurosci. Res. 45: 117–128
Bredesen DE and Rabizadeh S (1997) p75NTR and apoptosis: Trk-dependent and Trk-independent effects. Trends Neurosci. 20: 287–290
Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS and Bredesen DE (1998) The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature 395: 801–804
Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, Trifiro MA, Pinsky L, Wellington CL, Salvesen GS, Hayden MR and Bredesen DE (1999) Kennedy's disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J. Neurochem. 72: 185–195
Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, Bredesen DE, Edery P and Mehlen P (2000) The RET proto-oncogene induces apoptosis: a novel mechanism for Hirschsprung disease. EMBO J. 19: 4056–4063
Llambi F, Causeret F, Bloch-Gallego E and Mehlen P (2001) Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 20: 2715–2722
Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE and Mehlen P (2001) The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc. Natl. Acad. Sci. USA 98: 3416–3421
Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM and Cheresh DA (2001) Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J. Cell. Biol. 155: 459–470
Bredesen DE, Mehlen P and Rabizadeh S (2004) Apoptosis and dependence receptors: a molecular basis for cellular addiction. Physiol. Rev. 84: 411–430
Mehlen P and Bredesen DE (2004) The dependence receptor hypothesis. Apoptosis 9: 37–49
Mah SP, Zhong LT, Liu Y, Roghani A, Edwards RH and Bredesen DE (1993) The protooncogene bcl-2 inhibits apoptosis in PC12 cells. J. Neurochem. 60: 1183–1186
Bredesen DE (1996) Genetic control of neural cell apoptosis. Perspect. Dev. Neurobiol. 3: 101–109
Salvesen GS and Dixit VM (1997) Caspases: intracellular signaling by proteolysis. Cell 91: 443–446
Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW and Vogelstein B (1990) Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 247: 49–56
Cho KR and Fearon ER (1995) DCC: linking tumor suppressor genes and altered cell surface interactions in cancer? Curr. Opin. Genet. Dev. 5: 72–78
Fearon ER (1996) DCC: Is there a connection between tumorigenesis and cell guidance molecules? Biochim. Biophys. Acta 1288: M17–M23
Tanaka K, Oshimura M, Kikuchi R, Seki M, Hayashi T and Miyaki M (1991) Suppression of tumorigenicity in human colon carcinoma cells by introduction of normal chromosome 5 or 18. Nature 349: 340–342
Klingelhutz AJ, Hedrick L, Cho KR and McDougall JK (1995) The DCC gene suppresses the malignant phenotype of transformed human epithelial cells. Oncogene 10: 1581–1586
Klingelhutz AJ, Smith PP, Garrett LR and McDougall JK (1993) Alteration of the DCC tumor-suppressor gene in tumorigenic HPV-18 immortalized human keratinocytes transformed by nitrosomethylurea. Oncogene 8: 95–99
Velcich A, Corner G, Palumbo L and Augenlicht L (1999) Altered phenotype of HT29 colonic adenocarcinoma cells following expression of the DCC gene. Oncogene 18: 2599–2606
Kato H, Zhou Y, Asanoma K, Kondo H, Yoshikawa Y, Watanabe K, Matsuda T, Wake N and Barrett JC (2000) Suppressed tumorigenicity of human endometrial cancer cells by the restored expression of the DCC gene. Br. J. Cancer 82: 459–466
Mehlen P and Fearon ER (2004) Role of the dependence receptor DCC in colorectal cancer pathogenesis. J. Clin. Oncol. 22: 3420–3428
Fazeli A, Dickinson SL, Herminston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M and Weinberg RA (1997) Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature 386: 796–804
Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JKV, Markowitz S, Hamilton SR, Kern SE, Kinzler KW and Vogelstein B (1996) Evaluation of candidate tumor suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet. 13: 343–346
Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, Bredesen DE, Scoazec JY and Mehlen P (2004) Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 431: 80–84
Hedrick L, Cho KR, Fearon ER, Wu TC, Kinzler KW and Vogelstein B (1994) The DCC gene product in cellular differentiation and colorectal tumorigenesis. Genes Dev. 8: 1174–1183
Chuong C-M, Jiang T-X, Yin E and Widelitz RB (1994) cDCC (chicken homologue to a gene deleted in colorectal carcinoma) is an epithelial adhesion molecule expressed in the basal cells and involved in epithelial–mesenchymal interaction. Dev. Biol. 164: 383–397
Tessier-Lavigne M and Goodman CS (1996) The molecular biology of axon guidance. Science 274: 1123–1133
Cook G, Tannahill D and Keynes R (1998) Axon guidance to and from choice points. Curr. Opin. Neurobiol. 8: 61–72
Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS-Y, Culotti JG and Tessier-Lavigne M (1996) Deleted in colorectal cancer (DCC) encodes a netrin receptor. Cell 87: 175–185
Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC and Tessier-Lavigne M (1996) Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87: 1001–1014
Stein E, Zou Y, Poo M and Tessier-Lavigne M (2001) Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science 291: 1976–1982
Liu J, Yao F, Wu R, Morgan M, Thorburn A, Finley Jr RL and Chen YQ (2002) Mediation of the DCC apoptotic signal by DIP13 alpha. J. Biol. Chem. 277: 26281–26285
Zou H, Henzel WJ, Liu X, Lutschg A and Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90: 405–413
Corset V, Nguyen-Ba-Charvet KT, Forcet C, Moyse E, Chedotal A and Mehlen P (2000) Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature 407: 747–750
Shewan D, Dwivedy A, Anderson R and Holt CE (2002) Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat. Neurosci. 5: 955–962
Hong K, Hinck L, Nishiyama M, Poo MM, Tessier-Lavigne M and Stein E (1999) A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 97: 927–941
Matsunaga E, Tauszig-Delamasure S, Monnier PP, Mueller BK, Strittmatter SM, Mehlen P and Chedotal A (2004) RGM and its receptor neogenin regulate neuronal survival. Nat. Cell Biol. 6: 749–755
Meyerhardt JA, Look AT, Bigner SH and Fearon ER (1997) Identification and characterization of neogenin, a DCC-related gene. Oncogene 14: 1129–1136
Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB and Knowles BB (1997) The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature 386: 838–842
Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL and Tessier-Lavigne M (1997) Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature 386: 833–838
Williams ME, Strickland P, Watanabe K and Hinck L (2003) UNC5H1 induces apoptosis via its juxtamembrane region through an interaction with NRAGE. J. Biol. Chem. 278: 17483–17490
Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL, Verdi JM and Barker PA (2000) NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron 27: 279–288
Thiebault K, Mazelin L, Pays L, Llambi F, Joly MO, Scoazec JY, Saurin JC, Romeo G and Mehlen P (2003) The netrin-1 receptors UNC5H are putative tumor suppressors controlling cell death commitment. Proc. Natl. Acad. Sci. USA 100: 4173–4178
Tanikawa C, Matsuda K, Fukuda S, Nakamura Y and Arakawa H (2003) p53RDL1 regulates p53-dependent apoptosis. Nat. Cell. Biol. 5: 216–223
Takahashi M and Cooper G (1987) Ret transforming gene encodes a fusion protein homologous to tyrosine kinases. Mol. Cell. Biol. 7: 1378–1385
Takeichi M (1991) Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251: 1451–1455
Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson Jr EM and Milbrandt J (1996) Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 384: 467–470
Baloh RH, Tansey MG, Golden JP, Creedon DJ, Heuckeroth RO, Keck CL, Zimonjic DB, Popescu NC, Johnson Jr EM and Milbrandt J (1997) TrnR2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron 18: 793–802
Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F and Pachnis V (1994) Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367: 380–383
Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira S and Barbacid M (1996) Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 382: 70–73
Cacalano G, Farinas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A and Rosenthal A (1998) GFR{alpha}1 is an essential receptor component for GDNF in the develo. Neuron 21: 53–62
Santoro M, Wong WT, Aroca P, Santos E, Matoskova B, Grieco M, Fusco A and Di Fiore PP (1994) An epidermal growth factor receptor/ret chimera generates mitogenic and transforming signals: evidence for a ret-specific signalling pathway. Mol. Cell. Biol. 14: 663–675
Arighi E, Alberti L, Torriti F, Ghizzoni S, Rizzetti MG, Pelicci G, Pasini B, Bongarzone I, Piutti C, Pierotti MA and Borrello MG (1997) Identification of Shc docking site on Ret tyrosine kinase. Oncogene 14: 773–782
Lorenzo MJ, Gish GD, Houghton C, Stonehouse TJ, Pawson T, Ponder BA and Smith DP (1997) RET alternate splicing influences the interaction of activated RET with the SH2 and PTB domains of Shc and the SH2 domain of Grb2. Oncogene 14: 763–771
Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, Ponder MA, Telenius H, Tunnacliffo A and Ponder BAJ (1993) Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363: 458–460
Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, Holder S, Nihoul-Fekete C, Ponder BA and Munnich A (1994) Mutations of the RET proto-oncogene in Hirschsprung's disease. Nature 367: 378–380
Thibert C, Teillet MA, Lapointe F, Mazelin L, Le Douarin NM and Mehlen P (2003) Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science 301: 843–846
Charrier JB, Lapointe F, Le Douarin NM and Teillet MA (2001) Anti-apoptotic role of Sonic hedgehog protein at the early stages of nervous system organogenesis. Development 128: 4011–4020
Charrier JB, Teillet MA, Lapointe F and Le Douarin NM (1999) Defining subregions of Hensen's node essential for caudalward movement, midline development and cell survival. Development 126: 4771–4783
Rabizadeh S, Ye X, Sperandio S, Wang JJ, Ellerby HM, Ellerby LM, Giza C, Andrusiak RL, Frankowski H, Yaron Y, Moayeri NN, Rovelli G, Evans CJ, Butcher LL, Nolan GP, Assa-Munt N and Bredesen DE (2000) Neurotrophin dependence domain: a domain required for the mediation of apoptosis by the p75 neurotrophin receptor. J. Mol. Neurosci. 15: 215–229
Levi-Montalcini R (1966) The nerve growth factor: its mode of action on sensory and sympathetic nerve cells. Harvey Lect. 60: 217–259
Martin DP, Schmidt RE, deStefano PS, Lowry OH, Carter JG and Johnson EM (1988) Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell. Biol. 106: 829–844
Chao MV, Bothwell MA, Ross AH, Koprowski H, Lanahan AA, Buck CR and Sehgal A (1986) Gene transfer and molecular cloning of the human NGF receptor. Science 232: 518–521
Radeke MJ, Misko TP, Hsu C, Herzenberg LA and Shooter EM (1987) Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 325: 593–597
Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF and Chao MV (1991) High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor (see comments). Nature 350: 678–683
Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV and Anderson DJ (1994) p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron 12: 733–745
Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR and Greenberg ME (1997) Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science 275: 628–630
Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR and Miller FD (1998) p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell. Biol. 143: 1691–1703
Vaux DL, Cory S and Adams JM (1988) bcl-2 gene promotes haemopoietic cell survival and co-operates with c-myc to immortalise pre-B cells. Nature 335: 440–442
Sorg BA, Smith MM and Campagnoni AT (1987) Developing expression of the myelin proteolipid protein and basic protein mRNAs in normal and dysmyelinating mutant mice. J. Immunol. 49: 1146–1154
Frade JM, Rodriguez-Tebar A and Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166–168
Casaccia-Bonnefil P, Carter BD, Dobrowsky RT and Chao MV (1996) Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383: 716–719
Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC and Longo FM (1997) Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J. Neurosci. 17: 7594–7605
Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y and Nagata S (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66: 233–243
Ruan Y, Rabizadeh S, Camerini D and Bredesen DE (1997) Expression of CD40 induces neural apoptosis. J. Neurosci. Res. 50: 383–390
Naumann T, Casademunt E, Hollerbach E, Hofmann J, Dechant G, Frotscher M and Barde YA (2002) Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain. J. Neurosci. 22: 2409–2418
Bredesen DE (1994) Neuronal Apoptosis: Genetic and Biochemical Modulation. Apoptosis II: The Molecular Basis of Apoptosis in Disease Cold Spring Harbor: Cold Spring Harbor Laboratory Press pp. 397–421
Pflug BR, Onoda M, Lynch JH and Djakiew D (1992) Reduced expression of the low affinity nerve growth factor receptor in benign and malignant human prostate tissue and loss of expression in four human metastatic prostate tumor cell lines. Cancer Res. 52: 5403–5406
Stanford JL, Just JJ, Gibbs M, Wicklund KG, Neal CL, Blumenstein BA and Ostrander EA (1997) Polymorphic repeats in the androgen receptor gene: molecular markers of prostate cancer risk. Cancer Res. 57: 1194–1198
Ware CF, VanArsdale S and VanArsdale TL (1996) Apoptosis mediated by the TNF-related cytokine and receptor families. J. Cell. Biochem. 60: 47–55
Longo FM, Manthorpe M, Xie YM and Varon S (1997) Synthetic NGF peptide derivatives prevent neuronal death via a p75 receptor-dependent mechanism. J. Neurosci. Res. 48: 1–17
Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM, Kilpatrick TJ and Bartlett PF (2000) Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J. Biol. Chem. 275: 30537–30545
Wang JJ, Rabizadeh S, Tasinato A, Sperandio S, Ye X, Green M, Assa-Munt N, Spencer D and Bredesen DE (2000) Dimerization-dependent block of the proapoptotic effect of P75(NTR). J. Neurosci. Res. 60: 587–593
Rabizadeh S, Ye X, Wang JJ and Bredesen DE (1999) Neurotrophin dependence mediated by p75NTR: contrast between rescue by BDNF and NGF. Cell. Death Differ. 6: 1222–1227
Kong H, Kim AH, Orlinick JR and Chao MV (1999) A comparison of the cytoplasmic domains of the Fas receptor and the p75 neurotrophin receptor. Cell Death Differ. 6: 1133–1142
Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D, Suvanto P, Hanaoka T, Li Y, Irie S, Greene LA and Sato TA (2000) NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J. Biol. Chem. 275: 17566–17570
Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, Wang JJ, Leo E, Zapata J, Hauser CA, Reed JC and Bredesen DE (1999) TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J. Biol. Chem. 274: 30202–30208
Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G and Barde YA (1999) The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J. 18: 6050–6061
Frankowski H, Castro-Obregon S, del Rio G, Rao RV and Bredesen DE (2002) PLAIDD, a type II death domain protein that interacts with p75 neurotrophin receptor. J. NeuroMol. Med. 1: 153–170
Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M and Schecterson LC (2003) Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J. Neurosci. 23: 5425–5436
Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL and Petersen CM (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427: 843–848
Rabizadeh S and Bredesen DE (2003) Ten years on: mediation of cell death by the common neurotrophin receptor p75(NTR). Cytokine Growth Factor Rev. 14: 225–239
Wang KC, Kim JA, Sivasankaran R, Segal R and He Z (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74–78
Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Brufsky A, Talcott J, Hennekens CH and Kantoff PW (1997) The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc. Natl. Acad. Sci. USA 94: 3320–3323
Giancotti FG and Ruoslahti E (1990) Elevated levels of the alpha 5 beta 1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell 60: 849–859
Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH and Bredesen DE (2000) A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor (see comments). Nat. Med. 6: 397–404
Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S and Nicholson DW (1999) Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97: 395–406
Galvan V, Saganich M, Schroeder B, Gorostiza OF, Logvinova A, Banwait S, Jin K, Greenberg DA, Mucke L, Heinemann S, Koo EH and Bredesen DE (2004) Reversal of AD-like pathology in APP transgenic mice by mutation of ASP664 In Program Number: 488 4 (San Diego, CA: Society for Neuroscience) (Abstract)
Kibbey MC, Jucker M, Weeks BS, Neve RL, Van Nostrand WE and Kleinman HK (1993) beta-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc. Natl. Acad. Sci. USA 90: 10150–10153
Kusiak JW, Lee LL and Zhao B (2001) Expression of mutant amyloid precursor proteins decreases adhesion and delays differentiation of Hep-1 cells. Brain Res. 896: 146–152
Lu DC, Shaked GM, Masliah E, Bredesen DE and Koo EH (2003) Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein precursor complexes. Ann. Neurol. 54: 781–789
Terry R, Peck A, DeTeresa R, Schechter R and Horoupian D (1981) Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann. Neurol. 10: 184–192
Masliah E, Terry RD, DeTeresa RM and Hansen LA (1989) Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. 103: 234–239
La Spada AR and Clark AW (1997) Inherited neurodegenerative disorders caused by CAG/polyglutamine tract expansions: symposium introduction. Brain Pathol. 7: 877–880
Yee KT, Simon HH, Tessier-Lavigne M and O’Leary DM (1999) Extension of long leading processes and neuronal migration in the mammalian brain directed by the chemoattractant netrin-1. Neuron 24: 607–622
Bloch-Gallego E, Ezan F, Tessier-Lavigne M and Sotelo C (1999) Floor plate and netrin-1 are involved in the migration and survival of inferior olivary neurons. J. Neurosci. 19: 4407–4420
Baloh RH, Enomoto H, Johnson Jr EM and Milbrandt J (2000) The GDNF family ligands and receptors – implications for neural development. Curr. Opin. Neurobiol. 10: 103–110
Pelet A, Geneste O, Edery P, Pasini A, Chappuis S, Atti T, Munnich A, Lenoir G, Lyonnet S and Billaud M (1998) Various mechanisms cause RET-mediated signaling defects in Hirschsprung's disease. J. Clin. Invest. 101: 1415–1423
Placzek M, Dodd J and Jessell TM (2000) Discussion point. The case for floor plate induction by the notochord. Curr. Opin. Neurobiol. 10: 15–22
Charron F, Stein E, Jeong J, McMahon AP and Tessier-Lavigne M (2003) The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 113: 11–23
Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M and Poo MM (2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat. Neurosci. 5: 1302–1308
Hopker VH, Shewan D, Tessier-Lavigne M, Poo M and Holt C (1999) Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature 401: 69–73
Poinat P, De Arcangelis A, Sookhareea S, Zhu X, Hedgecock EM, Labouesse M and Georges-Labouesse E (2002) A conserved interaction between beta1 integrin/PAT-3 and Nck-interacting kinase/MIG-15 that mediates commissural axon navigation in C. elegans. Curr. Biol. 12: 622–631
Hell K, Saleh M, Crescenzo GD, O’Connor-McCourt MD and Nicholson DW (2003) Substrate cleavage by caspases generates protein fragments with Smac/Diablo-like activities. Cell Death Differ. 10: 1234–1239
Llambi F, Laurenco FC, Gozuacik D, Guix C, Pays L, Del Rio G, Kimchi A and Mehlen P (2005) The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J. 24: 1192–1201
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Bredesen, D., Mehlen, P. & Rabizadeh, S. Receptors that mediate cellular dependence. Cell Death Differ 12, 1031–1043 (2005). https://doi.org/10.1038/sj.cdd.4401680
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401680
Keywords
This article is cited by
-
Reciprocal antagonism between the netrin-1 receptor uncoordinated-phenotype-5A (UNC5A) and the hepatitis C virus
Oncogene (2017)
-
A Christianson syndrome-linked deletion mutation (∆287ES288) in SLC9A6 disrupts recycling endosomal function and elicits neurodegeneration and cell death
Molecular Neurodegeneration (2016)
-
Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not
Nature (2010)
-
Proteolytic cleavages give receptor tyrosine kinases the gift of ubiquity
Oncogene (2009)
-
Mitochondrial membrane permeabilization in neuronal injury
Nature Reviews Neuroscience (2009)
