
Abstract
Recent reports have brought back the acetylcholine synthesizing enzyme, choline acetyltransferase in the mainstream research in dementia and the cholinergic anti-inflammatory pathway. Here we report, a specific strategy for the design of novel ChAT ligands based on molecular docking, Hologram Quantitative Structure Activity Relationship (HQSAR) and lead optimization. Molecular docking was performed on a series of ChAT inhibitors to decipher the molecular fingerprint of their interaction with the active site of ChAT. Then robust statistical fragment HQSAR models were developed. A library of novel ligands was generated based on the pharmacophoric and shape similarity scoring function, and evaluated in silico for their molecular interactions with ChAT. Ten of the top scoring invented compounds are reported here. We confirmed the activity of α-NETA, the only commercially available ChAT inhibitor, and one of the seed compounds in our model, using a new simple colorimetric ChAT assay (IC50 ~ 88 nM). In contrast, α-NETA exhibited an IC50 of ~30 μM for the ACh-degrading cholinesterases. In conclusion, the overall results may provide useful insight for discovering novel ChAT ligands and potential positron emission tomography tracers as in vivo functional biomarkers of the health of central cholinergic system in neurodegenerative disorders, such as Alzheimer’s disease.
Similar content being viewed by others
Introduction
Dementia is a leading cause of death affecting more than 47.5 million people worldwide with additional 7.7 million new cases every year and expected to increase to 75.6 million by 20301. Alzheimer’s disease (AD) alone accounts for nearly 60–70% cases of dementia. Other forms include vascular dementia, dementia with Lewy bodies (DLB) and frontotemporal dementia and Down syndrome (DS).
One of the common hallmarks of AD, DLB and DS disorders is an early selective degeneration of cholinergic neurons2,3,4. Expression of the acetylcholine (ACh)-synthesizing enzyme, Choline acetyltransferase (ChAT; choline O-acetyltransferase, EC: 2.3.1.6) defines theses neurons. This enzyme catalyzes reversibly the transfer of acetyl group from acetyl-Co A to choline for synthesis of acetylcholine (ACh). The central cholinergic system consists of four “ChAT-containing” neuronal nuclei (Ch1-Ch4) in the basal forebrain5. Ch1 and Ch2 innervate the hippocampal complex, Ch3 the olfactory bulb and Ch4 the rest of cerebral cortex and amygdala5. Ch4-neurons are located in the nucleus basalis of Meynert (NBM). Histopathological analyses show 75–98% reduced levels of ChAT in brain regions that become affected early and severely in dementia6,7, such as the regions of medial temporal lobe8, involved in memory consolidation. This leads to the use of cholinesterase inhibitors with the rational of prolonging the action of ACh at the downstream targets of the remaining cholinergic projections. The targets of these drugs are acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). These two enzymes are responsible for degrading ACh, released into synaptic cleft and/or into extracellular fluids. Cholinesterase inhibitors are as today the main therapeutic agents with proven clinical symptomatic effect albeit modestly. Up to date, other therapeutic strategies have failed to show clinical effect.
An unmet need in the dementia field is the lack of a biomarker addressing the health of CNS, in particular of the cholinergic neuronal projections. In this context, in vivo brain imaging by positron emission tomography (PET) with specific tracers are gaining important clinical applications and is already proven to be invaluable translational research tool for understanding early pathological events. In the past decade a number of radiolabeled amyloid beta (Aβ) imaging agents have been developed as derivatives of specific dyes such as Congo red and thioflavin-T. They are targeted at fibrillar deposits of Aβ peptides in the AD brain. Pittsburgh compound B (PIB) is such an example of the preparation of a11C-labled Aβ tracer that is frequently used in clinical diagnosis of AD9. There are also currently several tracers under development that are targeting pathological aggregates of tau protein, which is another key feature of the AD brain.
Nonetheless, these tracers are capable to document the chronic build-up of Aβ or tau deposits in the brain rather than the acute phase events that are affecting the function and health of CNS. Thereby the core cholinergic enzyme ChAT might be of high interest for development of an in vivo functional bio-imaging marker. Recent research has extended the role of cholinergic signaling from merely in neurotransmission to anti-inflammatory pathways10 and diverse biological processes and disorders such as cancer11. Research on targeting ChAT for development of new ligands is thus of interest in elucidating the role of cholinergic signaling, and thereby need to be revived.
Despite the fact that this enzyme was discovered about a century ago12, remarkably few ChAT ligands are known, all of which act as inhibitors of the enzyme. These include derivatives of naphthylvinylpyridine, stilbazole, alkylaminoethyl esters and 2-(α-naphthoyl) ethyltrimethylammonium iodide (α-NETA)13. The major limitation of the most studied class of naphthylvinylpyridines compounds is that a quaternary amine is part of the structure, which poses in most cases poor brain permeability and thus limited applicability14.
Advances in computational methodologies have significantly improved discovery of new leads, and their transformation into clinically useful drugs15. This together with the recently resolved crystal structure of human ChAT, showing histidine (HIS324) as the major catalytic amino acid residue at the center of catalytic tunnel7,16, have provide the opportunity to perform systematic analysis for discovery of new potential ChAT ligands.
The Quantitative Structure Activity Relationship (QSAR) studies are popular due to their manifold contribution in successful design of several drug candidates in account of saving time, money and human resources17. Generally speaking, in silico QSAR methods are based on assumption that certain molecular descriptors correlate with the variation in the biological activity of a series of structurally similar compounds18. The major challenge in developing QSAR models is to identify specific sets of molecular descriptors which correlate well with the biological activities, such as enzyme inhibition.
Hologram QSAR (HQSAR) is a fragment based QSAR method, in which the structure of a molecule is subdivided into various components, such as bonds, chirality, donor/acceptor etc. Combinations of these components are then used to design a hologram, as structural fingerprints correlating with the biological activity of dataset molecules and to generate statistical models for predicting the activity of novel compounds with similar scaffold19,20.
In the present study a specific strategy is reported for the design of novel ChAT ligands based on HQSAR and molecular docking analyses on a dataset of 26 ChAT inhibitors with reported IC50. The results provide insight into the contribution of specific structural moieties of the compounds towards their activity on ChAT, which allowed us to select sixteen most potent compounds for use as reference and seed structures to generate novel ligands based on the pharmacophoric and shape similarity scoring function. The designed analogues were analyzed for their molecular interactions with the active site amino acid residues of ChAT using molecular docking and the top scoring compounds with overall high score are reported here. Additionally, we also performed in vitro evaluation of α-NETA, one of the only commercially available potent ChAT ligand using our in house developed colorimetric assays of ChAT, AChE and BuChE. Overall the results may be useful for design and discovery of novel and potent ChAT ligands and development of ChAT PET tracers for early diagnosis of Alzheimer Disease and other neurodegenerative disorders.
Results
Development of highly predictive Hologram QSAR models
HQSAR is a fragment based QSAR method which correlates structure based molecular fingerprints with the biological activity of dataset molecules and results in statistical models for predicting the activity of novel compounds with similar scaffold19,20.
First, structures along with biological activity of dataset of 26 known potent ChAT ligands (Table 1) were extracted from literature. Then statistical HQSAR models were prepared by using various components such as atoms (A), bonds (B), connections (C), hydrogens (D), chirality (Ch), donor-acceptor (DA). This approach implemented different combinations of the components (A/B, A/B/C, A/B/C/H, A/B/C/Ch, A/B/C/H/Ch, A/C/DA, A/B/C/H/DA, A/B/H and A/B/H/DA) with fragment size of 4–7 atoms, by partial least square regression, and were cross-validated by leave-one-out (LOO) methods21.
The results are summarized in Table 2.The best statistical parameters were obtained for the model no. 04, which was based on A/B/C/Ch fragments combination. This model resulted in LOO cross validated (CV) correlation coefficient (q2) of 0.809 and non-cross validated correlation coefficient (r2) of 0.977, along with the best hologram length of 151 and six components. The predictive strength of best HQSAR model (A/B/C/Ch) was then assessed by predicting the pIC50 value of dataset compounds. The actual pIC50, the predicted pIC50 and the CV-predicted pIC50values for the 26 compounds are given in Table 1. The correlations between the actual and the predicted pIC50 values are graphically depicted in Fig. 1. The high correlation between actual pIC50 and predicted pIC50 values indicated the good predictive ability of the developed HQSAR models. Indeed, the beauty and strength of HQSAR lies in its capability to provide information about individual atomic contribution to the biological activity using a color scale. The atomic contribution map for most active and least active compound is depicted in Fig. 2. As depicted, specific color is assigned to each atom according to its contribution towards the activity. Negative gradient contribution is assigned by red, red-orange, and orange color, while gradient of positive atomic contribution is assigned by yellow, yellow-green, and green. The atoms with neutral contribution are represented as white (see color key in Fig. 2). The N-methyl group on the pyridine ring was found essential, while the linker between the naphthyl group and the pyridine ring had high contribution for activity of the most active compound from the dataset (Fig. 2A). Removal of the N-methyl group led to loss of the activity as represented by the least active compound (Fig. 2B).

Plot of the actual pIC50 values versus the corresponding predicted pIC50 (A) and the cross validated (CV)-predicted pIC50 (B). The predicted pIC50 and CV-predicted pIC50value were obtained using best HQSAR model (A/B/C/Ch). The actual value corresponds to experimentally determined pIC50 using biological assay.

The N-methyl group on the pyridine ring is shown as green, which indicates a high positive contribution of this moiety for activity of the most active compound from the dataset (A). Removal of this group led to the loss of the activity as represented by the least active compound (B).
Bio-molecular docking revealed the structural basis of the binding affinity of ChAT ligands
In order to probe the mechanism of interaction of the ChAT ligands, all the compounds from the dataset were docked into the active site of the enzyme using Surflex-Dock GeomX (SFXC) module of SYBYL-X2.1.1 suite22. Docking was performed utilizing the crystal structure of ChAT (PDB ID: 2FY3)7.The docking site was defined using coordinates of co-crystallized choline molecule. As a first step of docking, a validation of docking protocol was performed by re-docking the co-crystallized ligand in the active site of the enzyme. The RMSD between co-crystallized conformation and docked conformation has a value of 0.63 Å which indicated the suitability of docking protocol.
All of the 26 compounds in the dataset were docked into the active site of ChAT, and the docking score (Total-Score), which represent –logKd are given in Table 3. The compounds with –logKdvalue larger than 6 were deemed to be the most active, represented by the compounds 1, 13, 18, 20, and 24, among which compound 24 exhibited the highest Total-Score (−logKd of 8.12).
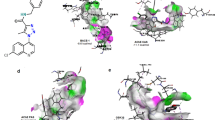
The 3D docked pose and 2D ligand interaction diagram of the compounds 1, 13, and 18 is shown in Fig. 3(A–C). As illustrated in the Fig. 3A, the naphthyl group of compound 1form π-π interactions with the TYR552 amino acid residue in the active site of ChAT. The nitrogen of the pyridine group was involved in a cation-π interaction with the HIS324 which is the catalytic amino acid responsible for transfer of acetyl group from A-CoA to choline. In addition, The TYR85, ASN95, SER96, VAL555, SER538, and GLY561 residues seemed to form a pocket to accommodate naphthyl group.

Three-dimensional docking poses of the compounds 1, 13 & 18 (A–C, respectively). The residues forming the potential interactions are given as stick model. The 2D ligand-interaction diagram illustrates the major interactions between the ligand and the active sites amino acid residues of ChAT.
The top scoring pose for the compound 13 (Fig. 3B) on the other hand indicated that the naphthyl group was directly involved in π-π interactions with the main catalytic amino acid residue HIS324, while the pyridine ring was extended away from the catalytic pocket and was surrounded by TYR85, LEU90 and ASN88 residues. The indole group of compound 18 (Fig. 3C), however like the compound 1 formed π-π interactions with TYR552, while HIS324 was one of residues that together with PRO98, GLY553, GLY561, VAL555, THR539 amino acid residues surrounded the indole moiety of the compound 18. The pyridine nucleus was in vicinity of SER438, ILE330, SER440, and GLN541 residues, which might indicate that they are involved in 3D orientation of the active compound.
The binding mode of compound 20 differed relative to the aforementioned compounds (Fig. 4A). In this case, the pyridine nucleus seemed tightly sandwiched between theTYR552 and HIS324 residues by π-π and π-cation interactions, most likely indicating their role in stabilizing the active conformation. The Phenyl-moiety of this compound was accommodated in a cavity formed by SER440, ASP328, ILE330, GLY329, and GLN541 residues.

Three-dimensional docking poses of the compounds 20, 23 & 24 (A–C). The residues forming the potential interactions are given as stick model. The 2D ligand-interaction diagram illustrates the major interactions between the ligand and the active sites amino acid residues of ChAT.
In the case of compound 23 (Fig. 4B), the azanaphthyl group interacted with both the TYR552 (π-π interaction) and HIS324 residues (π-cation interaction). For compound 24, the most active compounds with a pIC50 value of 6.28 (Fig. 4C), the docking pose once again includes interaction with these two amino acid residues. In this case, a 4-azanaphthyl group seemed become stabilized in 3D (Cartesian) space through formation of a π-π interaction with HIS324 and TYR436, and π-cation interactions with HIS324 and TYR552 residues. In contrast to all other active compounds, a hydroxyl group was present, which formed a hydrogen bond with SER438 residue at a distance of 2.30 Å. This additional interaction might be the reason behind the overall high activity of compound 24 toward ChAT.
The compound 26, (2-(α-naphthoyl) ethyltrimethylammonium iodide) was of particular interest since this is the only ChAT ligand that currently is commercially available, and is known as (α-NETA)13. For α-NETA, the naphthyl group formed π-π interaction with TYR552, while the quaternary trimethyl ammonium moiety was closely surrounded by HIS324, PRO98, ASP328 residues. The naphthyl moiety was instead accommodated in a pocket consisting of ASN95, PRO554, GLY553, THR539, and SER538 residues.
Thus the overall pattern seemed to suggest that the TYR552 and HIS324 amino acid residues are of outmost importance for stabilization of an active conformation of ligands of ChAT.
In vitro assessment of α-NETA as a potent selective ChAT inhibitor
As noted, α-NETA was the only compound available commercially. This allowed us to challenge and somewhat validate our approach. Purified recombinant human ChAT protein was prepared and used to test the activity of α-NETA in a recently developed in-house colorimetric assay of ChAT. In these in vitro analyses, α-NETA exhibited potent inhibitory activity of ChAT, with an estimated IC50 of 88 nM (Fig. 5B). Since the ChAT counteracting, enzymes, AChE and BuChE apparently share affinity for acetylcholine, we deemed important to determine the selectivity of α-NETA. The determined IC50 values were 34.18 μM for AChE and 33.30 μM for BuChE. This indicated that α-NETA had 388 fold higher selectivity for ChAT compared to AChE, and ~378 fold relative to BuChE (Fig.5B).

Three-dimensional docking poses of α-NETA (A) and its corresponding 2D ligand-interaction diagram. The 2D ligand-interaction diagram illustrates the major interactions between the ligand and the active sites amino acid residues of ChAT. (B) shows the in vitro dose-response curves for α-NETA against ChAT, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). α-NETA was pre-incubated at the specified concentrations for 30 min with samples. The enzyme activities were measured as described in the methods section. The effect of α-NETA on the cholinergic enzymes was compared to the activity of a control sample that was preincubated with the buffer. The IC50 values were calculated using GraphPad Prism.
Design of novel library of ChAT ligands by Lead optimization
Next the chemical structure of the aforementioned top scoring compounds was used to generate novel analogues, using Muse® Invent™ molecular design package. Briefly, the algorithm uses seed structures of the lead compounds as input, which are modified stepwise into novel analogues and rank them according to choice of scoring function23. For this procedure all compounds with a pIC50 value greater than five were selected, resulting in a set of 16 references and seed structures. This algorithm produces not only new structures with high pharmacophoric and 3D shape similarity relative to the parent structures, but also generate new compounds with moderate structural similarity to the reference compounds24. Through stepwise cycles this algorithm may hence generate completely new class of ligands. Here, this procedure generated a library of 996 novel potential ChAT ligands through 50 stepwise generated selection cycles.
To get insight into their interaction with ChAT’s active site amino acid residues, the generated library were subjected to molecular docking using the approach described before. The docking score for ten top-scoring compounds along with other parameters is given in the Table 4. The other parameters were those determining the BBB permeability and included hydrogen bond acceptor count, hydrogen bond donor count, rotatable bond count, total polar surface area (TPSA), and a calculated partition coefficient (ALogP).
The 3D docked pose of these ten top scoring compounds are illustrated in the Fig. 6.

The compounds are rendered as ball and stick.
Discussion
Here we described generation of novel ChAT ligands from a dataset of 26 active compounds. In the course, predictive HQSAR models based on the 26 active compounds was developed, which revealed the fragments essential for the activity of these ChAT ligands. Bio-molecular docking analysis then demonstrated the most frequent amino acids residues on the active site of ChAT together with functional groups in the active compounds. In this context, the Tyr552 and His324 were shown to be the two amino acids forming π-π and/or π-cation interactions with certain functional moiety of the ChAT ligands.
ChAT is the enzyme responsible for synthesis of acetylcholine, one of the most important signaling substances widely used by nature in diverse physiological processes. ChAT defines cholinergic neurons which are selectively affected in AD and DLB disorders7, and may hence be a potential biomarker of neurodegenerative dementia disorders. Development of novel potent ChAT ligands may thereby be useful as PET tracers to study the health of CNS and neurodegeneration and/or for diagnostic purposes.
However, none of these compounds are useful as PET tracer of cholinergic network in the brain due to their instability and/or BBB impermeability. Thereby we used the insights from the HQSAR and consequential molecular docking and selected sixteen most potent ChAT ligands as seed structure and reference compounds. A virtual library of 996 new ligands was generated and the top scoring analogues are reported here. Importantly, the new ligands had high shape and pharmacophore similarity but relatively low structural resemblance to the parent compounds.
In the current analyses Hologram QSAR was employed. This is a specialized fragment based methods where the binding affinity of the compounds are determined on the basis of their molecular fragments19,20. The predictive power of developed models was based on LOO method, which is one of the most used methods. The best model developed contained the combination of A/B/C/Ch components, which gave highest cross validated correlation coefficient (a Q2 value of 0.809). Our Hologram QSAR results are in close agreement with an earlier study based on CoMFA and CoMSIA modeling25. For instance in agreement with our findings, their results indicated that a pyridine ring, which is one of the most common functional group among the parent compounds, had major contribution towards the affinity for ChAT. Nonetheless, Hologram QSAR modeling has several advantages compared to CoMFA and CoMSIA modeling. Hologram QSAR models in this study are not only stable and highly predictive but also avoid additional steps, such as alignment and approximation of biologically active conformation of the compound which are necessary in CoMFA and CoMSIA modeling.
In our modeling the crystal structure of a recombinant human ChAT(PDB id: 2FY3) that has quite recently reported together with co-crystallized choline as natural ligand into the active site of ChAT was used7. This information was crucial and allowed validation of our docking protocol by redocking of choline, resulting in optimal root mean square deviation (RMSD) between its actual conformation and docked conformation. As was noted, the biomolecular docking of the most active compounds reported in literature revealed that HIS324 is one of the most important amino acid residues for binding of these ligands to the active site of ChAT. Interestingly, HIS324 is located in the middle of catalytic tunnel of the enzyme, and is reported to be responsible for transfer of acetyl group from acetyl-CoA to choline and synthesis of acetylcholine.
The compound α-NETA was the only parent compound which was commercially available. Although it has been reported as a ChAT inhibitor, there seem to be a controversy because it was initially tested on acetyltranferase found in seminal fluids, which are considered to be carnitine acetyltransferase rather than ChAT. In our re-docking analyses, α-NETA had a Total-score (−logKd) of 5.3 and a reported pIC50 of 5.05 (an IC50 of ~9 μM) on purified human placental ChAT13.To check our model the IC50 of α-NETA by a recently reported high-throughput colorimetric ChAT assay, using recombinant human ChAT protein was determined. An IC50 of 88 nM was found, which suggest ~100 folds higher potency for α-NETA as ChAT inhibitor in our system. Furthermore, α-NETA is reported to produce 10% inhibition of AChE at 1 mM concentration13. In our hand, α-NETA produced a 10% inhibition of AChE at a concentration of 10 μM and BuChE at a concentration of 3 μM. However, α-NETA exhibited an IC50 of 48.60 μM against AChE and 33.30 μM for BuChE, once again using a high-throughput colorimetric AChE and/or BuChE assay. Nonetheless based on IC50 ratios, α-NETA exhibited ~388 fold and ~378 fold higher selectivity for ChAT in our in vitro analyses compared to AChE and BuChE, respectively. Thus our report confirms that α-NETA is a highly selective ChAT inhibitor.
In any case, α-NETA had a Total-score of 5.3, while the top ten new designed analogues reported in the current study had a Total-score of over 8 which may suggest a much higher potency than α-NETA. This warrants further development of these analogues as new classes of ChAT ligands. In addition, we included several filtering criteria such as BBB permeability, accessibility for radiolabeling and pan assay interference compounds (PAINS) filter26, which altogether may prove them useful as ChAT PET tracers.
In addition an updated protocol for the colorimetric ChAT assay which can easily be adopted to run on 384-wells microtiter plates, with proven suitability as high throughput in vitro screening tool of new ChAT ligands was applied. Nonetheless, it should be noted that the PAINS filter criteria is important since it should reduce the selection of hits that may act as unspecific multi-target inhibitors of diverse enzymes26.The described ChAT assay uses two axillary enzymes, namely horseradish peroxidase and choline oxidase as detection system. Thereby the PAINS filter is important to reduce possible interferences of the hits with our in vitro screening assay.
The ease of radiolabeling is an essential requirement for developing a PET tracer. Keeping this in view, we analyzed the designed novel compounds for their radiolabeling feasibility. As shown in Fig. 7, it was observed that most of the designed compounds contain a chemical group which can be easily radiolabeled with 11C which is the most common radiolabeling isotope.

The possible radiolabeling position is highlighted in red. The structures of α-NETA and Acetylcholine are also given for structural similarity comparison. Marvin was used for calculating pKa/Log D @ pH 7.4 values for the compounds, Marvin 15.4.13.0, 2015, ChemAxon (http://www.chemaxon.com).
In conclusion, we report here a novel strategy to design novel ChAT ligands which can be developed into PET tracers with a potential to monitor the therapy and also probe the mechanism of neurodegeneration in AD and other dementia disorders.
Methods
Computational details
The molecular modeling studies including molecular docking and HQSAR analysis were performed using SYBYL-X 2.1.1 molecular modeling suite22 installed on Linux based Dell Precision T7610 workstation [Intel® Xeon® E5-2643 CPU @ 3.3 GHz; 16 GB RAM, 2 TB hard disk]. Muse® Invent™ molecular design workflow was utilized for ligand optimization and design of novel analogs.
Dataset
The dataset compounds were obtained from the literature with their ChAT inhibitory potency determined using the same protocol to obtain a good QSAR model. The given molar inhibitory concentration was converted to negative logarithmic scale (PIC50) of the compounds and was used as a dependent variable for HQSAR. In addition, we also included α-NETA13 in the dataset molecule as it is the only commercially available, potent ChAT inhibitor. The 2D structure of the molecules was drawn, converted to 3D and followed by energy minimization with gradient descent method. The energy was minimized using Confort module of SYBYL X 2.1.1. The final 3D global minima conformation of the molecules was used for molecular docking studies as well for development of HQSAR models.
Molecular docking
Docking was performed utilizing the crystal structure of ChAT (PDB ID: 2FY3)7 to get insight into the binding mode of dataset compounds in the active site of ChAT and performed with the Surflex-Dock module interfaced in SYBYL-X 2.1.1 which is a fully automatic flexible molecular docking algorithm with a combination of empirical scoring function and a surface based molecular similarity based search engine27. Prior to docking, the 3D structure of receptor was prepared as it is very common that the crystallographic data can have several problems in the structures which are needed to be fixed before molecular simulation28. The process of protein preparation includes addition of hydrogens, repairing side chain, treating termini, fixing atom type, setting protonation state, fixing bond order, adding charge, and fixing side chain amide. Finally the prepared structure is minimized in order to remove the strain produced during the earlier protein preparation steps. The ‘protomol’ which is the defined binding pocket of receptor was generated using the co-crystallized ligand in the active site of ChAT. The prepared dataset compounds were docked into the active site of ChAT using Surflex-Dock GeomX (SFXC) approach and the compounds were ranked using Total_Score (−logKd). Similar strategy was used for docking analysis of novel designed compounds.
HQSAR studies
Hologram QSAR (HQSAR) offers high quality QSAR models very rapidly and easily. In order to gain information about the individual contribution of molecular fragments of the potent molecule and to use the information along with the information obtained from molecular docking study to design novel potent analogs, HQSAR analysis was performed on the dataset compounds. HQSAR is based on the assumption that structure is a key determinant of all molecular property and the structural information encoded in the 2D molecular fingerprints can be thus successfully used for predict the activity of a molecule. As compared to traditional 2D fingerprints, molecular hologram contains additional information such as branched and cyclic fragments and stereochemistry of the molecule. Also, the molecular representation is purely binary in the case of hashed fingerprints while in the case of hologram, the bins contain information about the number of fragments hashed into each bin. The QSAR models were developed using various combinations of molecular fragments and fragment generation parameters for each hologram length in HQSAR module of SYBYL X 2.1.1 using partial least square (PLS) method21. In PLS method, the basic aim is to avoid over-fitted QSAR model by selecting the optimum number of components from PLS analysis with high cross validation correlation coefficient (q2) and low standard error of prediction. The generated models were cross validated using leave one out (LOO) and various statistical parameters such as optimum number of components (N) which provides highest q2 value, cross validation correlation coefficient (q2), coefficient of determination (r2). The various developed models on the basis of components for fragment generation are given in Table 3.
Lead optimization and design of new analogues
The Muse® Invent™ molecular design workflow was used to design several analogs based on the initial starting structures of active ligands from dataset. It contains a scoring function based on the pharmacophoric and shape based structural similarity24,29. The active compounds with pIC50 > 5 were used as not only reference molecules but also as seed structures. The other parameters were kept as default including 39 questionable structures and 99 undesirable structures to exclude. The top scored individuals from the population survives based on the cutoff score and further used to produce more generations until the optimization criteria are met. The scoring function, TriposScore based on ligands try to optimize the shape and pharmacophoric similarity to a set of lead structures. It also takes into account cLogP, MW, TPSA, Lipinski Counts, and Rotatable Bond Counts30.
Purification of recombinant human ChAT
DYT media (16 g/l Tryptone, 10 g/l yeast extract, 5 g/l NaCl, 100 μg/ml ampicillin, 34 μg/ml chloramphenicol) was inoculated with a preculture of E. Coli BL21 Rosetta2 transformed with pProEXHTa-ChAT (a generous gift from Brian Shilton, Department of biochemistry, University of western Ontario, London, Canada). The bacteria were grown at 37 °C and 200 rpm until the optical density at 600 nm reached 0.5. After which 0.5 mM IPTG was added and His6-ChAT was expressed for circa 16 h at 18 °C. The bacteria was harvested and stored at −80 °C. His6-ChAT was purified with “Ni-NTA fast start Kit” (Qiagen) following the manufacturer’s instructions. The elution buffer was exchanged to storage buffer (10 mMTris pH 7.4, 500 mMNaCl, 10% (v/v) glycerol) using Amicon Ultra concentrators (Merck Millipore) with a molecular cutoff of 30 kDa. The protein preparation was aliquoted, frozen at dry ice and stored at −80 °C. The absence of contaminating proteins was determined using sodium dodecyl sulfate PAGE and Coomassie staining. The total protein concentration was measured with BioRad DC protein Assay (BioRad).
Experimental enzymatic activity assays
A modified version of Ellman’s colorimetric assay was used for the enzymatic activity of BuChE and AChE, as described previously31,32. For BuChE activity, 50 μL/well of a 1:400 diluted solution of a pooled human plasma sample was used. For AChE activity, the 1:400 diluted pooled human plasma samples had been supplied with 50 ng/ml of purified AChE protein (Sigma). These were preincubated with 50 μL/wells of different concentrations of α-NETA for 30 minutes at room temperature. Then, 100 μL of a cocktail (Na/K phosphate buffer, containing 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, final concentration 0.4 mM) and butyrylthiocholine iodide (Sigma, final concentration 5 mM) or acetylthiocholine iodide (Sigma, final concentration 0.5 mM) were added and the changes in absorbance was monitored at one minutes intervals at 412 nm wavelength. For the AChE activity, the cocktail also contained the selective BuChE inhibitor ethopropazine (Sigma, final concentration 0.1 mM).
ChAT activity was measured with a new colorimetric assay as described previously33, using recombinant ChAT protein. Briefly, 20 μL/wells of 1.5 μg/ml of the recombinant ChAT was incubated with 20 μL/well of different concentrations of α-NETA for 30 minutes at room temperature in dilution buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1.0 mM EDTA, 0.05% (v/v) Triton X-100). Then 60 μl of a cocktail-A [dilution buffer containing choline chloride (Sigma, final concentration 250 μM), eserine (E8625, Sigma-Aldrich, final concentration 60 μM), acetyl coenzyme-A (A2181, Sigma-Aldrich, final concentration 50 μM), phosphotransacetylase (P2783, Sigma-Aldrich, final concentration 1.02 U/ml), lithium potassium acetyl-phosphate (#01409, Sigma-Aldrich, final concentration 12 mM)] was added to the samples. The final concentration of ChAT in the wells was 0.3 μg/mL.
In separate wells, 100 μl of a serial two-fold dilution of choline chloride (500–0 μM) was applied in triplicates, which were used as standards for determining choline concentration in the wells after reaction with ChAT. The plate was incubated for 20 minutes at 37 °C.
Then 50 μl of a cocktail-B [phosphate buffered saline, containing 0.93 U/ml choline oxidase (C5896, Sigma-Aldrich), 1/5000 U streptavidin-horseradish peroxidase, 6.3 mM phenol, and 3 mM 4-aminoantipyrine (A4382, Sigma-Aldrich)] was added to each sample including the standards. Absorbance was then monitored using a microplate spectrophotometer reader (Infinite M1000, Tecan) at 500 nm wavelength. ChAT activity (nmol/min/mg of recombinant protein) was calculated according to the following formula: ChAT activity [ChBL-ChS]/t/m, where ChBL is the measured number of mole of choline in control wells lacking the inhibitor and ChAT, ChS is the measured number of mole of choline in the sample wells, the t is the incubation time and m is the mass of ChAT protein added per sample well. Inhibition is given as compared to a ChAT sample incubated with only buffer.
Additional Information
How to cite this article: Kumar, R. et al. Novel ligands of Choline Acetyltransferase designed by in silico molecular docking, hologram QSAR and lead optimization. Sci. Rep. 6, 31247; doi: 10.1038/srep31247 (2016).
References
WHO 10 facts on dementia < http://www.who.int/features/factfiles/dementia/en/> (2015).
Mesulam, M. M. Cholinergic Circuitry of the Human Nucleus Basalis and Its Fate in Alzheimer’s Disease. J. Comp. Neuro. 521, 4124–4144 (2013).
Bohnen, N. I. et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch. Neurol. 60, 1745–1748 (2003).
Yates, C. M., Simpson, J., Maloney, A. F., Gordon, A. & Reid, A. H. Alzheimer-like cholinergic deficiency in Down syndrome. Lancet 2, 979 (1980).
Mesulam, M. M. The cholinergic innervation of the human cerebral cortex. Prog. Brain Res. 145, 67–78 (2004).
Wilcock, G. K., Esiri, M. M., Bowen, D. M. & Smith, C. C. Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 57, 407–417 (1982).
Kim, A. R., Rylett, R. J. & Shilton, B. H. Substrate binding and catalytic mechanism of human choline acetyltransferase. Biochemistry 45, 14621–14631 (2006).
Esiri, M. M., Pearson, R. C., Steele, J. E., Bowen, D. M. & Powell, T. P. A quantitative study of the neurofibrillary tangles and the choline acetyltransferase activity in the cerebral cortex and the amygdala in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 53, 161–165 (1990).
Nordberg, A., Rinne, J. O., Kadir, A. & Langstrom, B. The use of PET in Alzheimer disease. Nat. Rev. Neurol. 6, 78–87 (2010).
Tracey, K. J. Reflex control of immunity. Nat. Rev. Immunol. 9, 418–428 (2009).
Cheng, K. et al. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver. Physiol. 295, G591–G597 (2008).
Nachmansohn, D. & Machado, A. L. The formation of acetylcholine. A new enzyme: “Choline Acetylase”. J. Neurophysiol. September 1, 397–403 (1943).
Sastry, B. V., Jaiswal, N., Owens, L. K., Janson, V. E. & Moore, R. D. 2-(alpha-Naphthoyl)ethyltrimethylammonium iodide and its beta-isomer: new selective, stable and fluorescent inhibitors of choline acetyltransferase. J. Pharmacol. Exp. Ther. 245, 72–80 (1988).
Mehta, N., Musso, D. & White, H. Water soluble choline acetyltransferase inhibitors: SAR studies. Eur. J. Med. Chem. 20, 443–446 (1985).
Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug. Discov. 1, 882–894 (2002).
Green, K. D., Porter, V. R., Zhang, Y. & Garneau-Tsodikova, S. Redesign of cosubstrate specificity and identification of important residues for substrate binding to hChAT. Biochemistry 49, 6219–6227 (2010).
Kumar, R. & Kumar, M. 3D-QSAR CoMFA and CoMSIA studies for design of potent human steroid 5α-reductase inhibitors. Med. Chem. Res. 22, 105–114 (2013).
Malla, P., Kumar, R. & Kumar, M. Validation of formylchromane derivatives as protein tyrosine phosphatase 1B inhibitors by pharmacophore modeling, atom-based 3D-QSAR and docking studies. Chem. Biol. Drug. Des. 82, 71–80 (2013).
Zhang, C., Du, C., Feng, Z., Zhu, J. & Li, Y. Hologram quantitative structure activity relationship, docking, and molecular dynamics studies of inhibitors for CXCR4. Chem. Biol. Drug. Des. 85, 119–136 (2015).
Primi, M. C. et al. Convergent QSAR studies on a series of NK receptor antagonists for schizophrenia treatment. J. Enzyme Inhib. Med. Chem. 1–12 (2015).
Kumar, R., Kumar, A., Jain, S. & Kaushik, D. Synthesis, antibacterial evaluation and QSAR studies of 7-[4-(5-aryl-1, 3, 4-oxadiazole-2-yl) piperazinyl] quinolone derivatives. Eur. J. Med. Chem. 46, 3543–3550 (2011).
Radosevic-Stasic, B. et al. Immunological consequences of lesions of nucleus basalis in rats. Int. J. Neurosci. 51, 325–327 (1990).
Gao, J., Wu, X. H., Dong, W. L. & Wang, S. Q. A series of propofol analogs design by targeting pentameric ligand-gated ion channel in silico method. Protein Pept. Lett. 20, 1238–1245 (2013).
Damewood, J. R. Jr., Lerman, C. L. & Masek, B. B. NovoFLAP: A ligand-based de novo design approach for the generation of medicinally relevant ideas. J. Chem. Inf. Model. 50, 1296–1303 (2010).
Chandrasekaran, V., McGaughey, G. B., Cavallito, C. J. & Bowen, J. P. Three-dimensional quantitative structure-activity relationship (3D-QSAR) analyses of choline acetyltransferase inhibitors. J. Mol. Graph. Model. 23, 69–76 (2004).
Everett, J. R. Academic drug discovery: current status and prospects. Expert Opin. Drug Discov. 10, 937–944 (2015).
Jain, A. N. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 46, 499–511 (2003).
Warren, G. L., Do, T. D., Kelley, B. P., Nicholls, A. & Warren, S. D. Essential considerations for using protein–ligand structures in drug discovery. Drug Discov. Today 17, 1270–1281 (2012).
Liu, Q., Masek, B., Smith, K. & Smith, J. Tagged fragment method for evolutionary structure-based de novo lead generation and optimization. J. Med. Chem. 50, 5392–5402 (2007).
Bös, F., Uhrig, U., Masek, B. B. & Damewood, J. R. Muse + TriposScore: a ligand-based de novo design approach. j. Cheminform. 3, P26–P26 (2011).
Darreh-Shori, T., Brimijoin, S., Kadir, A., Almkvist, O. & Nordberg, A. Differential CSF butyrylcholinesterase levels in Alzheimer’s disease patients with the ApoE epsilon4 allele, in relation to cognitive function and cerebral glucose metabolism. Neurobiol. Dis. 24, 326–333, 2006).
Darreh-Shori, T. et al. Inhibition of acetylcholinesterase in CSF versus brain assessed by 11C-PMP PET in AD patients treated with galantamine. Neurobiol. Aging 29, 168–184 (2008).
Vijayaraghavan, S. et al. Regulated Extracellular Choline Acetyltransferase Activity- The Plausible Missing Link of the Distant Action of Acetylcholine in the Cholinergic Anti-Inflammatory Pathway. PLoS One 8, e65936 (2013).
Acknowledgements
This study was supported by grants from KI Foundations; Olle Engkvist Byggmästare Foundation; Åke Wibergs Foundation; Åhlén-Foundation; Gunvor and Josef Anérs Foundation; Magnus Bergvalls Foundation; Demens Foundation (Demensfonden); Gun and Bertil Stohnes Foundation; Ragnhild & Einar Lundströms Foundation; Foundation for Sigurd & Elsa Goljes Memory; Tore Nilsson Foundation; the Foundation for Old Servants; Alzheimer’s Association, USA. We are thankful to Emmy Rannikko for performing the enzyme purification and the in vitro assays. We gratefully acknowledge Dr. Brian H. Shilton (University of Western Ontario, ON, Canada) for the generous gift of the hChAT-containing plasmid.
Author information
Authors and Affiliations
Contributions
R.K. and T.D.-S. designed, performed the study and wrote the initial draft of the manuscript. B.L. provided critical input and evaluated the possibility of radiolabeling the invented compounds. All the authors read and approved the final draft of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kumar, R., Långström, B. & Darreh-Shori, T. Novel ligands of Choline Acetyltransferase designed by in silico molecular docking, hologram QSAR and lead optimization. Sci Rep 6, 31247 (2016). https://doi.org/10.1038/srep31247
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31247
This article is cited by
-
Significance of Choline Acetyltransferase Enzyme in Tackling Neurodegenerative Diseases
Current Molecular Biology Reports (2022)
-
Systemic administration of choline acetyltransferase decreases blood pressure in murine hypertension
Molecular Medicine (2021)
-
Visual hallucinations in Alzheimer's disease do not seem to be associated with chronic hypoperfusion of to visual processing areas V2 and V3 but may be associated with reduced cholinergic input to these areas
Alzheimer's Research & Therapy (2019)
-
Novel CMKLR1 Inhibitors for Application in Demyelinating Disease
Scientific Reports (2019)
-
Identification of TAAR5 Agonist Activity of Alpha-NETA and Its Effect on Mismatch Negativity Amplitude in Awake Rats
Neurotoxicity Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
