
Abstract
PSD-95 associated PSD proteins play a critical role in regulating the density and activity of glutamate receptors. Numerous previous studies have shown an association between the genes that encode these proteins and schizophrenia (SZ) and autism spectrum disorders (ASD), which share a substantial portion of genetic risks. We sequenced the protein-encoding regions of DLG1, DLG2, DLG4, DLGAP1, DLGAP2, and SynGAP in 562 cases (370 SZ and 192 ASD patients) on the Ion PGM platform. We detected 26 rare (minor allele frequency <1%), non-synonymous mutations, and conducted silico functional analysis and pedigree analysis when possible. Three variants, G344R in DLG1, G241S in DLG4, and R604C in DLGAP2, were selected for association analysis in an independent sample set of 1315 SZ patients, 382 ASD patients, and 1793 healthy controls. Neither DLG4-G241S nor DLGAP2-R604C was detected in any samples in case or control sets, whereas one additional SZ patient was found that carried DLG1-G344R. Our results suggest that rare missense mutations in the candidate PSD genes may increase susceptibility to SZ and/or ASD. These findings may strengthen the theory that rare, non-synonymous variants confer substantial genetic risks for these disorders.
Similar content being viewed by others


Introduction
Schizophrenia (SZ) is a debilitating disorder that affects approximately 1% of the population. A large portion of its heritability, which is estimated at 80%1, remains to be explained. Results from multiple large-scale genome-wide association studies as well as whole-genome/whole-exome sequencing support a polygenic model for explaining the susceptibility to the disorder. In this model, deleterious rare variants exert significantly larger effects than common single nucleotide polymorphisms (SNPs)2,3,4.
The postsynaptic density (PSD) is a protein complex localized at the postsynaptic plasma membrane of excitatory synapses. The PSD is essential for protein trafficking in neurons and synaptic plasticity5, processes commonly associated with the pathogenesis of SZ6. Scaffolding proteins, the primary components of the PSD structure, interact closely with glutamate receptors and play a major role in the dynamic regulation of their signaling activities7. PSD-95, which is a key protein in this subgroup, is a member of the membrane-associated guanylate kinase (MAGUK) family and is encoded by the disks large homolog 4 (DLG4) gene. Its linkage to SZ has been well established through both variant association8 and expression studies9,10,11,12,13,14. DLG1 and DLG2, which encode two other MAGUK family proteins, synapse-associated protein 97 (SAP97) and postsynaptic density protein 93 (PSD-93), respectively, have been similarly linked to SZ9,15,16,17,18,19,20. The products of DLGAP1 and DLGAP2 are guanylate kinase-associated protein (GKAP) family proteins that bind to the MAGUKs, mediating their interaction with other components of the PSD complex21,22. Resequencing studies have implicated these genes as susceptibility genes for SZ23,24. SynGAP1, another major scaffolding protein, has multiple protein-protein interacting motifs that enable it to act as a structural and regulatory anchor in synaptic homeostasis6. Its association with SZ has been shown in human expression studies and animal models10,25,26.
Recently, many whole-genome/whole-exome sequencing studies focusing on deleterious rare mutations, including copy number variants (CNVs), have frequently identified the PSD gene group, especially the PSD-95-related subgroup. In a study conducted by Purcell et al. who analyzed the exome sequences of 2536 SZ cases and 2543 controls for the burden of rare, disruptive mutations, the PSD, activity-regulated cytoskeleton-associated scaffolding protein, and PSD-95 gene sets were associated with SZ (p = 0.0808, p = 0.0016, p = 0.0017, for singletons, respectively)2. Multiple de novo CNVs spanning the coding regions of DLG1, DLG2, and DLGAP1 have been discovered in European and Asian SZ patients27. A Swedish study of 4719 SZ cases and 5917 controls found a significantly increased burden of large CNVs (>500 kb) in genes present in the PSD, especially in the 3q29/DLG1 locus, which has been implicated in previously conducted genome-wide association studies28.
Autism spectrum disorders (ASD) are a range of conditions characterized by persistent deficits in social communication and interaction, as well as restricted, repetitive patterns of behavior, interests, or activities. Both ASD and SZ belong to a group of distinct clinical entities known as neurodevelopmental disorders, as defined in DSM-V29. It has been indicated by clinical and epidemiologic studies that neurodevelopmental disorders have a high comorbidity rate, overlapping signs and symptoms, and significant similarities in genetic background30,31,32. Furthermore, various previous researches provide strong evidences of common underlying molecular pathways and shared genetic causes between ASD and SZ4,33,34,35,36,37. A recent review of targeted large-scale resequencing studies has pointed out that genetic evidence converges on three functional pathways, one of which is synaptic function. This review also predicted that PSD genes such as DLG4 and SynGAP1 will be identified as key nodes in the connected network38. A similar study utilizing the network-based analysis of genetic associations system identified a large biological network of genes that are affected by rare de novo CNVs in autism, with DLG4, DLG1, and DLG2 as important nodes in the cluster39. Individually, DLGAP2 and SynGAP1 are established risk genes for ASD40,41,42,43,44,45, and DLG1, DLG2, and DLG4 have also been implicated in various studies46,47,48,49.
Based on the results of these studies, we selected six candidate genes with the most evidence implicating an association with SZ and ASD: DLG1, DLG2, DLG4, DLGAP1, DLGAP2, and SynGAP1. The exonic regions of these genes were sequenced to look for rare, protein-altering point mutations.
Materials and Methods
Participants
Two independent sample sets were used in this study (Table 1). The first set, comprising 370 SZ patients (mean age = 49.73 ± 14.75 years; males = 52.97%) and 192 ASD patients (mean age = 16.34 ± 8.36 years; males = 77.60%), was sequenced for rare point mutations. The second, larger set, comprising 1315 SZ patients (mean age = 47.41 ± 15.35 years; males = 53.92%), 382 ASD patients (mean age = 19.61 ± 10.71 years; males = 77.75%), and 1793 controls (mean age = 45.11 ± 14.61 years; males = 51.25%), was used for association analysis of selected variants detected in the first set.
All participants in this study were recruited in the Nagoya University Hospital and its associated Institutes. Patients were included in the study if they (1) met DSM-5 criteria for SZ or ASD and (2) were physically healthy. Controls were selected from the general population and had no personal or family history of psychiatric disorders (first-degree relatives only based on the subject’s interview). The selection was based on the following: (1) questionnaire responses from the subjects themselves during the sample inclusion step; or (2) an unstructured diagnostic interview conducted by an experienced psychiatrist during the blood collection step. All subjects were unrelated, lived in the central area of the Honshu island of Japan, and self-identified as members of the Japanese population. The Ethics Committees of the Nagoya University Graduate School of Medicine approved this study. All experiments were performed in accordance with the Committee’s guidelines and regulations. Written informed consent was obtained from all participants. In addition, each patient’s capacity to provide consent was confirmed by a family member when needed. Individuals with a legal measure of reduced capacity were excluded.
Resequencing and Data Analysis
Genomic DNA was extracted from whole blood or saliva using the QIAGEN QIAamp DNA blood kit or tissue kit (QIAGEN Ltd., Germany). Custom amplification primers were designed to cover coding exons and flanking intron regions of the selected genes with Ion AmpliSeq Designer (Thermo Fisher Scientific, USA). Sample amplification and equalization were achieved using Ion AmpliSeq Library Kits 2.0 and the Ion Library Equalizer Kit, respectively (Thermo Fisher Scientific, USA). Amplified sequences were ligated with Ion Xpress Barcode Adapters (Thermo Fisher Scientific, USA). Emulsion PCR and subsequent enrichment were performed using the Ion OneTouch Template Kit v2.0 on Ion OneTouch 2 and Ion OneTouch ES, respectively (Thermo Fisher Scientific, USA). The final product was then sequenced on the Ion PGM sequencing platform (Thermo Fisher Scientific, USA). Raw data output from the sequencer was deposited in the DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp) under the accession number DRA004490, and uploaded to the Torrent Server (Life Technologies, USA) for variant calling, with NCBI GRCh37 as a reference. The resulting VCF files were analyzed by Ingenuity Variant Analysis (QIAGEN Ltd., Germany) for annotation and visualization.
Association Analysis
Missense mutations, small insertions/deletions, and splicing site variations with a minor allele frequency <1% were selected from the annotated data. The mutation calls were then validated for confidence by Sanger sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA). Genotyping prioritization was based on whether the mutation was 1) located in a functional domain or motif of the protein, according to the Human Protein Reference Database (http://www.hprd.org), Pfam (http://pfam.xfam.org/), and existing literature21,50,51,52,53,54,55,56,57,58,59, 2) functionally important, such as causing a frame shift, stop gain, or cysteine gain/loss, 3) novel, as in not documented in the NCBI dbSNP database (Build 137) (http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project (http://www.1000genomes.org/), the Exome Variant Server of NHLBI GO Exome Sequencing Project (ESP6500SI-V2) (http://evs.gs.washington.edu/EVS/), or the Human Genetic Variation Database of Japanese genetic variation consortium (http://www.genome.med.kyoto-u.ac.jp/SnpDB), and 4) predicted to be deleterious by in silico analytic methods. In addition to PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) that were originally incorporated in the Ingenuity Variant Caller, we also employed PROVEAN (http://provean.jcvi.org/index.php), PMut (http://www.ngrl.org.uk/Manchester/page/pmut), Mutation Taster (http://www.mutationtaster.org/), and PANTHER (http://pantherdb.org/) for enhanced prediction of the consequences of protein alterations.
Custom TaqMan SNP genotyping assays were designed and ordered from Applied Biosystems. Allelic discrimination analysis was performed on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, USA). Differences in allele and genotype frequencies of the mutations were compared between SZ patients/controls and ASD patients/controls using Fisher’s exact test (two-tailed), with a threshold of significance set at p < 0.05.
Additional Analysis for Amino Acid Changes
Conservation status of genotyping candidates in 11 common species was investigated using HomoloGene (http://www.ncbi.nlm.nih.gov/homologene). Possible 3D changes caused by mutations in the protein structure were predicted and modelled with I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) and UCSF Chimera (http://www.cgl.ucsf.edu/chimera/).
Results
Resequencing and Genetic Association Analyses
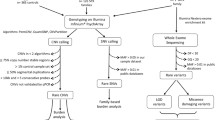
Thirty-seven rare, non-synonymous mutations were called by Ingenuity Variant Analysis during resequencing. Among them, 26 were validated via the Sanger method (Table 2). All variants were heterozygous. The carriers of four variants had pedigree DNA available. Sanger sequencing revealed that all four were inherited. Based on the selection criteria mentioned in Materials and Methods, G344R in DLG1, G241S in DLG4, and R604C in DLGAP2 were selected for association analysis (Fig. 1). The DLG4-G241S and DLGAP2-R604C variants were not found in any of the samples used for genotyping, whereas an additional DLG1-G344R variant carrier was detected in the SZ sample group (Table 3).

1. Protein sequence and domain data was obtained from Human Protein Reference Database. 2. Mutations validated in association analysis are marked in red.
Protein 3D Structure Analysis
3D modeling of the wild-type and mutated protein sequences indicated that for the DLGAP2-R604C variant, the additional cysteine gained from the mutation significantly changes the secondary and tertiary structures by adding a local β strand (Fig. 2).

α-helixes are marked in red and β-strands in purple.
Evolutionary Conservation Analysis
Results obtained from HomoloGene showed that the amino acids corresponding to the three mutations in DLG1, DLG4, and DLGAP2 were highly conserved among different species (Table S1).
Clinical Information of Mutation Carriers
Detailed descriptions of the clinical information can be found in the Supplement. Interestingly, the variant DLGAP2-R604C in one ASD patient was inherited from a parent who is also affected with ASD.
Discussion
Both SZ and ASD are disorders involving polygenic inheritance, with rare variants having a much higher impact on susceptibility than common variants. Recent large-scale genetic studies have reported that ultra-rare and private non-synonymous mutations are highly enriched in patient populations, especially in sets of genes with functions closely involved in neurodevelopment2,60,61,62,63. DLG4-G241S and DLGAP2-R604C were only present in single cases among a collective sample size of 562 patients during resequencing as well as in 1697 patients and 1793 controls, and DLG1-G344R was present in two SZ cases from the same sample sets. Therefore, they may confer a much higher risk than regular rare mutations discovered with the criterion of a minor allele frequency of <1%.
The second PDZ domain (PDZ2) of SAP-97, where DLG1-G344R is located, folds into a compact globular domain comprising six β-strands and two α-helices, which is a typical architecture for PDZ domains. During synaptic transmission, SAP-97 interacts with key protein partners such as ligand-binding units in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)53,64 and N-methyl-D-aspartate receptor (NMDAR)57 through the PDZ2 domain to regulate glutamate signaling and neuronal growth, which are major factors in the pathogenesis of SZ and ASD. The same domain also functions as a binding site for receptors for the neurotrophic growth factors corticotropin-releasing hormone (CRF)65 and epidermal growth factor receptor (ErbB1)66, which are linked to SZ.
The first PDZ domain (PDZ1) of PSD-95, where DLG4-G241S is located, similarly binds to NMDAR57. PDZ1 is also the site at which PSD-95 interacts with other PSD proteins such as SynGAP67. Mutated PDZ domains have been linked to defective PSD clustering and dendrite spine morphology in cultured cells67, as well as disrupted glutamate signaling and learning ability in animal models68. Interestingly, one study showed an association of this domain with Angelman Syndrome, a genetic disorder exhibiting a high occurrence rate in patients with autism, due to its functional relevance in the TrkB-PSD-95 signaling pathway69.
Cysteine is a ‘special’ amino acid that forms disulfide bonds between cysteine residues. These bonds are the basis of secondary and quaternary structures and are critical for the stabilization of tertiary structures of a protein70,71. The presence of DLGAP2-R604C introduces a new cysteine to the protein sequence and is highly likely to cause the formation or breaking of a disulfide bond that in turn disrupts the normal folding of DLGAP2.
The Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) integrates the exome sequencing data from 60,706 unrelated individuals from various studies and populations, which was reprocessed through the same pipeline, and jointly variant-called. While individuals in this dataset aren’t necessarily healthy controls since they only removed subjects with pediatric diseases, it is a useful reference set of allele frequencies due to its scale and data consistency. We searched ExAC for the frequencies of variants we detected in our study (Table S2). It should be noted that DLG1-G344R and DLG4-G241S did not exist in the database, while DLGAP2-R604C was found twice in European (non-Finnish) subjects.
Several limitations should be considered when interpreting the results of our study. First, our relatively small sample set did not have sufficient power to detect statistical significance in an association analysis72. Second, we did not conduct molecular biological analysis of the detected mutations. The in vitro and in vivo impacts of these mutations on the pathophysiology of the disorders need to be examined in future research. In addition, our stringent criteria for selection of variants for further analyses may have left out potentially interesting targets, such as DLG4-D375G, which is located in the PDZ domain of the encoded protein and was predicted by four in silico tools to be pathological. In addition, R72H and D703N in DLGAP2 are not present in a known functional domain but were predicted to be pathological by all five tools. These variants may be good candidates for a follow-up study (Fig. 1). Finally, our sequencing did not cover the promoter, untranslated regions, or intronic regions of the target genes, which may contain important mutations at regulatory sites.
Conclusion
In this study, we sequenced the exonic regions of PSD-95 and related genes in SZ and ASD patients using the Ion PGM platform and discovered 26 rare, non-synonymous variants. We then conducted an association analysis in a much larger sample set for three of these variants to investigate their relationship with SZ and/or ASD. Although statistical significance was not obtained, the observation that these mutations were only detected in cases, together with the structural relevance and in silico prediction results, indicates that they may impact the susceptibility of carriers to these disorders.
Additional Information
How to cite this article: Xing, J. et al. Resequencing and Association Analysis of Six PSD-95-Related Genes as Possible Susceptibility Genes for Schizophrenia and Autism Spectrum Disorders. Sci. Rep. 6, 27491; doi: 10.1038/srep27491 (2016).
Accession codes
References
Sullivan, P. F., Kendler, K. S. & Neale, M. C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 60, 1187–1192 (2003).
Purcell, S. M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014).
Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Sullivan, P. F., Daly, M. J. & O’Donovan, M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 13, 537–551 (2012).
Okabe, S. Molecular anatomy of the postsynaptic density. Mol Cell Neurosci 34, 503–518 (2007).
Verpelli, C., Schmeisser, M. J., Sala, C. & Boeckers, T. M. Scaffold proteins at the postsynaptic density. Adv Exp Med Biol 970, 29–61 (2012).
Elias, G. M. & Nicoll, R. A. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol 17, 343–352 (2007).
Cheng, M. C. et al. Genetic and functional analysis of the DLG4 gene encoding the post-synaptic density protein 95 in schizophrenia. PLoS One 5, e15107 (2010).
Kristiansen, L. V., Beneyto, M., Haroutunian, V. & Meador-Woodruff, J. H. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry 11, 737–747, 705 (2006).
Funk, A. J., Rumbaugh, G., Harotunian, V., McCullumsmith, R. E. & Meador-Woodruff, J. H. Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport 20, 1019–1022 (2009).
Kristiansen, L. V. & Meador-Woodruff, J. H. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res 78, 87–93 (2005).
Toro, C. & Deakin, J. F. NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr Res 80, 323–330 (2005).
Clinton, S. M. & Meador-Woodruff, J. H. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology 29, 1353–1362 (2004).
Ohnuma, T. et al. Gene expression of PSD95 in prefrontal cortex and hippocampus in schizophrenia. Neuroreport 11, 3133–3137 (2000).
Mulle, J. G. et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet 87, 229–236 (2010).
Sato, J., Shimazu, D., Yamamoto, N. & Nishikawa, T. An association analysis of synapse-associated protein 97 (SAP97) gene in schizophrenia. J Neural Transm 115, 1355–1365 (2008).
Uezato, A. et al. Further evidence for a male-selective genetic association of synapse-associated protein 97 (SAP97) gene with schizophrenia. Behav Brain Funct 8, 2 (2012).
Toyooka, K. et al. Selective reduction of a PDZ protein, SAP-97, in the prefrontal cortex of patients with chronic schizophrenia. J Neurochem 83, 797–806 (2002).
Dracheva, S., McGurk, S. R. & Haroutunian, V. mRNA expression of AMPA receptors and AMPA receptor binding proteins in the cerebral cortex of elderly schizophrenics. J Neurosci Res 79, 868–878 (2005).
MacLaren, E. J., Charlesworth, P., Coba, M. P. & Grant, S. G. Knockdown of mental disorder susceptibility genes disrupts neuronal network physiology in vitro . Mol Cell Neurosci 47, 93–99 (2011).
Kim, E. et al. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol 136, 669–678 (1997).
Takeuchi, M. et al. SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem 272, 11943–11951 (1997).
Li, J. M. et al. Genetic analysis of the DLGAP1 gene as a candidate gene for schizophrenia. Psychiatry Res 205, 13–17 (2013).
Li, J. M. et al. Role of the DLGAP2 gene encoding the SAP90/PSD-95-associated protein 2 in schizophrenia. PLoS One 9, e85373 (2014).
Sodhi, M. S., Simmons, M., McCullumsmith, R., Haroutunian, V. & Meador-Woodruff, J. H. Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol Psychiatry 70, 646–654 (2011).
Guo, X. et al. Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology 34, 1659–1672 (2009).
Kirov, G. et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 17, 142–153 (2012).
Szatkiewicz, J. P. et al. Copy number variation in schizophrenia in Sweden. Mol Psychiatry 19, 762–773 (2014).
Arlington, V. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5thed. American Psychiatric Publishing (2013).
Moreno-De-Luca, A. et al. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. The Lancet. Neurology 12, 406–414 (2013).
Reiss, A. L. Childhood developmental disorders: an academic and clinical convergence point for psychiatry, neurology, psychology and pediatrics. Journal of child psychology and psychiatry, and allied disciplines 50, 87–98 (2009).
Capute & Accardo’s 3e Vol II,The Spectrum of Neurodevelopmental Disabilities,Capute & Accardo’s Neurodevelopmental Disabilities in Infancy and Childhood: Neurodevelopmental Diagnosis and Treatment. 61–104 (2008).
Crespi, B. J. & Crofts, H. J. Association testing of copy number variants in schizophrenia and autism spectrum disorders. J Neurodev Disord 4, 15 (2012).
Ku, C. S. et al. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry 18, 141–153 (2013).
Martin, J. et al. Biological Overlap of Attention-Deficit/Hyperactivity Disorder and Autism Spectrum Disorder: Evidence From Copy Number Variants. J Am Acad Child Psy 53, 761–770 (2014).
McCarthy, S. E. et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatr 19, 652–658 (2014).
Daniel Moreno-De-Luca, A. M.-D.-L., Joseph F. Cubells, Stephan & J. Sanders. Cross-Disorder Comparison of Four Neuropsychiatric CNV Loci. Current Genetic Medicine Reports 2, 151–161 (2014).
Krumm, N., O’Roak, B. J., Shendure, J. & Eichler, E. E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37, 95–105 (2014).
Gilman, S. R. et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907 (2011).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372 (2010).
Chien, W. H. et al. Deep exon resequencing of DLGAP2 as a candidate gene of autism spectrum disorders. Mol Autism 4, 26 (2013).
Lo-Castro, A. & Curatolo, P. Epilepsy associated with autism and attention deficit hyperactivity disorder: is there a genetic link? Brain Dev 36, 185–193 (2014).
Berryer, M. H. et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat 34, 385–394 (2013).
Hamdan, F. F. et al. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry 69, 898–901 (2011).
Cook, E. H., Jr. De novo autosomal dominant mutation in SYNGAP1. Autism Res 4, 155–156 (2011).
Willatt, L. et al. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet 77, 154–160 (2005).
Quintero-Rivera, F., Sharifi-Hannauer, P. & Martinez-Agosto, J. A. Autistic and psychiatric findings associated with the 3q29 microdeletion syndrome: case report and review. Am J Med Genet A 152A, 2459–2467 (2010).
Egger, G. et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 15, 117–127 (2014).
Feyder, M. et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am J Psychiatry 167, 1508–1517 (2010).
Leonard, A. S., Davare, M. A., Horne, M. C., Garner, C. C. & Hell, J. W. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem 273, 19518–19524 (1998).
Cai, C., Coleman, S. K., Niemi, K. & Keinanen, K. Selective binding of synapse-associated protein 97 to GluR-A alpha-amino-5-hydroxy-3-methyl-4-isoxazole propionate receptor subunit is determined by a novel sequence motif. J Biol Chem 277, 31484–31490 (2002).
Kim, E. et al. Plasma membrane Ca2+ ATPase isoform 4b binds to membrane-associated guanylate kinase (MAGUK) proteins via their PDZ (PSD-95/Dlg/ZO-1) domains. J Biol Chem 273, 1591–1595 (1998).
Zhang, L. et al. Structure-function analysis of SAP97, a modular scaffolding protein that drives dendrite growth. Mol Cell Neurosci 65, 31–44 (2015).
Hong, X., Avetisyan, M., Ronilo, M. & Standley, S. SAP97 blocks the RXR ER retention signal of NMDA receptor subunit GluN1-3 through its SH3 domain. Biochim Biophys Acta 1853, 489–499 (2015).
Zhu, J. et al. Guanylate kinase domains of the MAGUK family scaffold proteins as specific phospho-protein-binding modules. EMBO J 30, 4986–4997 (2011).
Wu, H. et al. Intramolecular interactions regulate SAP97 binding to GKAP. EMBO J 19, 5740–5751 (2000).
Niethammer, M., Kim, E. & Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 16, 2157–2163 (1996).
Cousins, S. L., Kenny, A. V. & Stephenson, F. A. Delineation of additional PSD-95 binding domains within NMDA receptor NR2 subunits reveals differences between NR2A/PSD-95 and NR2B/PSD-95 association. Neuroscience 158, 89–95 (2009).
Tong, J., Yang, H., Eom, S. H., Chun, C. & Im, Y. J. Structure of the GH1 domain of guanylate kinase-associated protein from Rattus norvegicus. Biochem Biophys Res Commun 452, 130–135 (2014).
Loohuis, L. M. et al. Genome-wide burden of deleterious coding variants increased in schizophrenia. Nat Commun 6, 7501 (2015).
De Rubeis, S. & Buxbaum, J. D. Recent advances in the genetics of autism spectrum disorder. Curr Neurol Neurosci Rep 15, 36 (2015).
Griswold, A. J. et al. Targeted massively parallel sequencing of autism spectrum disorder-associated genes in a case control cohort reveals rare loss-of-function risk variants. Mol Autism 6, 43 (2015).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
von Ossowski, I. et al. Crystal structure of the second PDZ domain of SAP97 in complex with a GluR-A C-terminal peptide. FEBS J 273, 5219–5229 (2006).
Dunn, H. A., Walther, C., Godin, C. M., Hall, R. A. & Ferguson, S. S. Role of SAP97 protein in the regulation of corticotropin-releasing factor receptor 1 endocytosis and extracellular signal-regulated kinase 1/2 signaling. J Biol Chem 288, 15023–15034 (2013).
Yokomaku, D. et al. ErbB1 receptor ligands attenuate the expression of synaptic scaffolding proteins, GRIP1 and SAP97, in developing neocortex. Neuroscience 136, 1037–1047 (2005).
Nonaka, M., Doi, T., Fujiyoshi, Y., Takemoto-Kimura, S. & Bito, H. Essential contribution of the ligand-binding beta B/beta C loop of PDZ1 and PDZ2 in the regulation of postsynaptic clustering, scaffolding, and localization of postsynaptic density-95. J Neurosci 26, 763–774 (2006).
Nagura, H. et al. Impaired synaptic clustering of postsynaptic density proteins and altered signal transmission in hippocampal neurons, and disrupted learning behavior in PDZ1 and PDZ2 ligand binding-deficient PSD-95 knockin mice. Mol Brain 5, 43 (2012).
Cao, C. et al. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol 11, e1001478 (2013).
Cohen, F. E. General-Principles of Protein-Structure. Journal of the American Society of Nephrology 5, 1842–1842 (1995).
Ptitsyn, O. B. Physical Principles of Protein-Structure and Protein Folding. Journal of Biosciences 8, 1–13 (1985).
Hong, E. P. & Park, J. W. Sample size and statistical power calculation in genetic association studies. Genomics Inform 10, 117–122 (2012).
Acknowledgements
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Ministry of Health, Labour and Welfare of Japan; “Integrated research on neuropsychiatric disorders” carried out under the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development, AMED; the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from AMED; Innovative Areas “Glial assembly: a new regulatory machinery of brain function and disorders”; and Innovative Areas “Comprehensive Brain Science Network”. We would like to thank the patients and their families for participating in this study.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: J.X., H.K., C.W., K.I., I.K., Y.A., A.Y., Y.N., T.S., T.O.I., Y.T., Y.U., T.O., T.I., B.A., D.M. and N.O. Performed the experiments: J.X., H.K., C.W., K.I., I.K., Y.A., A.Y., Y.N., T.S., T.O.I., Y.T. and Y.U. Analyzed the data: J.X., H.K., C.W., K.I., I.K., Y.A., A.Y., Y.N., T.S., T.O.I., Y.T., Y.U., T.O., T.I., B.A., D.M. and N.O. Contributed reagents/materials/analysis tools: J.X., T.O., T.I., B.A., D.M. and N.O. Wrote/reviewed the paper: J.X., H.K., C.W., K.I., I.K., B.A., D.M. and N.O.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xing, J., Kimura, H., Wang, C. et al. Resequencing and Association Analysis of Six PSD-95-Related Genes as Possible Susceptibility Genes for Schizophrenia and Autism Spectrum Disorders. Sci Rep 6, 27491 (2016). https://doi.org/10.1038/srep27491
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27491
This article is cited by
-
A novel DLG4 variant causes DLG4-related synaptopathy with intellectual regression
Human Genome Variation (2024)
-
Schizophrenia-associated SAP97 mutations increase glutamatergic synapse strength in the dentate gyrus and impair contextual episodic memory in rats
Nature Communications (2022)
-
Developmental dynamics of RNA translation in the human brain
Nature Neuroscience (2022)
-
Combined cellomics and proteomics analysis reveals shared neuronal morphology and molecular pathway phenotypes for multiple schizophrenia risk genes
Molecular Psychiatry (2021)
-
Neonatal Rotenone Administration Induces Psychiatric Disorder-Like Behavior and Changes in Mitochondrial Biogenesis and Synaptic Proteins in Adulthood
Molecular Neurobiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
