
Abstract
The distributions of chemical elements within cells are of prime importance in a wide range of basic and applied biochemical research. An example is the role of the subcellular Zn distribution in Zn homeostasis in insulin producing pancreatic beta cells and the development of type 2 diabetes mellitus. We combined transmission electron microscopy with micro- and nano-synchrotron X-ray fluorescence to image unequivocally for the first time, to the best of our knowledge, the natural elemental distributions, including those of trace elements, in single organelles and other subcellular features. Detected elements include Cl, K, Ca, Co, Ni, Cu, Zn and Cd (which some cells were supplemented with). Cell samples were prepared by a technique that minimally affects the natural elemental concentrations and distributions and without using fluorescent indicators. It could likely be applied to all cell types and provide new biochemical insights at the single organelle level not available from organelle population level studies.
Similar content being viewed by others


Introduction
The chemical compositions of organelles within cells and the relationship of the chemical compositions to the organelle morphologies and functions, are key for understanding biochemical processes in healthy and diseased cells. Specifically, the study of the biological roles, buffering, trafficking and compartmentalization of elements, especially Cu and Zn, in eukaryotic cells, under normal and pathologic conditions, is an active area of research at the forefront of biochemistry1,2,3,4,5,6,7. However, progress in the field has been hampered by limitations of the techniques commonly used for chemical analysis. These techniques can be divided to two groups, macro-analytical techniques, which analyze aggregates of organelles and micro-analytical techniques, which analyze single organelles.
Macro-analytical techniques8,9 involve isolating a large number of organelles of a certain type from cells by lysis and fractionation. The chemical composition of the organelle aggregate is then usually determined by inductively coupled plasma - mass spectrometry (ICP-MS). This analysis represents the average composition of a population of organelles and cannot reveal compositional differences between single organelles. In addition, chemical elements not covalently bonded to atoms in cell structures are especially susceptible to lysis and fractionation and their natural concentrations could be altered significantly in the process.
Most relevant micro-analytical techniques lack the required spatial resolution and/or elemental detection sensitivity to measure the chemical compositions of single organelles, including trace elements, with the exception of the nucleus10,11. The most widely used technique in this category is fluorescence optical microscopy of cells loaded with fluorescent indicators (that fluoresce when binding to their target elements). In addition to the relatively low spatial resolution of optical microscopy, the technique is limited as well by two properties of the fluorescent indicators: They are not entirely specific to their target elements and they bind primarily to the free or loosely bound fraction of the target elements in the cells. The latter means that by binding to their target elements, the fluorescent indicators potentially alter the natural elemental distributions.
An example is the fluorescent indicators used for detecting the biochemically essential element Zn. They have moderate Zn binding affinities, which are significantly lower than those of typical Zn-binding enzymes, such as carbonic anhydrase12. Thus, the fluorescent indicators detect chelatable Zn that is loosely bound to intracellular Zn-binding proteins that have lower affinities for Zn. However, most cellular Zn is reported to be bound to metallothioneins and other proteins which have much higher affinities for Zn13 and therefore cannot be detected by Zn fluorescent indicators. While fluorescent indicators with higher Zn affinity are theoretically feasible, they are likely to scavenge Zn from Zn-binding and transporting proteins, thereby altering the natural distribution of Zn. Therefore, commonly used Zn fluorescent indicators are unable to detect cytosolic Zn, unless labile Zn concentrations exceed the buffering capacity of the abundant buffering proteins4,14,15. The situation is the same for many other biochemically essential elements in cells, e.g., Fe and Cu, which are tightly bound to buffering proteins as well. This property limits the utility of fluorescent indicators for many applications, especially as tracers of elemental distributions in single organelles.
Additional micro-analytical techniques include conventional and scanning transmission electron microscopy (TEM and STEM) and nano-secondary ionization mass spectrometry (NanoSIMS). The former techniques have superb spatial resolution, at the sub-nanometre (nm) level, but their elemental detection limits are not sensitive enough to detect most trace elements. The latter technique has high spatial resolution, down to 50 nm and high sensitivity, which enables the detection of some trace elements (e.g., P and S). At the subcellular level, it is primarily used to study metabolism in isotopically labeled cells16,17, hence it could be a complementary technique to the method presented here. Two limitations of NanoSIMS that need to be considered are potential isobaric interferences (i.e., interferences by atoms and/or molecules of the same nominal mass as the isotope of interest) and the fact that it is destructive.
Synchrotron X-ray fluorescence (SXRF) chemical imaging, combined with appropriate sample preparation methods and high spatial resolution structural imaging techniques, is a promising micro-analytical technique for analyzing elemental distributions in single organelles. It measures the chemical compositions of samples without the need for using fluorescent indicators or isotopically labeling the cells, hence it avoids the limitations of biased fluorescent indicator binding and altering the natural elemental distributions. However, like with other analytical techniques, determining elemental concentrations with SXRF may be limited by background and/or elemental interferences. The mostly flat background is due to scattered primary X-ray photons (the tail of the Compton peak). Elemental interference is due to X-ray fluorescence peaks of other elements that partially or fully overlap peaks of the elements of interest. In order to improve elemental detection limits, one takes steps to decrease both sources of interference. Minimizing the scattering background is done by placing the detector in the plane of the synchrotron storage ring at 90° to the primary X-ray beam (scattering is minimized when the angle between the beam electric vector, which is the direction of the beam polarization vector and the scattering vector is 0°, as in this configuration) and selecting the thinnest possible sample substrate, usually a few 100 nm thick (a thicker substrate would be a major source of scattered primary X-ray photons in thin samples). Minimizing elemental interference is done by using sample preparation procedures that reduce the concentrations of potentially interfering elements in the sample.
Earlier studies with the SXRF microprobe, with spatial resolutions down to ~150 nm, of chemically treated and untreated, chemically fixed, whole cells (a few micrometre (μm) thick) were able to image elemental distributions in some subcellular features18,19,20,21. Subcellular structures were imaged as well in thin (300 nm) sections of chemically treated and untreated cells that were prepared by high-pressure freezing22.
The introduction of SXRF nanoprobes in recent years23, with spatial resolutions in the range of tens of nm, coupled with improved elemental detection sensitivity, brought researchers closer to imaging trace element distributions in single organelles. When nano-SXRF was combined with the relatively low spatial resolution of optical microscopy for structural imaging, the chemical compositions of organelles were imaged in thick (whole cell) sections of chemically treated cells prepared by freeze-drying24 or by chemical fixation25 (the primary X-ray beam is likely to transverse a number of organelles in thick samples). In a novel study that combined nano-SXRF with X-ray ptychography (for structural imaging), the chemical compositions of organelles were imaged in whole, frozen-hydrated, green algae cells at cryogenic temperatures26. In addition, the chemical compositions of isolated single, freeze-dried, melanosomes (organelles where melanin is synthesized, stored and transported) were imaged as well27.
We present here, to the best of our knowledge, the first unequivocal imaging of natural trace element distributions in single organelles in cells. This was achieved by combining the highest available spatial resolution nano-SXRF with TEM and improved sample preparation protocol, of minimally chemically treated cells, that minimized the potential for altering the natural elemental distributions. The method opens up new possibilities in subcellular biochemistry for studying variabilities among single organelles and other subcellular compartments, which are usually analyzed at the organelle population level.
Results
Analytical techniques
In order to map elemental distributions in single organelles and other subcellular features, we combined the high spatial resolution of TEM, for cellular ultrastructure imaging, with the high spatial resolution and high sensitivity of micro- and nano-SXRF, for chemical imaging. The hard X-rays commonly used in SXRF can detect Al and heavier elements with a detection limit of ~1 part per million (ppm), depending on experimental conditions. In addition, under normal operating conditions, these two techniques are non-destructive, which allows repeated analyses of the same samples.
Cells and preparation
Cells of the insulin producing mouse pancreatic beta cell line MIN628 were grown for 72 hours in a cell culture medium. For a subset of the cells, the medium was supplemented with 1 μmole/l CdCl2 in order to introduce the biochemically non-essential element Cd and examine its intracellular distribution. The cells were then prepared using high pressure freezing, followed by freeze substitution. This technique is preferable to the commonly used chemical fixation in terms of superior preservation of both the cell’s original ultrastructure29,30 and chemical composition30, especially with regard to elements bound to soluble compounds31. In order to minimize the possibility of altering the natural elemental distributions in the cells during preparation, especially with respect to biochemically important trace elements, we took a number of additional steps: (1) Cells were only fixed with glutaraldehyde. The commonly used fixer osmium tetroxide (OsO4) was specifically avoided to prevent Os Lα1,2 lines, with energies 8.912, 8.841 keV, respectively, interfering with the Kα1,2 lines of Zn, 8.639, 8.616 keV, respectively31. (2) Cells were stained only with tannic acid, which provided contrast for ultrastructure imaging, but does not contain any heavy elements, with potentially interfering elemental X-ray lines, as major components. The commonly used staining materials lead citrate and uranyl acetate were avoided due to interferences of the Pb and U Mα1 lines, 2.346 and 3.171 keV, respectively, with the Kα1,2 lines of biochemically important S, 2.308 and 2.307 keV and the Lα1,2 lines of Cd, 3.134 and 3.127 keV. (3) Cells were embedded in HM20 Lowicryl resin, which can be used at cryogenic temperatures and better preserves ultrastructure and intrinsic chemical composition compared with most other resins30. (4) To prevent diffusion of atoms out of/into the cells, ethylene glycol was used in ultramicrotoming the cells instead of water31. Trace element concentrations in the chemicals used to prepare the samples are given in Supplementary Table S1.
The cryo-fixed and plasticized cell samples were ultramicrotomed to a thickness of 350 nm. This thickness was chosen to optimize the requirements of SXRF and TEM, as well as the need to image single organelles. Thicker samples are preferable for SXRF imaging since the elemental X-ray signal intensity increases with sample thickness. At the same time, 350 nm is the maximum thickness that can be imaged by TEM and maintain spatial resolution. In addition, in order to chemically analyze single organelles by SXRF, the primary X-ray beam needs to traverse only one organelle at each point on the sample. Considering the tens to hundreds of nm size range and spatial distribution of organelles in the cells (Figs 1 and 2, S1), it is reasonable that this requirement was met in most cases.

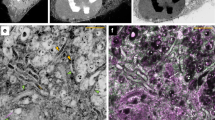
Complementary TEM and SXRF micro/nanoprobe Cu Kα distribution maps of MIN6 cells 1–3.
The combined information from the two imaging techniques gives a detailed ultrastructural and chemical picture of the subcellular environment at the single organelle level. (a) TEM map of cell 1. This cell was grown in a medium with no Cd. It serves as a reference for the sample preparation and imaging procedure we used. Scale bar = 2 μm. (b) The corresponding SXRF microprobe map to a. Scan step size = 0.1 μm, dwell time = 1 s. Same scale as a. The two Au nanoparticles that are seen in the map, as red pixels in the upper and lower left parts, are off the reported concentration range. (c) TEM image of cells 2 and 3. These cells were grown in a medium supplemented with 1 μmole/l CdCl2. Red rectangles outline the areas imaged with the SXRF nanoprobe at high-resolution. Scale bar = 2 μm. (d) The SXRF microprobe map corresponding to c. Scan step size = 0.25 μm, dwell time = 1 s. Same scale as c. (e–f) Enlargements of the high-resolution (h. r.) scan areas in the TEM image of cells 3 and 2 (c), respectively. (g–h) The SXRF nanoprobe maps corresponding to e–f. Scan step size = 0.05 μm, dwell time = 5 s. One μm scale bar (below g) applies to (e–h). Elemental spatial density scale bar (below g) applies to (b,d,g–h) and the measured range, in units of nmole/cm2, is indicated below each map. Contrast of the SXRF images (b,d,g–h) has been enhanced. A – autophagosome-like feature, E – edge of the cell, h – heterochromatin, N – nucleus, circles outline mitochondria. Additional concentration information, including uncertainties, appears in Supplementary Table S2.

Complementary TEM and SXRF nanoprobe images of MIN 6 cell 4.
The cell was grown in a medium supplemented with 1 μmole/l CdCl2. (Compare with untreated cell 5, Supplementary Fig. S1, Table S3). In this case, the 350 nm thick cell section was mounted on a non-coated Au TEM finder grid and was sandwiched between two Formvar layers (each ~100 nm thick). (a) TEM image of the cell. The red rectangle outlines the area imaged with the SXRF nanoprobe. Scale bar = 1 μm. (b) Enlargement of the high-resolution scan area in the TEM image a. (c–j) SXRF nanoprobe elemental distribution maps corresponding to b of, respectively, Cl, K, Ca, Co, Ni, Cu, Zn, Cd. All elemental spectra were collected simultaneously. With the exception of Cd, all elements were mapped by their Kα lines. Cadmium was mapped by its Lα lines. Scan step size = 0.04 μm, dwell time = 8 s. One μm scale bar (above b) applies to (b–j). Elemental spatial density scale bar with the measured range, in units of nmole/cm2, appears below each elemental map. Contrast of the SXRF images (c–j) has been enhanced. The Cl concentration, Kα1,2 = 2.622, 2.621 keV, respectively, in c may be overestimated due to the presence of an unidentified strong line at ~2.400 keV. Due to lack of suitable Cd standard, there could be a systematic uncertainty of up to a factor of 2 in the inferred Cd concentrations (j, see Methods). A – autophagosome-like feature, N – nucleus, arrows point to vesicles, potentially insulin producing, circles/ellipses outline mitochondria. Additional concentration information, including uncertainties, appears in Supplementary Table S2.
Transmission electron microscopy (TEM) and synchrotron X-ray fluorescence (SXRF) imaging
The cell sections were placed on Au TEM finder grids coated with carbon and Formvar. Whole sections were imaged by TEM and suitable cells for SXRF analysis were selected. The criteria for suitable cells were good preservation of cellular ultrastructure, no ice damage and presence of a variety organelle types and other subcellular features (Figs. 1, 2, S1). The chemical compositions of the selected cells were then imaged with a 10 keV primary X-ray beam using the microprobe32 at beamline 2-ID-D of the Advanced Photon Source (APS) at Argonne National Laboratory (ANL). The microprobe has a 150 × 150 nm2 beam spot size on the sample and a flux of 4 × 109 photons/s/0.01% BW (at 10 keV). These properties enable one to image whole cell sections in relatively short time. The whole cell scans give the chemical compositions of the sections and reveal significant elemental distributions within the sections (Figs. 1b, d). Areas within the cell sections that met the criteria above and gave good SXRF spectra were then selected for high-resolution nanoprobe SXRF imaging (the whole cell scans also helped with navigation on the cell sections during the high-resolution scans).
High spatial resolution and high sensitivity imaging of the preselected subcellular areas were performed with the ANL’s Center for Nanoscale Materials (CNM) nanoprobe33 (beamline 26-ID-C at the APS). The nanoprobe offers the highest available SXRF spatial resolution, with a 40 × 40 nm2 beam spot size and a flux of 1.8 × 109 photons/s/0.01% BW (at 9.75 keV) (work is currently being done to improve the spatial resolution even further34,35). Figure 1 shows complementary TEM, microprobe and nanoprobe SXRF Cu Kα images of the same cells. The Cu distribution in the micro- and nano-SXRF maps, with spatial resolutions of <150 nm and 50 nm, respectively, can be clearly correlated with the ultrastructure seen in the TEM images. For example, high Cu content spots in the micro-SXRF image of cell 1 (Fig. 1b) correlate with mitochondria and heterochromatin (that is known to have Cu-containing proteins) in the cell’s TEM image (Fig. 1a). The high-resolution scans of areas in cells 2 and 3 (Figs. 1g, h) show very detailed maps that correlate with fine ultrastructure in Figs. 1e, f, including the nuclei, mitochondria, autophagosome-like features and the edge of one of the cells. The nano-SXRF maps even reveal additional ultrastructure not seen in the TEM maps, as in the nuclei of the two cells. In addition, the Cu concentrations in cells 2 and 3 measured with the nanoprobe (Figs. 1g, h) agree with those measured with the microprobe (Fig. 1d). This demonstrates the robustness of the SXRF results obtained in two beamlines.
The TEM and high-resolution SXRF images of cell 4 are shown in Fig. 2. In this case, the sensitivity of the high-resolution SXRF imaging, with spatial resolution of 0.04 μm, is demonstrated by the distribution maps of seven other elements in addition to Cu: Cl, K, Ca, Co, Ni, Zn and Cd (elemental concentrations were below the TEM chemical imaging detection limit). All elemental spectra were collected simultaneously. The detection of Cd was especially challenging, due to its suggested low concentration in single organelles under these exposure conditions36 and since unlike the other elements above, which were detected by their K lines, it was detected by the much weaker L lines. However, due to lack of suitable Cd standard, there could be a systematic uncertainty of up to a factor of 2 in the inferred Cd concentrations (see Methods). In addition, the measured Cl concentration in this cell, using Kα1,2 = 2.622, 2.621 keV, respectively, may be overestimated due to the presence of an unidentified strong line at ~2.400 keV. (This unidentified line masked the S lines, with Kα1,2 = 2.308, 2.307 keV, respectively and prevented unequivocal S detection).
As stated above, the cryo-fixation/freeze substitution method used to prepare the samples is the best for preserving the cell’s ultrastructure and elemental distributions. Hence, it is expected that the elemental distribution maps (Figs. 1, 2, S1) represent the natural distributions in living MIN6 cells. To evaluate whether our results are biologically realistic, we used the elemental spatial densities per pixel as a rough comparison with other SXRF studies25,27,37,38,39,40,41. These studies were conducted at different synchrotrons, used a variety of cell types, sample preparations and experimental conditions (all imaged cells at lower spatial resolutions than presented here). The comparison shows that our measured ranges for all elements are comparable to the ranges reported in the studies above. The only exception may be Co, for which our values are higher. This may indicate that our cells were contaminated with Co. However, since only a few studies measured Co by SXRF in single cells, it is unclear at this time whether our cells were contaminated or not. We tested as well whether treating cells with Cd (1 μmole/l CdCl2) affects other elements’ distributions and spatial densities. For this purpose, the data of cell 4 (Fig. 2, Table S2), which was treated with Cd, were compared with the data of cell 5 (Fig. S1, Table S3), which was not treated. Using again the range of elemental spatial densities per pixel, the comparison shows that the elemental concentration ranges in both cells are comparable, with the exception of Cd, as expected. This indicates that the elemental distributions and concentrations at the cellular level were not affected by exposure to Cd at the level of 1 μmole/l CdCl2. In addition, the cellular ultrastructure of the cells we analyzed appears free of processing artifacts in the TEM images. However, despite the agreement with other studies, considering the natural variability of cell elemental compositions, it cannot be ruled out that the original elemental compositions and distributions of the cells we analyzed were altered to some extent.
Discussion
The comparison between the TEM images and high-resolution SXRF elemental maps in Fig. 2 reveals some interesting initial observations regarding the natural elemental distributions between the different organelles. Divalent metals that are known to bind to proteins, mainly Co, Cu, Zn, Cd and potentially Ni, have similar distributions. They are concentrated in areas rich in proteins, such as the nucleus and mitochondria, while having lower concentrations in the autophagosome/lysosome-like structures. The likely reason for the latter is that the affinity of divalent metals for proteins is significantly lower in lower pH environments, such as that in the autophagosome/lysosome, which leads to their dissociation from the proteins. Chlorine and K, on the other hand, have lower affinities for cellular proteins. The Cl− and K+ ions are mainly present in cells in free solution. This is reflected in their more even distribution between the organelles. The chemical behavior of Ca is in between those of the two groups of elements above. The concentrations of the less tightly bound elements, Cl and K, are more susceptible to alteration during sample preparation, which could lead to differences between measured and natural concentrations and distributions.
These observations agree qualitatively with the results of Matsuyama et al.25. These authors combined fluorescence optical microscopy and nano-SXRF, with spatial resolution of 70 nm, to map elemental distributions in whole, chemically fixed, NIH/3T3 cells. Similar to the results presented here, Matsuyama et al. found as well that Cl, Ca, Cu and Zn (as well as P, S and Fe which are not reported here) were concentrated in the nucleus and that Zn was concentrated as well in mitochondria.
The method presented here enables simultaneous mapping of subcellular distributions of multiple elements non-destructively at high spatial resolution and high sensitivity and without the need for fluorescent indicators. It offers higher spatial resolution than fluorescence optical microscopy and since it does not use fluorescent indicators, avoids biases in the way they bind to the target elements and minimizes the potential for altering the natural elemental distributions. This was enabled by improved sample preparation technique, imaging thin (350 nm) cell sections, which ensure that in most cases the primary X-ray beam transverses only one organelle at each spot on the sample, combining TEM, micro- and nano-SXRF imaging and the high spatial resolution (≤40 nm) and elemental detection sensitivity of the nanoprobe. We demonstrated the capabilities of the method by mapping unequivocally for the first time, to the best of our knowledge, natural elemental distributions, including those of some trace elements, between single organelles and other subcellular compartments.
An example of where the method presented here could provide insights not available from other analytical techniques is the role of Zn in insulin producing pancreatic beta cells. Zinc plays a prominent role in the proper function of these cells through several unique mechanisms4,42,43,44,45,46. It is co-secreted with insulin and therefore a high cellular turnover of Zn has to be maintained. Zinc homeostasis is achieved through the interaction between Zn and several specialized transporter (ZnT and ZIP family) and buffering proteins, mainly metallothionein, as well as regulated transcription of the proteins involved in Zn trafficking and buffering5,47,48,49,50,51,52,53,54,55,56,57,58,59. The relevance of Zn homeostasis to the development of diabetes mellitus has been highlighted by numerous studies. They linked a common, non-silent genetic variation in the gene encoding the beta cell specific Zn transporter protein ZnT8 to beta cell dysfunction and type 2 diabetes mellitus60,61,62,63,64,65,66. The exact mechanism by which alterations in this Zn transporter protein leads to beta cell dysfunction has remained elusive so far, despite intense efforts to decipher its action4,45,67,68. Specifically, the mechanism of Zn transfer from its main buffering protein, metallothionein, to ZnT8 and other Zn acceptor proteins is being studied extensively. The mechanism by which the presence of Zn in most mammalian insulin producing vesicles facilitates the proper maturation of insulin precursors to biologically active insulin is an additional area of active investigation42,43. A better understanding of the distributions of protein-bound Zn and other, potentially interfering, divalent metals in various subcellular compartments of beta cells, which could be studied using the method presented here, is essential for understanding the role of Zn homeostasis in the development of diabetes mellitus.
Two recent papers69,70 showed that analyzing single cells reveals important metabolomic information that is not available from cell population level studies. Similarly, the method presented here, which could likely be applied to every cell type, opens up new possibilities for studies at the single organelle level, as in the Zn homeostasis example above. Such studies will provide new insights which are not available from organelle population level studies and help answer many important biological questions related to the biochemistry of living cells.
Methods
Cells and culture
The MIN6 cells were a gift from Dr. Junichi Miyazaki (Osaka University Medical School, Osaka, Japan). They were grown and maintained as described previously28,36. Briefly, cells were plated on 6 well cell culture dishes (Corning) at a density of 0.8 × 106 cells per well. The cell culture medium was composed of DMEM (Invitrogen) supplemented with 15% heat inactivated FBS (Hyclone), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), 2 mmole/l glutamine (Invitrogen) and 50 μM beta mercaptoethanol (Invitrogen). Cells between passages 28 and 35 were used for the experiments. Cells were grown at 5% CO2 and 37 °C. Culture medium was exchanged at 48 hours following cell seeding. At that time, 1.0 μmole/l CdCl2 or vehicle (PBS) were added.
Sample preparation and transmission electron microscopy (TEM) imaging
Cells were harvested 72 hours after being placed into planchettes (Ted Pella), cryo-fixed using a high pressure freezing machine (HPM 010; RMC) and placed immediately into cryo-tubes containing a frozen cocktail of 0.25% glutaraldehyde and 0.1% tannic acid in anhydrous acetone. All frozen samples were placed into an automatic freeze substitution device (ASF2, Leica). The samples were freeze substituted at −80 °C for 48 hrs, then the temperature was raised to −50 °C. The samples were washed 3 times with acetone, infiltrated with successive increasing concentrations (25%, 50%, 75%, each 8 hrs long; 100% overnight) of Lowicryl HM20 resin, followed by three incubations, 1 hr each, with 100% resin. The samples were UV polymerized at −50 °C for 20 hrs. 350 nm sections were cut with an ultramicrotome (UC6, Leica) and placed on a gold, Formvar-carbon coated, London-finder grid (EMS). Images were collected on an electron microscope operated at 300 kV (Tecnai TF30; FEI) with a 4K ultrascan digital camera (Gatan). Cells were selected for SXRF imaging based on two criteria: (a) showing no ice damage and (b) showing variety of well-preserved organelles.
Synchrotron X-ray fluorescence (SXRF) imaging
The SXRF chemical imaging was performed at the Advanced Photon Source (APS) at Argonne National Laboratory (ANL). Whole, TEM pre-selected, cell sections were imaged with the microprobe at beamline 2-ID-D. Based on the TEM and microprobe SXRF images, areas within cell sections, with a variety of well-preserved organelles and good SXRF spectra, were selected for high-resolution SXRF imaging with the Center for Nanoscale Materials (CNM) nanoprobe (beamline 26-ID-C). The SXRF spectra from both beamlines were quantified and visualized with the MAPS software package71.
The primary X-ray beam energy in both beamlines was 10 keV. All elements analyzed, with the exception of Cd, were detected by their K lines. The standards used for these elements were Standard Reference Materials (SRM) 1832 and 1833 by the National Institute of Standards and Technology (NIST). Cadmium was detected by its L lines. We used standards with varying Cd/Zn ratios to improve Cd quantification by correcting (1) relative Cd L line intensities and (2) normalization of Cd L line intensities to Zn K line intensities. The standards were prepared by depositing drops of liquids with varying Cd/Zn ratios on Formvar-coated TEM slot grids (EMS) (one Cd/Zn ratio per grid). When the liquids evaporated, they left residues on the Formvar with different Cd/Zn ratios. The standards worked well for correcting relative Cd L line intensities. However, The Cd/Zn ratios varied spatially on the grids and did not match the nominal ratios in the liquids, so they could not be used to normalize the Cd/Zn concentrations. As a consequence, there could be a systematic uncertainty of up to a factor of 2 in the inferred Cd concentrations in the cells. We are planning to reduce the uncertainty in quantifying the Cd concentration in future measurements by using improved standards.
Additional Information
How to cite this article: Kashiv, Y. et al. Imaging trace element distributions in single organelles and subcellular features. Sci. Rep. 6, 21437; doi: 10.1038/srep21437 (2016).
References
Lee, S. M., Chen, H., Dettmer, C. M., O’Halloran, T. V. & Nguyen, S. T. Polymer-caged lipsomes: a pH-responsive delivery system with high stability. Journal of the American Chemical Society 129, 15096–15097 (2007).
Davis, A. V. & O’Halloran, T. V. A place for thioether chemistry in cellular copper ion recognition and trafficking. Nat Chem Biol 4, 148–151 (2008).
Kim, B. E., Nevitt, T. & Thiele, D. J. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol 4, 176–185 (2008).
El Muayed, M. et al. Acute cytokine-mediated downregulation of the zinc transporter ZnT8 alters pancreatic beta-cell function. The Journal of endocrinology 206, 159–169 (2010).
Kim, A. M., Vogt, S., O’Halloran, T. V. & Woodruff, T. K. Zinc availability regulates exit from meiosis in maturing mammalian oocytes. Nat Chem Biol 6, 674–681 (2010).
Bernhardt, M. L., Kong, B. Y., Kim, A. M., O’Halloran, T. V. & Woodruff, T. K. A zinc-dependent mechanism regulates meiotic progression in mammalian oocytes. Biology of reproduction 86, 114 (2012).
Kong, B. Y., Bernhardt, M. L., Kim, A. M., O’Halloran, T. V. & Woodruff, T. K. Zinc maintains prophase I arrest in mouse oocytes through regulation of the MOS-MAPK pathway. Biology of reproduction 87, 11, 11–12 (2012).
Thiers, R. E. & Vallee, B. L. Distribution of metals in subcellular fractions of rat liver. Journal of Biological Chemistry 226, 911–920 (1957).
Oyoo-Okoth, E. et al. Parasites modify sub-cellular partitioning of metals in the gut of fish. Aquat Toxicol 106–107, 76–84 (2012).
Chen, K. G. et al. Melanosomal sequestration of cytotoxic drugs contributes to the intractability of malignant melanomas. Proceedings of the National Academy of Sciences of the United States of America 103, 9903–9907 (2006).
McRae, R., Lai, B. & Fahrni, C. J. Subcellular redistribution and mitotic inheritance of transition metals in proliferating mouse fibroblast cells. Metallomics: integrated biometal science 5, 52–61 (2013).
Bozym, R. A., Thompson, R. B., Stoddard, A. K. & Fierke, C. A. Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem Biol 1, 103–111 (2006).
Jacob, C., Maret, W. & Vallee, B. L. Control of zinc transfer between thionein, metallothionein and zinc proteins. Proceedings of the National Academy of Sciences of the United States of America 95, 3489–3494 (1998).
Laychock, S. G., Duzen, J. & Simpkins, C. O. Metallothionein induction in islets of Langerhans and insulinoma cells. Mol Cell Endocrinol 165, 179–187 (2000).
Li, X., Chen, H. & Epstein, P. N. Metallothionein protects islets from hypoxia and extends islet graft survival by scavenging most kinds of reactive oxygen species. The Journal of biological chemistry 279, 765–771 (2004).
Guerquin-Kern, J. L., Wu, T. D., Quintana, C. & Croisy, A. Progress in analytical imaging of the cell by dynamic secondary ion mass spectrometry (SIMS microscopy). Biochimica et biophysica acta 1724, 228–238 (2005).
Steinhauser, M. L. & Lechene, C. P. Quantitative imaging of subcellular metabolism with stable isotopes and multi-isotope imaging mass spectrometry. Seminars in cell & developmental biology 24, 661–667 (2013).
Twining, B. S. et al. Quantifying Trace Elements in Individual Aquatic Protist Cells with a Synchrotron X-ray Fluorescence Microprobe. Analytical Chemistry 75, 3806–3816 (2003).
Kemner, K. M. et al. Elemental and Redox Analysis of Single Bacterial Cells by X-ray Microbeam Analysis. Science 306, 686–687 (2004).
Yang, L. et al. Imaging of the intracellular topography of copper with a fluorescent sensor and by synchrotron x-ray fluorescence microscopy. Proceedings of the National Academy of Sciences of the United States of America 102, 11179–11184 (2005).
Finney, L. et al. X-ray fluorescence microscopy reveals large-scale relocalization and extracellular translocation of cellular copper during angiogenesis. Proceedings of the National Academy of Sciences of the United States of America 104, 2247–2252 (2007).
Chen, K. G. et al. Influence of melanosome dynamics on melanoma drug sensitivity. Journal of the National Cancer Institute 101, 1259–1271 (2009).
Holt, M., Harder, R., Winarski, R. & Rose, V. Nanoscale Hard X-Ray Microscopy Methods for Materials Studies. Annual Review of Materials Research 43, 183–211 (2013).
Ortega, R., Cloetens, P., Devès, G., Carmona, A. & Bohic, S. Iron Storage within Dopamine Neurovesicles Revealed by Chemical Nano-Imaging. PloS one 2, e925 (2007).
Matsuyama, S. et al. Trace element mapping of a single cell using a hard x-ray nanobeam focused by a Kirkpatrick-Baez mirror system. X-Ray Spectrometry 38, 89–94 (2009).
Deng, J. et al. Simultaneous cryo X-ray ptychographic and fluorescence microscopy of green algae. Proceedings of the National Academy of Sciences 112, 2314–2319 (2015).
Gorniak, T. et al. Support and challenges to the melanosomal casing model based on nanoscale distribution of metals within iris melanosomes detected by X-ray fluorescence analysis. Pigment cell & melanoma research 27, 831–834 (2014).
Miyazaki, J.-I. et al. Establishment of a Pancreatic β Cell Line That Retains Glucose-Inducible Insulin Secretion: Special Reference to Expression of Glucose Transporter Isoforms. Endocrinology 127, 126–132 (1990).
Gilkey, J. C. & Staehelin, L. A. Advances in ultrarapid freezing for the preservation of cellular ultrastructure. Journal of Electron Microscopy Technique 3, 177–210 (1986).
Quintana, C. Cryofixation, cryosubstitution, cryoembedding for ultrastructural, immunocytochemical and microanalytical studies. Micron 25, 63–99 (1994).
Nagata, T. X-ray microanalysis of biological specimens by high voltage electron microscopy. Progress in Histochemistry and Cytochemistry 39, 185–319 (2004).
Cai, Z. et al. A hard x-ray scanning microprobe for fluorescence imaging and microdiffraction at the advanced photon source. AIP Conference Proceedings 507, 472–477 (2000).
Winarski, R. P. et al. A hard X-ray nanoprobe beamline for nanoscale microscopy. J Synchrotron Radiat 19, 1056–1060 (2012).
Yan, H. et al. Two dimensional hard x-ray nanofocusing with crossed multilayer Laue lenses. Opt. Express 19, 15069–15076 (2011).
Shirato, N. et al. Elemental fingerprinting of materials with sensitivity at the atomic limit. Nano letters 14, 6499–6504 (2014).
El Muayed, M. et al. Accumulation of cadmium in insulin-producing beta cells. Islets 4, 405–416 (2012).
Corezzi, S. et al. Synchrotron-based X-ray fluorescence imaging of human cells labeled with CdSe quantum dots. Analytical biochemistry 388, 33–39 (2009).
James, S. A. et al. Quantification of ZnO Nanoparticle Uptake, Distribution and Dissolution within Individual Human Macrophages. ACS Nano 7, 10621–10635 (2013).
Sheridan, E. J. et al. Synchrotron X-ray fluorescence studies of a bromine-labelled cyclic RGD peptide interacting with individual tumor cells. Journal of Synchrotron Radiation 20, 226–233 (2013).
Grubman, A. et al. X-ray fluorescence imaging reveals subcellular biometal disturbances in a childhood neurodegenerative disorder. Chem Sci 5, 2503–2516 (2014).
Leonardo, T. et al. Determination of elemental distribution in green micro-algae using synchrotron radiation nano X-ray fluorescence (SR-nXRF) and electron microscopy techniques - subcellular localization and quantitative imaging of silver and cobalt uptake by Coccomyxa actinabiotis. Metallomics : integrated biometal science 6, 316–329 (2014).
Huang, X. F. & Arvan, P. Formation of the insulin-containing secretory granule core occurs within immature beta-granules. The Journal of biological chemistry 269, 20838–20844 (1994).
Huang, X. F. & Arvan, P. Intracellular transport of proinsulin in pancreatic beta-cells. Structural maturation probed by disulfide accessibility. The Journal of biological chemistry 270, 20417–20423 (1995).
Chimienti, F., Favier, A. & Seve, M. ZnT-8, a pancreatic beta-cell-specific zinc transporter. Biometals: an international journal on the role of metal ions in biology, biochemistry and medicine 18, 313–317 (2005).
Pound, L. D. et al. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. The Biochemical journal 421, 371–376 (2009).
O’Halloran, T. V., Kebede, M., Philips, S. J. & Attie, A. D. Zinc, insulin and the liver: a menage a trois. The Journal of clinical investigation 123, 4136–4139 (2013).
Huber, A. M. & Gershoff, S. N. Effect of zinc deficiency in rats on insulin release from the pancreas. J Nutr 103, 1739–1744 (1973).
Jiang, L. J., Maret, W. & Vallee, B. L. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America 95, 3483–3488 (1998).
Habeebu, S. S., Liu, J., Liu, Y. & Klaassen, C. D. Metallothionein-null mice are more sensitive than wild-type mice to liver injury induced by repeated exposure to cadmium. Toxicol Sci 55, 223–232 (2000).
Jiang, L. J., Vasak, M., Vallee, B. L. & Maret, W. Zinc transfer potentials of the alpha - and beta-clusters of metallothionein are affected by domain interactions in the whole molecule. Proceedings of the National Academy of Sciences of the United States of America 97, 2503–2508 (2000).
Kim, B. J. et al. Zinc as a paracrine effector in pancreatic islet cell death. Diabetes 49, 367–372 (2000).
Lichtlen, P. et al. Target gene search for the metal-responsive transcription factor MTF-1. Nucleic Acids Res 29, 1514–1523 (2001).
Hasumi, M. et al. Regulation of metallothionein and zinc transporter expression in human prostate cancer cells and tissues. Cancer Letters 200, 187–195 (2003).
Huang, L., Kirschke, C. P., Zhang, Y. & Yu, Y. Y. The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. The Journal of biological chemistry 280, 15456–15463 (2005).
He, L. et al. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Mol Pharmacol 70, 171–180 (2006).
Hogstrand, C., Zheng, D., Feeney, G., Cunningham, P. & Kille, P. Zinc-controlled gene expression by metal-regulatory transcription factor 1 (MTF1) in a model vertebrate, the zebrafish. Biochem Soc Trans 36, 1252–1257 (2008).
Li, Y., Kimura, T., Huyck, R. W., Laity, J. H. & Andrews, G. K. Zinc-induced formation of a coactivator complex containing the zinc-sensing transcription factor MTF-1, p300/CBP and Sp1. Molecular and cellular biology 28, 4275–4284 (2008).
Mocchegiani, E., Giacconi, R. & Malavolta, M. Zinc signalling and subcellular distribution: emerging targets in type 2 diabetes. Trends Mol Med 14, 419–428 (2008).
Lichten, L. A., Ryu, M. S., Guo, L., Embury, J. & Cousins, R. J. MTF-1-mediated repression of the zinc transporter Zip10 is alleviated by zinc restriction. PloS one 6, e21526 (2011).
Saxena, R. et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316, 1331–1336 (2007).
Boesgaard, T. W. et al. The common SLC30A8 Arg325Trp variant is associated with reduced first-phase insulin release in 846 non-diabetic offspring of type 2 diabetes patients–the EUGENE2 study. Diabetologia 51, 816–820 (2008).
Omori, S. et al. Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8 and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes 57, 791–795 (2008).
Wu, Y. et al. Common variants in CDKAL1, CDKN2A/B, IGF2BP2, SLC30A8 and HHEX/IDE genes are associated with type 2 diabetes and impaired fasting glucose in a Chinese Han population. Diabetes 57, 2834–2842 (2008).
Xiang, J. et al. Zinc Transporter-8 Gene (SLC30A8) Is Associated with Type 2 Diabetes in Chinese. J Clin Endocrinol Metab 93, 4107–4112 (2008).
Majithia, A. R. et al. Association of the SLC30A8 missense polymorphism R325W with proinsulin levels at baseline and after lifestyle, metformin or troglitazone intervention in the Diabetes Prevention Program. Diabetologia 54, 2570–2574 (2011).
Shan, Z. et al. Interactions between Zinc Transporter-8 Gene (SLC30A8) and Plasma Zinc Concentrations for Impaired Glucose Regulation and Type 2 Diabetes. Diabetes (2013).
Fu, Y. et al. Down-regulation of ZnT8 expression in INS-1 rat pancreatic beta cells reduces insulin content and glucose-inducible insulin secretion. PloS one 4, e5679 (2009).
Wijesekara, N. et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia (2010).
Ibáñez, A. J. et al. Mass spectrometry-based metabolomics of single yeast cells. Proceedings of the National Academy of Sciences 110, 8790–8794 (2013).
Zenobi, R. Single-cell metabolomics: analytical and biological perspectives. Science 342, 1243259 (2013).
Vogt, S. MAPS: A set of software tools for analysis and visualization of 3D X-ray fluorescence data sets. J. Phys. IV France 104, 635–638 (2003).
Acknowledgements
We would like to thank Jorg Maser for encouragement and helpful discussions and Dubi Kaufmann for help with the software used to prepare the figures. This work was supported by grant 1K08ES020880-01 from the NIEHS/ NIH to ME. Work at the Advanced Photon Source and the Center for Nanoscale Materials was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
J.A., M.E. and Y.K. conceived and designed the experiment; M.E. cultured the cells, J.A. prepared the samples and performed the TEM work; B.L., V.R. and Y.K. performed the SXRF work; B.L., S.V., V.R. and Y.K. analyzed the SXRF data; B.L., J.A., M.E., S.V., V.R. and Y.K. discussed the results; Y.K. wrote the paper with contributions from B.L., J.A., S.V., V.R. and mainly from M.E.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kashiv, Y., Austin, J., Lai, B. et al. Imaging trace element distributions in single organelles and subcellular features. Sci Rep 6, 21437 (2016). https://doi.org/10.1038/srep21437
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21437
This article is cited by
-
Sensitive fluorescent chemosensor for Hg(II) in aqueous solution using 4’-dimethylaminochalcone
Journal of Fluorescence (2022)
-
Arabidopsis bZIP19 and bZIP23 act as zinc sensors to control plant zinc status
Nature Plants (2021)
-
Nano-imaging trace elements at organelle levels in substantia nigra overexpressing α-synuclein to model Parkinson’s disease
Communications Biology (2020)
-
X-ray Fluorescence Nanotomography of Single Bacteria with a Sub-15 nm Beam
Scientific Reports (2018)
-
The emerging role of zinc transporters in cellular homeostasis and cancer
Signal Transduction and Targeted Therapy (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
