
Abstract
Protein kinase C (PKC)-δ is proapoptotic in human keratinocytes, and is downregulated or inactivated in keratinocytes expressing the activated Ha-ras oncogene, making it a candidate tumor suppressor gene for squamous cell carcinoma (SCC). We evaluated the significance of PKC-δ loss in transformed human keratinocytes using tumorigenic HaCaT Ras II-4 cells that have significantly reduced PKC-δ levels. Re-expression of PKC-δ by retrovirus transduction caused an increase in apoptosis and growth inhibition in culture. The growth inhibition induced by PKC-δ could be partially reversed by Bcl-xL expression, indicating that apoptosis was in part responsible for PKC-δ-induced growth inhibition. PKC-δ re-expression suppressed the tumorigenicity of HaCaT Ras II-4 cells in nude mice (P<0.05), and the small tumors that did form contained elevated levels of activated caspase-3, indicating increased apoptosis. In addition, we found that 29% (12/42) of human Bowen's disease (squamous carcinoma in situ) or SCC cases had absent or reduced PKC-δ when compared to the surrounding normal epidermis. These results indicate that PKC-δ inhibits transformed keratinocyte growth by inducing apoptosis, and that PKC-δ may function as a tumor suppressor in human SCCs where its loss in cells harboring activated ras could provide a growth advantage by conferring resistance to apoptosis.
Similar content being viewed by others


Introduction
Tumor suppressor genes are able to limit premalignant cell proliferation by a variety of mechanisms, most notably by the induction of growth arrest or apoptosis. Activation of protein kinase C (PKC)-δ, a widely expressed PKC isoform, can induce growth arrest in several cell types (Watanabe et al., 1992; Griffiths et al., 1996; Black, 2000; Frey et al., 2001). In addition, the cleavage and activation of PKC-δ is both necessary and sufficient for apoptosis induced by UV light in normal human keratinocytes (Denning et al., 1998, 2002; D’Costa and Denning, 2005). PKC-δ is activated proteolytically by caspase-3-mediated cleavage in human keratinocytes (Denning et al., 1998), as well as many other cell types undergoing apoptosis (Emoto et al., 1995; Ghayur et al., 1996; Reyland et al., 2000; Cataldi et al., 2002). Since apoptosis can be a protective mechanism from UV-induced skin cancer, PKC-δ activation may participate in the elimination of precancerous cells by promoting apoptosis (Ziegler et al., 1994; Jiang et al., 1999). Indeed PKC-δ has tumor suppressor properties in mouse skin since PKC-δ-overexpressing transgenic mice are resistant to skin chemical carcinogenesis, and treatment of mouse keratinocytes overexpressing PKC-δ with the tumor promoter TPA triggers apoptosis (Li et al., 1999; Reddig et al., 1999). The PKC-δ inhibitor rottlerin induces a transformed phenotype in HaCaT cells (Dietrich et al., 2001), and PKC-δ acts as a tumor suppressor in other cell types such as transformed colon epithelial and fibroblast cell lines (Lu et al., 1997; Perletti et al., 1999).
The expression of activated Ha-ras in mouse keratinocytes induces tyrosine phosphorylation and enzymatic inhibition of PKC-δ, and has been linked to a reduced differentiated phenotype (Denning et al., 1993, 1996; Joseloff et al., 2002). In addition, PKC-δ protein and activity are lost in chemically induced mouse skin tumors, where activating Ha-ras mutations are initiating events (Roop et al., 1986; Reddig et al., 1999). While ras gene mutations are found in 58% of human squamous cell carcinomas (SCCs) (Pierceall et al., 1991), it is not known if the proapoptotic PKC-δ is lost in human SCCs. In the HaCaT human keratinocyte cell line, expression of activated Ha-ras selectively reduces PKC-δ protein and mRNA levels without affecting other PKC isoform levels, and loss of PKC-δ in ras-transformed HaCaT cells is mediated by transforming growth factor (TGF)-α/epidermal growth factor receptor (EGFR) signaling (Geiges et al., 1995). Autocrine TGF-α/EGFR signaling is also responsible for the tyrosine phosphorylation and enzymatic inhibition of PKC-δ in ras-transformed mouse keratinocytes (Denning et al., 1996). Thus, loss of PKC-δ protein and/or activity is a common feature of ras-transformed skin tumors. In addition, inhibition of PKC-δ by rottlerin or a dominant/negative PKC-δ transforms EGFR-overexpressing rat 3Y1 fibroblasts in the absence of epidermal growth factor (EGF), further implicating PKC-δ as a suppressor of EGFR-dependent cell transformation (Hornia et al., 1999).
The human PKC-δ gene is located at chromosome 3p21.31, and this region is frequently lost in poorly differentiated SCCs and in 60% of Bowen's disease, as determined by interphase FISH and comparative genomic hybridization (Dobler et al., 1999; Popp et al., 2000, 2002). Thus, in addition to the loss of PKC-δ expression in ras-transformed keratinocytes via a TGFα/EGFR autocrine loop, the PKC-δ gene may be frequently deleted in SCCs.
In this study, we re-expressed PKC-δ in human HaCaT cells expressing activated ras (HaCaT Ras II-4 cells), and found that PKC-δ induced apoptosis and suppressed tumor formation in nude mice. We also evaluated the expression of PKC-δ in human skin SCCs, Bowen's disease, and normal human skin by immunohistochemical staining, and found that PKC-δ was lost in 12/42 (29%) of human skin cancers evaluated. These results indicate that the loss of PKC-δ expression is important for ras-mediated transformation, and supports a role of PKC-δ as a tumor suppressor in human skin SCCs.
Materials and methods
Immunohistochemistry
Tissue samples were fixed in 10% neutral buffered formalin and embedded in paraffin. Tissue sections were deparaffinized followed by antigen retrieval (10 mM citrate buffer, pH 6.0, microwave 500 W; 15 min). The samples were stained for PKC-δ (sc-937, 1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA), PCNA (BD Transduction Laboratories, Palo Alto, CA, USA 1:50), or active caspase-3 (R&D Systems, 1:500) by immunohistochemical methods using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). The level of PKC-δ staining was evaluated throughout the entire tumor section, and was classified as having lost PKC-δ if greater than 50% of the tumor cells had low reactivity that was comparable to the blocking peptide control. The tissue samples were also stained for phospho-EGFR (Y1068) (2236, Cell Signaling Technology Inc., Beverly, MA, USA) at a dilution of 1:100 overnight. To avoid any potential bias, the person reading the phospho-EGFR-stained tumors was blinded to the PKC-δ status of the tumor. All human tissue samples were obtained with approval from the Loyola University Medical Center's Institutional Review Board.
Virus construction
A retrovirus encoding PKCδ was constructed by cloning the full-length human PKC-δ cDNA (80048, American Tissue Culture Collection, Manassas, VA, USA) into the XbaI site of pBluescript KS (Denning et al., 2002), digesting it with KasI/BamHI to remove the initiation codon, and replacing it with double-stranded oligonucleotides encoding a Kozak sequence and an amino-terminal c-myc epitope tag to generate pKS-myc-PKCδ-1. The myc-tagged PKC-δ was removed by digesting with BamHI/NotI, and cloned into the BamHI/NotI sites of LZRS (Paul Khavari, Stanford University School of Medicine, Stanford, CA, USA) to generate LZRS-myc-PKC-δ. Active Ras (Q61) virus was constructed by inserting Ras (Q61) into the BamHI/XhoI sites of LZRS-Linker, and the dominant/negative PKCδ(K378A) was described previously (Denning et al., 2002; D’Costa and Denning, 2005). Retroviruses were generated by transfecting plasmids with calcium phosphate into the Phoenix Ampho retroviral packaging line (ATCC with permission Garry P Nolan, Stanford University Medical Center) and selecting with puromycin. Viruses were harvested from confluent dishes cultured at 32°C for 24–48 h. As a negative control, virus was generated from the empty LZRS-Linker vector (Denning et al., 2002).
Cell culture and retroviral infection
HaCaT, HaCaT Ras I-7, and HaCaT Ras II-4 cells (kindly provided by Norbert Fusenig and Mihaela Skobe) were grown in DMEM with 10% FBS. All cells were infected twice with Linker virus or PKC-δ virus by spin infections in six well dishes. The level of overexpression of PKC-δ was measured by flow cytometry prior to injection into nude mice and confirmed by Western blotting for PKC-δ. For some experiments, HaCaT cells were treated with EGF at 10 ng/ml for 2 days.
Growth assays
HaCaT and HaCaT Ras II-4 cells were plated at a density of 1 × 105 per well in six well dishes, and infected the next day twice with either Linker virus or PKC-δ virus. Viable cells were counted by trypan blue staining. Data from the HaCaT Ras II-4 growth curve in Figure 2c were fitted to an exponential curve using Microsoft Excel (R2=0.9996). Using the equation describing HaCaT Ras II-4 cell growth [y=(starting cell number)e0.4557x], where x equals the number of days, a new theoretical growth curve was calculated by subtracting 6% of the cell number per day, starting with day 1, and using the adjusted cell yield as the starting cell number for the next day's calculation. Thus, the calculated growth curve represents the growth rate of HaCaT Ras II-4 cell if they lost 6% of their cell number every day due to apoptosis.

PKC-δ inhibits the growth of HaCaT Ras II-4 cells. (a) HaCaT Ras II-4 cells infected with either Linker virus or PKC-δ virus were photographed. Note the increase in cell size induced by PKC-δ infection. (b) HaCaT cells were infected with PKC-δ and/or Bcl-xL viruses as indicated, and the viable cells counted at indicated days after infection (N=2, error bars denote range). (c) HaCaT Ras II-4 cells were infected with PKC-δ and/or Bcl-xL viruses as indicated, and the viable cells counted at indicated days after infection. The mock-6% group (dotted line) is a theoretical growth curve calculated by subtracting 6% of the cell yield per day from the mock-infected HaCaT Ras II-4 cells. (N=2, error bars denote range).
Flow cytometry
The levels of PKC-δ overexpression in nude mice were analysed by flow cytometry prior to injection. Cell cycle analysis of HaCaT Ras II-4 cells infected with empty Linker or PKC-δ virus was performed by propidium iodide staining and flow cytometry as described previously (Denning et al., 1998). The cells were washed twice with PBS and fed the day after infection, and then assayed 2 days after infection.
Tumor growth in nude mice
Athymic male (BALB/c) nude mice were obtained from Taconic (Germantown, NY, USA) and housed in laminar airflow cabinets under pathogen-free conditions with a 12 h light/12 h dark cycle. The mice were injected subcutaneously on either side with 5 × 106 cells in 200 μl per injection and the growth of tumors measured twice a week over 21 days. Tumor volume was calculated using the formula length × width × height, as measured using calipers.
Western blotting
Total cell lysates from cultured cells were prepared by lysing cells in 20 mM Tris-HCl, pH 7.5, 1% Triton X-100, and 1 × Complete Protease Inhibitor Cocktail (Boehringer Mannheim, Indianapolis, IN, USA). For tumor lysates, freshly harvested tissues samples were homogenized in ice-cold CHAPS buffer (20 mM Hepes, pH 7.4, 140 mM NaCl, 10 mM CHAPS, 2 mM EDTA, 1 mM EGTA, and Complete Protease Inhibitor Cocktail), and incubated on the thermoshaker for 30 min at 4°C. Lysates were spun in a microcentrifuge for 10 min at 4°C, and 20 μg protein from the supernatant taken for analysis. The proteins were run on SDS–polyacrylamide gels, transferred to nitrocellulose, and stained with Ponceau S to ensure equal protein loading. The membranes were blocked with 5% milk in 20 mM Tris-HCl, pH 7.6, 150 mM NaCl (TBS), stained with primary antibody and horseradish peroxidase-conjugated secondary antibody for 1 h each, and extensively washed in TBS, 0.05% Tween-20. Proteins were detected using the ECL detection kit (Amersham Life Sciences, Piscataway, NJ, USA). PKC-α (sc-208, 1:1000), PKC-δ (sc-937, 1:4000), PKC-ɛ (sc-214, 1:200), PKC-η (sc-215, 1:200), and PKC-ζ (sc-216, 1:1000) were detected by Western blotting using rabbit antibodies from Santa Cruz Biotech. An actin antibody (ICN Biomedicals Inc., Irvine, CA, USA) used at a dilution of 1:4000 served as a control for equal loading.
Results
PKC-δ levels are decreased in human keratinocytes expressing active ras
PKC-δ protein and mRNA are reported to be selectively lost in two ras-transfected and clonally selected human keratinocyte cell lines, HaCaT Ras I-7 and HaCaT Ras II-4 (Boukamp et al., 1990; Geiges et al., 1995). In Figure 1b, we found that PKC-δ protein levels were reduced but still detectable in HaCaT Ras I-7 and II-4 cells. In addition, Figure 1a shows that PKC-δ levels were decreased in a mass population of HaCaT cells infected with an active Ha-ras (Q61) retrovirus, indicating that the ras-induced loss of PKC-δ is not due to clonal variation. HaCaT cells treated with EGF also had a loss of PKC-δ protein (Figure 1b), confirming that EGFR signaling is involved in the loss of PKC-δ (Geiges et al., 1995; Denning et al., 1996). Using retroviral transduction, the full-length wild-type PKC-δ was re-expressed in a mass population of HaCaT Ras II-4 cells, as shown in Figure 1a (lane 4). As shown in Figure 1c, the levels of PKC-α, PKC-ɛ, and PKC-ζ were not significantly different between HaCaT and HaCaT Ras II-4 cells, consistent with previous results (Geiges et al., 1995). Furthermore, PKC-δ overexpression did not alter the levels of other PKC isoforms. PKC-η was not detected as PKC-η is only expressed in differentiating keratinocytes (data not shown) (Koizumi et al., 1993; Osada et al., 1993). PKC-δ re-expression was also determined by FACS analysis for rapid monitoring of expression levels (data not shown). PKC-δ was routinely overexpressed 10-fold above the Linker-infected control HaCaT Ras II-4 cells.

Active ras reduces PKC-δ protein levels. (a) HaCaT cells were infected with Linker virus or with active ras virus and cell lysates analysed by Western blotting for PKC-δ and actin as a loading control. Transformed HaCaT Ras II-4 cells infected with either Linker virus or PKC-δ virus were also analysed by Western blotting for PKC-δ and actin. Similar results were obtained in two additional experiments. (b) HaCaT cells cultured with 10 ng/ml EGF for 2 days, and ras-transformed HaCaT Ras I-7 and II-4 cell lines were analysed for PKC-δ levels by Western blot. Note the reduction in PKC-δ levels triggered by ras or EGF treatment. (c) HaCaT or HaCaT Ras II-4 cells were infected with either Linker (L) virus or PKC-δ (δ) virus and lysates analysed for the expression of PKC-α, PKC-δ, PKC-ɛ, PKC-ζ, and actin. Note the selective loss of PKC-δ in the HaCaT Ras II-4 cells. PKC-η was not detected.
Growth inhibition and induction of apoptosis in HaCaT Ras II-4 cells overexpressing PKC-δ
In addition to downregulating PKC-δ levels, expression of activated ras triggers the activation of multiple mitogenic and survival pathways (Raf/MAPK, PI3K/Akt, RalGDS/Ral) (Ewen, 2000). To determine if the downregulation of PKC-δ in tumorigenic HaCaT Ras II-4 cells was required for their enhanced growth rate and transformed phenotype, we restored the expression of PKC-δ in HaCaT Ras II-4 cells by retroviral transduction and analysed their growth properties. Figure 2a shows that re-expression of PKC-δ in HaCaT Ras II-4 cells caused them to appear larger compared to Linker-infected cells, with some cells becoming elongated. PKC-δ expression also inhibited the growth of HaCaT and HaCaT Ras II-4 cells in culture as compared to mock-infected controls (Figure 2b and c). Note that the HaCaT Ras II-4 cells grow faster than the parental HaCaT cells (compare Figure 2b with c). A cell cycle analysis was performed to determine if PKC-δ expression was arresting HaCaT Ras II-4 cells in a specific phase of the cell cycle. As shown in Figure 3, over-expression of PKC-δ triggered a modest but statistically significant (P<0.01) 6% increase in apoptosis over a 1-day period. While PKC-δ did not arrest the cells in any specific cell cycle phase, there was a reduced percentage of cells with S (P<0.05) and G2/M DNA content in cells expressing PKC-δ, suggesting that fewer cells were in cycle.

Cell cycle analysis of HaCaT Ras II-4 cells. (a) HaCaT Ras II-4 cells were infected with either Linker virus or PKC-δ virus and cell cycle analysis performed by propidium iodide staining and flow cytometry. The cells were extensively washed 1 day before analysis so that the floating (apoptotic) cells accumulating during 1 day could be assessed. (b) Quantitation of HaCaT Ras II-4 cells in various stages of the cell cycle (N=2, error bars denote range). Similar results were obtained in two experiments.
To determine if the growth-suppressive effects of PKC-δ expression in HaCaT Ras II-4 cells were due to the increase in apoptosis, we inhibited apoptosis in HaCaT and HaCaT Ras II-4 cells by Bcl-xL retroviral transduction. Bcl-xL expression inhibited UV-induced cell death in both the HaCaT cells (59% inhibition) and HaCaT Ras II-4 cells (87% inhibition), indicating that the Bcl-xL was functional (data not shown). As shown in Figures 2b and c, Bcl-xL caused a small increase in growth in HaCaT and HaCaT Ras II-4 cells, but reversed the growth-inhibitory effects of PKC-δ in both HaCaT cells and HaCaT Ras II-4 cells. The growth inhibition induced by PKC-δ was not completely reversed by Bcl-xL, indicating either that Bcl-xL expression did not completely block apoptosis, or that cell cycle inhibition may also be involved in the growth-suppressive effects of PKC-δ. To address the role of apoptosis further, we calculated the growth rate of HaCaT Ras II-4 cell, assuming that 6% of the cells underwent apoptosis per day, based on the apoptosis data from Figure 3. As shown in Figure 2c (dashed line), a mere 6% increase in daily apoptosis could completely account for the reduced growth rate of HaCaT Ras II-4 cells expressing PKC-δ.
PKC-δ suppressed the tumorigenicity of HaCaT Ras II-4 cells in nude mice
To determine if reduction of PKC-δ in ras-transformed HaCaT cells was required for their tumorigenicity, we re-expressed full-length PKC-δ in HaCaT Ras II-4 cells and assayed tumor formation in nude mice following subcutaneous injection. HaCaT Ras II-4 cells infected with Linker virus formed large tumors, while the parental HaCaT cells that were Linker-infected did not form palpable tumors (Figure 4). HaCaT Ras II-4 cells expressing PKC-δ formed either undetectable or significantly smaller tumors over a 21-day period. A similar inhibition of tumor growth was seen with HaCaT Ras I-7 cells infected with PKC-δ virus, although the HaCaT Ras I-7 cells formed only benign tumors (data not shown). Histopathologic analysis of the tumors stained with hematoxylin and eosin (Figure 5) showed that the HaCaT Ras II-4 cells all formed SCCs (5/5), while only two of seven HaCaT Ras II-4 cells infected with the PKC-δ virus formed SCCs, and three of seven formed cysts. Two of the seven HaCaT Ras II-4 cells infected with PKC-δ did not form palpable tumors, and nothing was detected at the site of injection by histology. These results are summarized in Table 1.

Tumor suppression of HaCaT Ras II-4 cells in nude mice by PKC-δ. Nude mice were injected subcutaneously with 5 × 106 cells per injection in 200 μl of DMEM. The size of the tumors was measured twice a week, and the tumor volume plotted. The sample groups studied were HaCaT (Linker infected, N=6), HaCaT Ras II-4 (Linker infected, N=5) and HaCaT Ras II-4 (PKC-δ infected, N=7). Suppression of tumor formation by PKC-δ was also observed in an additional experiment.

Histopathologic analysis of nude mice tumors. Tumors removed from nude mice were sectioned and stained by hematoxylin and eosin. The parental HaCaT cells infected with Linker virus and HaCaT Ras II-4 cells infected with PKC-δ virus formed benign cysts. The arrows point to the outer epithelia lining and the asterisks denote the fluid-filled or necrotic interior of the cysts. HaCaT Ras II-4 cells infected with Linker virus formed SCCs.
Since apoptosis was found to be partially responsible for PKC-δ-induced growth inhibition in culture, we examined the tumors and cysts recovered from nude mice for markers of apoptosis (active caspase-3) and cell proliferation (PCNA). As shown in Figure 6, the cysts formed by the parental HaCaT cells had rare cells with active caspase-3, even in areas of morphological terminal differentiation. In addition, only rare isolated cells in the Linker-infected HaCaT Ras II-4 tumors were undergoing apoptosis, as detected by active caspase-3 staining. In contrast, all the PKC-δ-infected HaCaT Ras-II-4 tumors had extensive areas of active caspase-3 staining, indicating a much higher apoptotic rate. Surprisingly, PCNA-positive nuclei were also much more prevalent in the PKC-δ-infected HaCaT Ras II-4 tumors than the HaCaT cysts or the HaCaT Ras II-4 tumors. This suggests that the PKC-δ-transduced HaCaT Ras II-4 cells may have an enhanced proliferative rate to compensate for the increased apoptosis. Note that the active caspase-3 staining in PKC-δ-infected HaCaT Ras II-4 tumors was confined to areas of morphological differentiation and did not colocalize with PCNA-positive areas.

Increased apoptosis and proliferation in PKC-δ-expressing tumors. Tumors from Linker-infected HaCaT cells, Linker-infected HaCaT Ras II-4 cells, and PKC-δ-infected HaCaT Ras II-4 cells were stained by immunohistochemistry with antibodies to active caspase-3 to detect apoptotic cells, or PCNA to detect proliferating cells. Note the enhanced active caspase-3 (arrows) and PCNA staining in the HaCaT Ras II-4 (PKC-δ) tumors.
Loss of PKC-δ in human SCCs
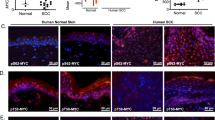
The loss of PKC-δ in ras-transformed HaCaT cells and chemically induced mouse papillomas (Geiges et al., 1995; Reddig et al., 1999), plus the ability of PKC-δ to induce growth arrest (Watanabe et al., 1992; Griffiths et al., 1996) and apoptosis (Li et al., 1999; Denning et al., 2002; Efimova et al., 2004; Sitailo et al., 2004), prompted us to explore the potential loss of PKC-δ in human SCCs. Normal human skin was stained for PKC-δ and 6/6 (100%) samples were positive for PKC-δ, as shown in Figure 7a. The staining for PKC-δ in normal skin was stronger in the basal layer, but was seen in all layers and could be completely prevented by preincubation of the antibody with a blocking peptide. To determine the expression level and pattern of PKC-δ in human SCCs, we stained 32 SCCs and 10 Bowen's disease (carcinomas in situ) samples and found that PKC-δ was lost in 12/42 (∼29%) of the skin tumors analysed. Tumors were classified as having lost PKC-δ if at least 50% of the tumor area had low or no PKC-δ immunoreactivity. Representative SCC tumors (case 282 and case 356) stained for PKC-δ are shown in Figure 7a. PKC-δ staining was lost in focal areas surrounded by large regions of the tumors that had only weak PKC-δ positively. Even areas with very low PKC-δ had some scattered strongly positive cells, indicating that the PKC-δ antibody could react in these tumors. Table 2 summarizes the PKC-δ status of the tumors stained. Although there was no correlation between PKC-δ loss and tumor histology, PKC-δ was reduced in four of 10 Bowen's disease samples and one solar keratosis (data not shown), both precancerous lesions, indicating that loss of PKC-δ can be an early event in human skin carcinogenesis.

Loss of PKC-δ in SCCs. (a) PKC-δ staining of paraffin-embedded normal human skin and human skin SCCs by immunohistochemistry. Note the reduced staining of PKC-δ in areas of the SCCs (case 282 and case 356). The blocking peptide for PKC-δ used as a control in upper right panel (+Peptide) shows that the staining for PKC-δ is specific. A low power image of Case 282 tumor is depicted in the lower panel of the figure. (b) Lysates were prepared from freshly excised human SCCs or normal skin (patient 3), and analysed for PKC-δ and actin levels by Western blotting. Note the loss of PKC-δ in the SCC lysates. (c) Phospho-EGFR staining of normal skin and two SCCs (case 282 and case 356). Note the membrane staining specific for the SCCs that have lost or reduced PKC-δ staining.
The loss of PKC-δ observed by immunohistochemistry was confirmed by Western blotting in an independent group of tumors. Figure 7b shows three randomly-selected human SCC tumors that had almost complete loss of PKC-δ. For patient 3, matched normal skin was also analysed and had abundant PKC-δ levels. As a loading control, actin levels were also assessed and were not different between the tumor lysates.
Since EGFR signaling is linked to the loss of PKC-δ in both human and mouse keratinocytes (Geiges et al., 1995; Denning et al., 1996), we also stained SCCs with PKC-δ loss for phospho-EGFR as an indicator of activated EGFR signaling. As shown in Figure 7c, while normal skin gave no membrane reactivity for phospho-EGFR, several of the tumors (five of eight, 62%) that had reduced PKC-δ also had membrane phospho-EFGR staining. Although the overall intensity of phospho-EGFR staining varied among normal and tumor samples, membrane staining was restricted to the SCCs and was not detected in normal human skin. Thus, the majority of human SCCs that have lost PKC-δ immunoreactivity also have activated (autophosphorylated) EGFR, consistent with the role for EGFR signaling in PKC-δ loss identified in cell culture studies (Figure 1; Geiges et al., 1995; Denning et al., 1996).
Discussion
Our data support a role for PKC-δ as a tumor suppressor in human SCCs of the skin, and is the first description of the loss of PKC-δ in human SCCs. The general properties of a tumor suppressor are its ability to induce apoptosis, trigger growth arrest and/or differentiation, and its loss in malignant and premalignant tumors (Sherr, 2004). PKC-δ fulfills these criteria since it is necessary and sufficient for UV apoptosis (Denning et al., 1998; Li et al., 1999; Leitges et al., 2001; Sitailo et al., 2004; D’Costa and Denning, 2005), can cause cell cycle arrest (Watanabe et al., 1992; Perletti et al., 1999), and can induce keratinocyte differentiation marker expression (Deucher et al., 2002). We found that re-expression of PKC-δ in ras-transformed HaCaT cells induces a moderate level of apoptosis (Figure 3), but this level of apoptosis compounded daily can dramatically inhibit overall cell growth and is sufficient to account for the growth-suppressive effects of PKC-δ (Figure 2c). In addition, we partially reversed PKC-δ growth inhibition by expressing the antiapoptotic Bcl-2 family member Bcl-xL.
Ectopic expression of tumor suppressors can also inhibit tumor formation, and transgenic mice overexpressing PKC-δ are resistant to skin tumor promotion by chemical carcinogens (Reddig et al., 1999). A recent study also demonstrated that overexpressing PKC-δ in HaCaT cells suppressed tumor formation in SCID mice (Papp et al., 2004). Since HaCaT cells and normal human keratinocytes express comparable levels of PKC-δ (LA Sitailo and MF Denning, unpublished observation), and oncogenic ras triggers the selective downregulation of PKC-δ in transformed HaCaT cells (Figure 1; Geiges et al., 1995), we restored PKC-δ expression in HaCaT Ras II-4 cells to determine the effect of PKC-δ loss in ras transformation. Our results in Figure 4 demonstrate that restoration of PKC-δ suppressed tumor formation in nude mice. However, inhibition of PKC-δ activity with a dominant/negative PKC-δ(K378A) was not sufficient to transform HaCaT cells (data not shown), indicating that additional oncogenic events are required for HaCaT cells to form tumors in nude mice. We also found that PKC-δ was lost in ∼30% of the human skin tumors that we studied (Table 2). The loss or inactivation of PKC-δ in human tumors is a prerequisite for it being a tumor suppressor in human skin carcinogenesis. The loss of PKC-δ in human skin cancers helps validate mouse models of chemical skin carcinogenesis where loss of PKC-δ has been previously observed (Reddig et al., 1999; Wheeler et al., 2002).
Keratinocytes express five PKC isoforms (α, δ, ɛ, η, and ζ) which have selective roles in skin carcinogenesis, functioning to enhance (i.e. PKC-ɛ) or suppress (i.e. PKC-δ, PKC-η) tumor formation (Reddig et al., 1999, 2000; Jansen et al., 2001; Chida et al., 2003; Wheeler et al., 2003, 2004, 2005; Papp et al., 2004). While there is selective loss of PKC-δ in ras-transformed HaCaT cells, loss of PKC-ɛ has been reported in both mouse and human SCCs (Wheeler et al., 2005), and thus multiple PKC isoform changes can occur during multi-step skin carcinogenesis.
The mechanism of PKC-δ loss in human SCCs is unknown, but is likely to involve ras/EGFR signaling. Ras activation has been reported in ∼50% of human SCC (Pierceall et al., 1991), and keratinocytes with active ras mutation secrete autocrine EGFR ligands such as TGF-α (Glick et al., 1991; Dlugosz et al., 1995). EGFR is required to maintain the proliferative population in the basal compartment of epidermal tumors (Hansen et al., 2000), and it provides an essential survival signal for skin tumor development (Sibilia et al., 2000). Furthermore, disruption of the EGFR receptor impairs the growth of squamous papillomas expressing the v-Ha-ras oncogene (Dlugosz et al., 1997). Membrane staining for phospho-EGFR was seen in the majority of human SCC tumors which had lost PKC-δ (Figure 7c), suggesting activation of the ras/TGF-α/EGFR autocrine feedback loop. Several studies have identified autocrine TGF-α and the EGFR as being necessary for the inhibition/loss of PKC-δ in neoplastic keratinocytes (Geiges et al., 1995; Denning et al., 1996). In addition, Src family tyrosine kinases can phosphorylate PKC-δ in response to EGFR activation and lead to inhibition or downregulation of PKC-δ (Denning et al., 1996; Zang et al., 1997; Joseloff et al., 2002). The exact mechanism of how PKC-δ is reduced in human SCC tumors with active ras/EGFR signaling is still unknown; however, HaCaT Ras I-7 and HaCaT Ras II-4 cells have lost PKC-δ mRNA expression, and thus inhibition of PKC-δ transcription may play a role in ras-transformed keratinocytes (Geiges et al., 1995). Chromosome loss at 3p21.31, where the human PKC-δ gene resides, is frequent in human SCC, and may explain the loss of PKC-δ in the subset of SCCs without activated ras/EGFR signaling (Dobler et al., 1999; Ashton et al., 2003). The loss of PKC-δ in ras-transformed keratinocytes was specific as no changes in other PKC isoforms were observed (Figure 1c) (Geiges et al., 1995). Furthermore, there was no compensatory increase in other PKC isoforms in the epidermis of PKC-δ null mice (Wheeler et al., 2002).
The tumor suppression mechanism of PKC-δ is likely to involve the induction of apoptosis. We found that re-expression of PKC-δ in HaCaT Ras II-4 cells inhibited the growth and increase apoptosis, both in culture (Figures 2 and 3) and in nude mice (Figure 4). Furthermore, the growth inhibition in culture could be partially reversed by overexpressing the antiapoptotic protein Bcl-xL. Bcl-xL did not significantly enhance the growth rate of mock-infected HaCaT cells or HaCaT Ras II-4 cells, indicating that Bcl-xL was not stimulating growth in the absence of high PKC-δ levels. Finally, we observed ∼6% increase in apoptotic cells over a 24-h period (Figure 3). While this increase in apoptosis is modest, it can entirely explain the growth suppression observed in Figure 2.
The mechanism of PKCδ-induced apoptosis has been extensively studied, and involves components of the intrinsic death effector pathway (Brodie and Blumberg, 2003). Expression of the PKC-δ catalytic fragment, which is generated by caspase-3 cleavage in response to UV radiation and other apoptotic stimuli, has been associated with both the mitochondria and nucleus (Denning et al., 2002; DeVries et al., 2002). Kinase activity is required for PKC-δ to induce apoptosis, and several apoptotic substrates for the PKC-δ catalytic fragment have been identified, including lamin B, phospholipid scramblase 1 and 3, DNA-dependent protein kinase, Rad9, p73β, and p38δ (Bharti et al., 1998; Cross et al., 2000; Frasch et al., 2000; Ren et al., 2002; Liu et al., 2003; Yoshida et al., 2003; Efimova et al., 2004). Despite the identification of these substrates, how the PKC-δ catalytic fragment initiates the activation of Bax, release of cytochrome c, and activation of multiple caspases is unclear (Sitailo et al., 2004). Studies overexpressing the full-length PKC-δ and activating with TPA have also found mitochondrial death effector pathway activation (Li et al., 1999; Fujii et al., 2000). Although the full-length PKC-δ we expressed was wild type, spontaneous activation can occur, and ras-transformed keratinocytes have elevated cellular diacylglycerol, an endogenous activator of PKC isoforms (Lee et al., 1992).
Surprisingly, we also found elevated PCNA staining in HaCaT Ras II-4 tumors expressing PKC-δ (Figure 6). Enhanced proliferation in tumors with enhanced apoptosis has also been reported for human SCC (Einspahr et al., 1999), and this strong correlation between proliferation and apoptosis suggests that the elevated apoptotic rate is driving increased proliferation in these tumors. It is also possible that the cells capable of forming tumors or cysts have somehow overcome the PKC-δ-induced growth inhibition, perhaps by acquiring additional growth-promoting mutations. Despite the enhanced PCNA staining in the HaCaT Ras II-4 tumors with PKC-δ, the tumors that did form were much smaller and most were benign cysts, indicating that the enhanced proliferation could not completely compensate for the elevated apoptotic rate.
Re-expression or activation of residual proapoptotic PKC-δ in SCC tumors has a potential therapeutic application. Pharmacological reversal of PKC-δ downregulation in neoplastic keratinocytes may be possible with inhibitors of EGFR signaling or Src activity, and the re-expression of PKC-δ could cause an inhibition of cell growth and induction of apoptosis. A small increase in apoptotic rate can cause relatively large growth-inhibitory effects in regressing skin tumors (Burns et al., 1976), and our results have demonstrated that even a modest increase in apoptosis induced by PKC-δ re-expression can inhibit the growth of transformed human keratinocytes. In summary, our findings establish the proapoptotic function of PKC-δ as a tumor suppressor mechanism that may be functioning in the pathogenesis of human SCCs.
Abbreviations
- SCC:
-
squamous cell carcinoma
- PKC-δ:
-
protein kinase C-delta
- EGF:
-
epidermal growth factor
- EGFR:
-
epidermal growth factor receptor
- TGF-α:
-
transforming growth factor-alpha
References
Ashton KJ, Weinstein SR, Maguire DJ, Griffiths LR . (2003). Arch Dermatol 139: 876–882.
Bharti A, Kraeft SK, Gounder M, Pandey P, Jin S, Yuan ZM et al. (1998). Mol Cell Biol 18: 6719–6728.
Black JD . (2000). Front Biosci 5: D406–D423.
Boukamp P, Stanbridge EJ, Foo DY, Cerutti PA, Fusenig NE . (1990). Cancer Res 50: 2840–2847.
Brodie C, Blumberg PM . (2003). Apoptosis 8: 19–27.
Burns FJ, Vanderlaan M, Sivak A, Albert RE . (1976). Cancer Res 36: 1422–1427.
Cataldi A, Miscia S, Centurione L, Rapino M, Bosco D, Grifone G et al. (2002). J Cell Biochem 86: 553–560.
Chida K, Hara T, Hirai T, Konishi C, Nakamura K, Nakao K et al. (2003). Cancer Res 63: 2404–2408.
Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D et al. (2000). Oncogene 19: 2331–2337.
D’Costa AM, Denning MF . (2005). Cell Death Differ 12: 224–232.
Denning MF, Dlugosz AA, Howett MK, Yuspa SH . (1993). J Biol Chem 268: 26079–26081.
Denning MF, Dlugosz AA, Threadgill DW, Magnuson T, Yuspa SH . (1996). J Biol Chem 271: 5325–5331.
Denning MF, Wang Y, Nickoloff BJ, Wrone-Smith T . (1998). J Biol Chem 273: 29995–30002.
Denning MF, Wang Y, Tibudan S, Nickoloff BJ, Qin JZ . (2002). Cell Death Differ 9: 40–52.
Deucher A, Efimova T, Eckert RL . (2002). J Biol Chem 277: 17032–17040.
DeVries TA, Neville MC, Reyland ME . (2002). EMBO J 21: 6050–6060.
Dietrich C, Gumpert N, Heit I, Borchert-Stuhltrager M, Oesch F, Wieser R . (2001). Biochem Biophys Res Commun 282: 575–579.
Dlugosz AA, Cheng C, Williams EK, Darwiche N, Dempsey PJ, Mann B et al. (1995). Cancer Res 55: 1883–1893.
Dlugosz AA, Hansen L, Cheng C, Alexander N, Denning MF, Threadgill DW et al. (1997). Cancer Res 57: 3180–3188.
Dobler M, Schuh J, Kiesewetter F, Schell H, Liehr T, Gebhart E . (1999). Int J Oncol 14: 571–576.
Efimova T, Broome AM, Eckert RL . (2004). Mol Cell Biol 24: 8167–8183.
Einspahr JG, Alberts DS, Warneke JA, Bozzo P, Basye J, Grogan TM et al. (1999). Neoplasia 1: 468–475.
Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M et al. (1995). EMBO J 14: 6148–6156.
Ewen ME . (2000). Prog Cell Cycle Res 4: 1–17.
Frasch SC, Henson PM, Kailey JM, Richter DA, Janes MS, Fadok VA et al. (2000). J Biol Chem 275: 23065–23073.
Frey MR, Leontieva O, Watters DJ, Black JD . (2001). Biochem Pharmacol 61: 1093–1100.
Fujii T, Garcia-Bermejo ML, Bernabo JL, Caamano J, Ohba M, Kuroki T et al. (2000). J Biol Chem 275: 7574–7582.
Geiges D, Marks F, Gschwendt M . (1995). Exp Cell Res 219: 299–303.
Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y et al. (1996). J Exp Med 184: 2399–2404.
Glick AB, Sporn MB, Yuspa SH . (1991). Mol Carcinogen 4: 210–219.
Griffiths G, Garrone B, Deacon E, Owen P, Pongracz J, Mead G et al. (1996). Biochem Biophys Res Commun 222: 802–808.
Hansen LA, Woodson RL, Holbus S, Strain K, Lo YC, Yuspa SH . (2000). Cancer Res 60: 3328–3332.
Hornia A, Lu Z, Sukezane T, Zhong M, Joseph T, Frankel P et al. (1999). Mol Cell Biol 19: 7672–7680.
Jansen AP, Verwiebe EG, Dreckschmidt NE, Wheeler DL, Oberley TD, Verma AK . (2001). Cancer Res 61: 808–812.
Jiang W, Ananthaswamy HN, Muller HK, Kripke ML . (1999). Oncogene 18: 4247–4253.
Joseloff E, Cataisson C, Aamodt H, Ocheni H, Blumberg P, Kraker AJ et al. (2002). J Biol Chem 277: 12318–12323.
Koizumi H, Kohno Y, Osada S, Ohno S, Ohkawara A, Kuroki T . (1993). J Invest Dermatol 101: 858–863.
Lee E, Punnonen K, Cheng C, Glick A, Dlugosz A, Yuspa SH . (1992). Carcinogenesis 13: 2367–2373.
Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G et al. (2001). J Clin Invest 108: 1505–1512.
Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH . (1999). Mol Cell Biol 19: 8547–8558.
Liu J, Chen J, Dai Q, Lee RM . (2003). Cancer Res 63: 1153–1156.
Lu Z, Hornia A, Jiang YW, Zang Q, Ohno S, Foster DA . (1997). Mol Cell Biol 17: 3418–3428.
Osada S, Hashimoto Y, Nomura S, Kohno Y, Chida K, Tajima O et al. (1993). Cell Growth Differ 4: 167–175.
Papp H, Czifra G, Bodo E, Lazar J, Kovacs I, Aleksza M et al. (2004). Cell Mol Life Sci 61: 1095–1105.
Perletti GP, Marras E, Concari P, Piccinini F, Tashjian Jr AH . (1999). Oncogene 18: 1251–1256.
Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN . (1991). Mol Carcinogen 4: 196–202.
Popp S, Waltering S, Herbst C, Moll I, Boukamp P . (2002). Int J Cancer 99: 352–360.
Popp S, Waltering S, Holtgreve-Grez H, Jauch A, Proby C, Leigh IM et al. (2000). J Invest Dermatol 115: 1095–1103.
Reddig PJ, Dreckschimdt NE, Ahrens H, Simsiman R, Tseng CP, Zou J et al. (1999). Cancer Res 59: 5710–5718.
Reddig PJ, Dreckschmidt NE, Zou J, Bourguignon SE, Oberley TD, Verma AK . (2000). Cancer Res 60: 595–602.
Ren J, Datta R, Shioya H, Li Y, Oki E, Biedermann V et al. (2002). J Biol Chem 277: 33758–33765.
Reyland ME, Barzen KA, Anderson SM, Quissell DO, Matassa AA . (2000). Cell Death Differ 7: 1200–1209.
Roop DR, Lowy DR, Tambourin PE, Strickland J, Harper JR, Balaschak M et al. (1986). Nature 323: 822–824.
Sherr CJ . (2004). Cell 116: 235–246.
Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM et al. (2000). Cell 102: 211–220.
Sitailo LA, Tibudan SS, Denning MF . (2004). J Invest Dermatol 123: 434–443.
Watanabe T, Ono Y, Taniyama Y, Hazama K, Igarashi K, Ogita K et al. (1992). Proc Natl Acad Sci USA 89: 10159–10163.
Wheeler DL, Li Y, Verma AK . (2005). Photochem Photobiol 81: 9–18.
Wheeler DL, Martin KE, Ness KJ, Li Y, Dreckschmidt NE, Wartman M et al. (2004). Cancer Res 64: 7756–7765.
Wheeler DL, Ness KJ, Oberley TD, Verma AK . (2003). Cancer Res 63: 6547–6555.
Wheeler DL, Reddig PJ, Dreckschmidt NE, Leitges M, Verma AK . (2002). Oncogene 21: 3620–3630.
Yoshida K, Wang HG, Miki Y, Kufe D . (2003). EMBO J 22: 1431–1441.
Zang Q, Lu Z, Curto M, Barile N, Shalloway D, Foster DA . (1997). J Biol Chem 272: 13275–13280.
Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J et al. (1994). Nature 372: 773–776.
Acknowledgements
We thank all members of the Skin Cancer Research Program for their help with this project, especially Jeffrey Panella for constructing the Ras (Q61) retroviral vector. We also thank Dr Divaker Choubey (Loyola University Medical Center, Maywood, IL, USA) for furnishing the Ras (Q61) cDNA, and Dr Norbert Fusenig (German Cancer Research Center, Heidelberg, Germany) and Mihaela Skobe (Harvard School of Medicine, Boston, MA, USA) for providing HaCaT cells and HaCaT Ras clones. This work was supported in part by a grant from the Potts Foundation (MFD), and NIH grants CA83784 (MFD) and AR47814 (BJN).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
D'Costa, A., Robinson, J., Maududi, T. et al. The proapoptotic tumor suppressor protein kinase C-δ is lost in human squamous cell carcinomas. Oncogene 25, 378–386 (2006). https://doi.org/10.1038/sj.onc.1209065
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1209065
