
Abstract
Schizophrenia may be related to dysfunctional glutamatergic activity, specifically hypofunction of the N-methyl-D-aspartate receptor (NMDAR). In addition, it has been proposed that NMDAR hypofunction may paradoxically cause an increase in glutamate release and hypermetabolism in corticolimbic regions. If a state of partial, chronic NMDAR blockade underlies schizophrenia, then schizophrenic volunteers (SV) may have greater glutamate release and associated elevations in regional cerebral blood flow (rCBF) than normal volunteers (NV), following drug-induced NMDAR antagonism. Therefore, we have given acute ketamine, a noncompetitive NMDAR antagonist, to NV (n=13) and medicated volunteers with schizophrenia (n=10) in conjunction with serial positron emission tomography blood flow studies. Drug administration caused marked rCBF elevations in frontal and cingulate regions in both groups. Contrasts between NV and SV ketamine groups showed that SV had greater relative blood flow increases in the anterior cingulate than NV. Maximum blood flow, and the area under the curve for blood flow in the anterior cingulate cortex, significantly correlated with changes in psychosis ratings in SV and NV (maximum rCBF only). These changes are consistent with a relatively hypoactive thalamic NMDAR and increased cortical glutamate neurotransmission at non-NMDARs in schizophrenia. We hypothesize that ketamine antagonizes an NMDAR-dependent inhibitory system that is partially compromised in subjects with schizophrenia. The ketamine-induced reduction of inhibition leads to a marked increase in glutamate release and hypermetabolism (elevated rCBF) in frontal and cingulate cortical regions. The loss of inhibition and increased glutamate release may cause the distorted thoughts and diminished cognitive abilities elicited by NMDAR blockade.
Similar content being viewed by others

INTRODUCTION
Based on the schizophrenia-like actions of phencyclidine in humans (Pearlson, 1981), researchers have hypothesized that an antagonism in glutamate transmission at the N-methyl-D-aspartate receptor (NMDAR) may be causally associated with schizophrenia (Olney et al, 1999; Tamminga, 1998; Javitt and Zukin, 1991). An antagonism at this location could, in turn, cause regional hypermetabolism (Rothman et al, 2003) from increased glutamate release (Gluck et al, 2002; Theberge et al, 2002; Bartha et al, 1997; Moghaddam et al, 1997; Moghaddam and Adams, 1998). The mechanism for this phenomenon may be attributed to disinhibition of inhibitory interneurons in the anterior thalamus, basal forebrain, or frontal cortex (Farber, 2003; Carter et al, 2004; Tomitaka et al, 2000). Loss of GABAergic inhibition may lead to excessive glutamate release in frontal and cingulate cortical regions. The resulting hyperglutamatergic state could promote stimulation of non-NMDA glutamatergic receptors, for example, kainate and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid). If schizophrenia is due in part to reduced glutamate action on an inhibitory neuron's NMDAR and increased action at cortical non-NMDARs, then drugs that block glutamate binding at the NMDAR may cause greater physiological changes in patients with schizophrenia than healthy controls. Those changes may manifest in regional cerebral blood flow (rCBF) patterns following ketamine administration.
Ketamine (2-chlorphenyl-2-methylamino-cyclohexanone) is a phencyclidine congener that has psychotomimetic properties in adult humans at subanesthetic doses (Malhotra et al, 1996; Vollenweider et al, 1997a; Lahti et al, 2001a; Newcomer et al, 1999; Zukin and Zukin, 1979). Since, ketamine exacerbates psychotic symptoms in schizophrenia and induces both psychotomimetic symptoms and cognitive disturbances characteristic of schizophrenia, it has been used as a model of the illness in both clinical and preclinical paradigms (Lahti et al, 2001b; Duncan et al, 1999; Jentsch et al, 1997). Ketamine is a non-competitive antagonist of the NMDA glutamate receptor. It provides a means of assessing the physiological consequences of NMDAR ionophore blockade in normal volunteers (NV) and schizophrenic volunteers (SV) in a safe and reliable manner (Carpenter Jr, 1999). As a surrogate measure of glutamate release (Rothman et al, 2003), rCBF should reflect neurotransmitter turnover in the cortex following ketamine infusions.
We carried out an analysis of ketamine-induced brain blood flow changes in persons with schizophrenia and healthy controls to ascertain whether the anterior cingulate cortex is more sensitive to NMDAR blockade in schizophrenic volunteers than healthy controls. We hypothesized that ketamine would cause greater behavioral and physiological changes in schizophrenic volunteers. Behavioral measures suggest that the two groups have similar ketamine-induced psychosis changes (Lahti et al, 2001b). However, this study with positron emission tomography (PET) suggests that volunteers with schizophrenia have an abnormally large cingulate blood-flow response to NMDAR blockade.
METHODS
A total of 23 normal volunteers (NV) were recruited via advertisements in local newspapers. Potential subjects were screened by telephone, using a structured interview, in order to exclude those with a medical or psychiatric condition and those with a family history of major psychiatric illness in a first-degree relative. Prospective volunteers were then interviewed in person by a trained research technician to exclude primary psychiatric diagnoses using the Structured Clinical Interview Diagnosis (SCID) (First et al, 1995). NV then conferred with a psychiatrist who answered questions concerning the research protocol. Only those normal volunteers who received ketamine are considered in this report.
A total of 10 schizophrenia volunteers (SV) were recruited from the voluntary inpatients residing on the Residential Research Unit (RRU) of the Maryland Psychiatric Research Center (MPRC). The RRU is a hospital ward where persons with schizophrenia participate in research protocols that require hospitalization. Prospective subjects are informed about the research nature of the program and encouraged to visit and inspect the facilities prior to admission. Patient volunteers have active but stable symptoms and require hospitalization. The diagnosis of schizophrenia was made by two experienced research clinicians using DSM-III-R criteria based on detailed history and mental state examination and all available historical information, gleaned from extensive contact with previous treatment facilities and family members. We evaluated schizophrenic volunteers with the SCID (First et al, 1995), the Schedule for the Deficit Syndrome (Kirkpatrick et al, 1989), the MPRC Involuntary Movement Scale, the Cannon-Spoor Premorbid Adjustment Scale (Cannon-Spoor et al, 1982), and the Prognostic Scale (Strauss and Carpenter Jr, 1974). Data from some of these patients were previously published (Lahti et al, 1995a).
All participants received a physical examination, EKG and laboratory analysis of plasma and urine samples, including HIV testing and an illicit drug screen. All subjects denied previous ketamine use when they were specifically asked. Subjects were not eligible for inclusion in this study if they suffered from any significant medical condition (including high blood pressure) or had recently used drugs or excessive alcohol. Additional exclusion criteria for the NV included any history of axis I or II psychiatric disorder or any first degree relative with a diagnosis of schizophrenia. NV were paid for their participation in this study. The demographic details of the volunteers are given in Table 1. Familial education levels were significantly different between groups (the t-statistic for the difference in means is 2.57, with df=21, p<0.02). All SV were taking clinically effective doses of haloperidol, 10–20 mg per day, during this study. No atypical neuroleptics were administered.
The NV described in this report are the same volunteer subjects described in a previous publication (Holcomb et al, 2001).
Consent Process
The physical and psychological effects of ketamine were fully explained to each volunteer during the consent process and were described in detail on the written consent form. Every volunteer was fully informed about the nature of the study and the potential risks associated with it. The consent process for the SV involved additional safeguards. A clinician selected those patients deemed to be competent and capable of appreciating the risks of the study. An independent designated State of Maryland Mental Health Administration employee confirmed that the volunteer was able to understand the nature of the research. When available, family members or caregivers were involved in the consent process. Patients were given detailed verbal descriptions of the procedures and risks that accompanied the study. They were informed that adverse effects could include temporary worsening of psychosis. At the end of the informed consent process, patients were required to demonstrate their understanding of the protocol by listing its key features and risks. The quality of the consent procedures was monitored by a designated RRU ombudsperson who worked closely with the patients throughout the duration of the research project.
The protocol for this study was reviewed, approved, and monitored by both the University of Maryland Human Volunteers Committee and the Johns Hopkins Joint Committee on Clinical Investigation.
Ketamine Administration
Prior to the image acquisition reported here, each volunteer was given ketamine (0.3 mg/kg). and assessed under research conditions without imaging. All subjects were familiar with the psychological effects of ketamine before the PET phase of the study. During PET scanning 13 NV and 10 volunteers with schizophrenia were administered ketamine (0.3 mg/kg).
Mental State Evaluation
The Brief Psychiatric Rating Scale (BPRS) (Overall and Gorham, 1962) was administered to subjects when they received ketamine both in the laboratory and in the scanner. Ratings were made before and repeatedly after drug administration. In the laboratory, the first post-drug rating was at 20 min. During the imaging procedure, the first post-drug BPRS was done 7-9 min after drug administration.
Scan Technique
Subjects were scanned using a GE 4096+ PET camera in conjunction with the bolus water method, 62.5 mCi of 15-oxygen water per tracer injection. Volunteers were positioned in the scanner using a thermoplastic facemask, with the most inferior plane placed 20 mm above the canthomeatal axis. Once the subjects were comfortable, three baseline scans (BL) were obtained at 10-min intervals. Subjects rested in the scanner with their eyes open. Scan data were acquired for 60 s, starting 20 s after tracer infusion. Seven minutes prior to the fourth scan, ketamine (0.3 mg/kg) was infused into an antecubital vein over 60 s. The first postinjection scan was acquired 6 min after the completion of the ketamine (or saline) infusion. Additional scans were obtained at 10-min intervals: 16, 26, and 36 min postinjection.
Image and Statistical Analysis
The methods used to analyze the scan data were described in detail in a previous publication (Holcomb et al, 2001). Blood flow data were normalized with respect to anatomical and mean flow rates for the whole brain (50 ml/100 gm/min), according to the algorithms applied by SPM99 software (Friston et al, 1994, 1996). In summary, three steps were used to analyze the differences between the ketamine-induced blood flow activation patterns in the two groups (SV-ketamine, NV-ketamine). (1) Ketamine-activated Volumes of Interest (VOIs) were identified with a voxel-based subtraction analysis (further defined below). (2) VOIs were compared across groups and over time with an analysis of variance (ANOVA). (3) Cmax (maximum rCBF response), Tmax (time to reach the maximum rCBF response), and AUC (area under the curve) values were calculated for each VOI.
Ketamine-activated VOIs were identified using SPM 99 software (Friston et al, 1994, 1996) to contrast the averaged BL scans with each of four post-ketamine scans (6, 16, 26, and 36 min) in the NV-ketamine group. Positive and negative contrasts were generated using a significance level of p<0.05, corrected for multiple comparisons. VOIs were first identified in the 6-min vs baseline contrast. Maxima obtained at subsequent time points (16, 26, and 36 min) were considered to be distinct if they were located more than one full width half maximum (FWHM) away from a previously identified VOI. This was done sequentially with respect to time: 16 min before 26 min, and 26 min before 36 min. Other candidate VOIs were selected from the double subtractions generated from each of the four time points. The double subtractions were: (Ketamine−Baseline, SV−NV) and (Ketamine−Baseline, NV−SV). Regions that were significant at p<0.001, uncorrected, were subject to further analysis. The average signal from each of these points, in both groups, was grown to a 3 × 3 × 3 mm VOI. This procedure generated individual values for each subject, for each VOI, at baseline and at the four post-drug time points.
The VOI created from the subtraction analyses described above were then compared across the groups and over time with a 3 (SV-ketamine, NV-ketamine, NV-saline) by 5 (BL, 6, 16, 26, and 36 min scans) repeated measures ANOVA with Greenehouse-Geisser correction and a quadratic contrast. A separate ANOVA was conducted for each VOI. These analyses were used to determine if the ketamine-activated VOIs have a significantly different pattern of change over time between the two groups.
The WinNonlin program (Pharsight Corp., Mountain View, CA) was used to identify the Cmax, Tmax, and AUC for each subject for each VOI. Independent t-tests were used to assess differences between the NV (ketamine) and the SV (ketamine) groups on the Tmax, Cmax, and AUC values for each VOI. The Cmax and the AUC for each VOI were correlated with the change in the BPRS score of each subject after ketamine (20 min), obtained during the behavioral studies prior to the scanning.
RESULTS
Ketamine-Induced rCBF Effects
Ketamine increased rCBF within medial frontal structures, principally in the anterior cingulate and medial frontal cortex, but also in the right inferior frontal region (Table 2). Figure 1 depicts areas of increased CBF, by region, across time at the four post-ketamine time points in the normal and the schizophrenia volunteers. Relative to the whole brain, CBF diminished as previously described (Holcomb et al, 2001), in SV and NV groups. No significant difference in group cerebellar rCBF was observed. Maximal rCBF changes following ketamine occurred at 6 min and were largely resolved by 36 min.

Ketamine elicited increases in ventral anterior cingulate CBF, in schizophrenic and healthy volunteers.
Ketamine-Induced rCBF Changes: NV Vs SV
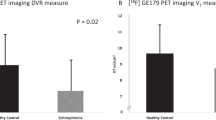
As predicted, there was a significant difference between the SV-Ketamine (SV-K) and NV-Ketamine (NV-K) in the anterior cingulate (8, 38, 14) VOI (F(1,21)=8.12, p=0.01). The difference between SV-K and NV-K in the inferior frontal cortex (44, 34, −16) VOI approached statistical significance (F(1,21)=3.85, p=0.063) (Table 2 and Figure 2).

Relative increases in rCBF with respect to time, following bolus ketamine administration.
rCBF Correlations with Behavioral Response
Behavioral data were collected in the laboratory prior to scanning. Each subject received ketamine and placebo in resting conditions. Ratings obtained during PET scans were not used because of the extensive intrusions associated with the imaging procedure between radioisotope injections. The Cmax and the AUC were correlated with the change in BPRS score in response to ketamine (Table 3). In the volunteers with schizophrenia, the Cmax and total BPRS score correlated with rCBF in the two maxima located in the anterior cingulate cortex (8, 38, 14 and −6, 40, −4). AUC only correlated with total BPRS at one of these points (−6, 40, −4). Cmax correlated with the psychosis subscale in the medial frontal cortex (10, 54, 18). In the NV ketamine group, the correlations between rCBF (Cmax at coordinates 10, 52, 16) and the Psychosis subscale of the BPRS were positive and significant (r=0.57, df=12, p=0.041) (Holcomb et al, 2001). There were no group differences in overall ketamine-induced psychosis changes. Furthermore, there were no correlations between medication status or demographic variables and ketamine-induced psychosis change. Although familial education levels differed significantly between groups, there was no association between this demographic measure and drug-related rCBF changes.
DISCUSSION
Previously we applied a kinetic analysis of ketamine action to sequential rCBF scans in healthy humans with the purpose of characterizing the drug's regional dynamic action (Holcomb et al, 2001). That analytic strategy provided a quantitative regional and time demonstration of rCBF actions of ketamine in healthy volunteers. Here we have used the same kinetic analysis to compare the rCBF response to ketamine between normal and schizophrenic volunteers. As predicted, the anterior cingulate showed a greater response to ketamine in schizophrenic than NV. These data suggest that the glutamatergic system of SV may be more sensitive to NMDAR antagonism than NV.
Results revealed a limited number of relative regional blood flow activations/reductions after a single bolus dose of ketamine. Relative to the whole brain, blood flow changes in NV were limited to increases in the ACC, medial frontal cortex (MFC), and right inferior frontal cortex (IFC), and reductions in the cerebellum. In response to bolus ketamine, the schizophrenia group showed a similar response but with a greater magnitude and duration of elevated ACC and IFC blood flow than the healthy group. Both groups, however, exhibited significant correlations between regional blood flow changes and ketamine-elicited subjective experience.
This PET study draws attention to three observations: (1) ketamine induces hypermetabolism in anterior cingulate and prefrontal cortical regions; (2) the drug-induced behavioral response is correlated with the metabolic (rCBF) changes in medial frontal and anterior cingulate cortex; and (3) SV exhibit greater relative blood flow increases in cingulate cortex than healthy subjects. Two previous studies have reported on the first two findings in normal volunteers (Breier et al, 1997; Vollenweider et al, 1997b). This discussion interprets previous findings and those associated with the group comparison in light of recent research on glutamate pharmacology and the NMDAR.
The physiological basis for ketamine-induced frontal/cingulate hypermetabolism may be the cause of the drug's psychotomimetic behavioral effects. Ketamine-induced changes in mental status (Lahti et al, 1995b; Krystal et al, 1994) and brain metabolism (Vollenweider et al, 1997b; Holcomb et al, 2001; Breier et al, 1997; Lahti et al, 1995a) have been hypothesized to reflect antagonism of NMDARs located on inhibitory interneurons (probably GABAergic) in the anterior thalamus. That antagonism, in turn, may cause increased glutamate-dependent excitation in downstream cortical regions like the ACC (Farber, 2003; Newcomer et al, 1999; Krystal et al, 1994, 2003; Konradi and Heckers, 2003). In support of this idea, preclinical studies have revealed that NMDAR antagonism results in (1) increased frontal extracellular glutamate (Moghaddam et al, 1997; Lorrain et al, 2003; Takahata and Moghaddam, 2003) and acetylcholine levels (Giovannini et al, 1994), and (2) increased spontaneous firing rate of neurons in the frontal cortex (Moghaddam and Jackson, 2003; Homayoun et al, 2004). The resulting excessive and disorganized activity of the frontal and cingulate regions could, in turn, diminish the brain's capacity to ‘make sense’ of the environment. This pharmacologically driven hyperactivity in the fronto-cingulate system is best accounted for by the large changes in glutamate release that accompany NMDAR antagonism.
Supporting evidence for frontal and cingulate glutamate release following NMDAR blockade in humans is found in neurochemical (magnetic resonance spectroscopy; MRS) and metabolic studies that reveal a strong stoichiometric relationship between the dynamics of the glutamate/glutamine cycle and brain metabolism and blood flow (Patel et al, 2003; Shulman et al, 2002; Rothman et al, 2003; Behar and Rothman, 2001; Shen et al, 1999). A recent MRS study showed increased ACC glutamatergic turnover with ketamine administration in healthy human subjects (Rowland et al, 2005). It is important to note that studies using interventions to reduce glutamate efflux (Anand et al, 2000) and mitigate glutamate action at non-NMDARs (Moghaddam and Adams, 1998; Carter et al, 2004) have shown promise in ameliorating some of the symptoms elicited by NMDAR antagonism.
The SV group demonstrated a significantly greater anterior cingulate blood flow response to bolus ketamine than the NV group. This observation is consistent with the hypothesis that schizophrenia is partly caused by hypofunctional NMDARs. If the anterior thalamus of SV contain relatively hypoactive NMDARs on inhibitory interneurons, then ketamine may generate a more sustained disinhibition of those GABAergic components (Farber et al, 2002; Sharp et al, 2001). Such a scenario might, in turn, promote a prolonged and vigorous medial frontal and anterior cingulate response to anterior thalamic glutamatergic input. Since GABAergic inhibitory interneurons may be more sensitive to NMDAR antagonism than pyramidal cells (Grunze et al, 1996), the loss of local circuit inhibition may have important consequences to frontal metabolic activity and information processing.
Several study weaknesses are acknowledged. The ketamine-induced response is measured against a baseline and not against a placebo infusion. Optimally each subject should be assessed against himself in a drug/placebo-paired design. This was not feasible in light of our goal to use repeat injections of a radiotracer, over an extended time period within the same day, in each volunteer. This omission prevented a direct assessment of placebo vs drug-induced blood flow changes in the each subject, in both groups. Previously published data from this laboratory indicate that haloperidol treatment diminishes anterior cingulate metabolism when compared to the untreated condition (Holcomb et al, 1996). Consequently, it is extremely unlikely that these neuroleptic-treated SZ volunteers would have a significantly greater resting anterior cingulate blood flow than NV. Preclinical animal studies give no indication that chronic (Duncan et al, 2003) haloperidol treatment is likely to make the anterior cingulate more responsive to ketamine. It is, instead, more likely to diminish the brain's hypermetabolic response.
Haloperidol's impact on the SV group's physiological response to ketamine is not clearly known at this time. Animal studies have provided some insights on this matter. Duncan et al (1998) has shown that haloperidol does not diminish ketamine-induced medial frontal hypermetabolism. Consistent with that report, Adams and Moghaddam (2001) have demonstrated that haloperidol does not ameliorate the hyperglutamatergic effects of phencyclidine. In contrast with haloperidol, clozapine has been shown to diminish the metabolic effects of ketamine (Duncan et al, 1998) and to partially reverse the cognitive impairments (Jentsch et al, 1997) and cortical hyper-responsivity to PCP (Arvanov and Wang, 1999) caused by prolonged phencyclidine administration. In volunteers with schizophrenia, a similar pattern of evidence has emerged. Haloperidol did not diminish the patient volunteers' subjective responses to ketamine (Lahti et al, 1995b), whereas clozapine significantly diminished the patients' psychotomimetic responses to ketamine (Malhotra et al, 1997).
Other studies of ketamine's behavioral and physiological effects have used prolonged infusions. In this study, ketamine bolus administration was used to assess the time course of drug action. It is not clear to what extent the bolus infusion causes a qualitatively different behavioral and neurophysiological responses from the continuous infusion (Newcomer et al, 1999; Krystal et al, 1994). The transient nature of the bolus infusion's effects will, however, reduce the likelihood of seeing sustained drug action in the two groups. It also compromises our ability to make strong inferences from other studies that have used continuous infusions. Nonetheless, the cingulate cortex changes described here are consistent with the NMDAR antagonist models developed by other groups (Moghaddam, 2003).
Future studies combining magnetic resonance spectroscopy, metabolic imaging (through PET), and functional MRI will be able to assess the relationships between the effects of ketamine on glutamatergic activity, brain blood flow, and behavioral changes. As therapeutic agents become available to test the role of NMDAR hypofunction in schizophrenia, it may become possible to use those tools to explicate the precise mechanisms of this illness.
References
Adams BW, Moghaddam B (2001). Effect of clozapine, haloperidol, or M100907 on phencyclidine-activated glutamate efflux in the prefrontal cortex. Biol Psychiatry 50: 750–757.
Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A et al (2000). Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gene Psychiatry 57: 270–276.
Arvanov VL, Wang RY (1999). Clozapine, but not haloperidol, prevents the functional hyperactivity of N-methyl-D-aspartate receptors in rat cortical neurons induced by subchronic administration of phencyclidine. J Pharmacol Exp Ther 289: 1000–1006.
Bartha R, Williamson PC, Drost DJ, Malla A, Carr TJ, Cortese L et al (1997). Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gene Psychiatry 54: 959–965.
Behar KL, Rothman DL (2001). In vivo nuclear magnetic resonance studies of glutamate-gamma-aminobutyric acid-glutamine cycling in rodent and human cortex: the central role of glutamine. J Nutr 131: 2498S–2504S.
Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D (1997). Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry 154: 805–811.
Cannon-Spoor HE, Potkin SG, Wyatt RJ (1982). Measurement of premorbid adjustment in chronic schizophrenia. Schizophr Bull 8: 470–484.
Carpenter Jr WT (1999). The schizophrenia ketamine challenge study debate. Biol Psychiatry 46: 1081–1091.
Carter K, Dickerson J, Schoepp DD, Reilly M, Herring N, Williams J et al (2004). The mGlu2/3 receptor agonist LY379268 injected into cortex or thalamus decreases neuronal injury in retrosplenial cortex produced by NMDA receptor antagonist MK-801: possible implications for psychosis. Neuropharmacology 47: 1135–1145.
Duncan GE, Leipzig JN, Mailman RB, Lieberman JA (1998). Differential effects of clozapine and haloperidol on ketamine-induced brain metabolic activation. Brain Res 812: 65–75.
Duncan GE, Miyamoto S, Leipzig JN, Lieberman JA (1999). Comparison of brain metabolic activity patterns induced by ketamine, MK- 801 and amphetamine in rats: support for NMDA receptor involvement in responses to subanesthetic dose of ketamine. Brain Res 843: 171–183.
Duncan GE, Miyamoto S, Lieberman JA (2003). Chronic administration of haloperidol and olanzapine attenuates ketamine-induced brain metabolic activation. J Pharmacol Exp Ther 305: 999–1005.
Farber NB (2003). The NMDA receptor hypofunction model of psychosis. In: Moghaddam B, Wolf ME (eds). Glutamate and Disorders of Cognition and Motivation. New York Academy of Sciences: New York. pp 119–130.
Farber NB, Kim SH, Dikranian K, Jiang XP, Heinkel C (2002). Receptor mechanisms and circuitry underlying NMDA antagonist neurotoxicity. Mol Psychiatry 7: 32–43.
First MB, Spitzer RL, Williams JBW, Gibbon M (1995). Structured Clinical Interview for DSMIV (SCID). American Psychiatric Association: Washington, D.C.
Friston KJ, Holmes A, Poline JB, Price CJ, Frith CD (1996). Detecting activations in PET and fMRI: levels of inference and power. Neuroimage 4: 223–235.
Friston KJ, Worsley KJ, Frackowiak RSJ, Mazziotta JC, Evans AC (1994). Assessing the significance of focal activations using their spatial extent. Hum Brain Mapp 1: 210–220.
Giovannini MG, Mutolo D, Bianchi L, Michelassi A, Pepeu G (1994). NMDA receptor antagonists decrease GABA outflow from the septum and increase acetylcholine outflow from the hippocampus: a microdialysis study. J Neurosci 14: 1358–1365.
Gluck MR, Thomas RG, Davis KL, Haroutunian V (2002). Implications for altered glutamate and GABA metabolism in the dorsolateral prefrontal cortex of aged schizophrenic patients. Am J Psychiatry 159: 1165–1173.
Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW et al (1996). NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 16: 2034–2043.
Holcomb HH, Cascella NG, Thaker GK, Medoff DR, Dannals RF, Tamminga CA (1996). Functional sites of neuroleptic drug action in the human brain: PET/FDG studies with and without haloperidol. Am J Psychiatry 153: 41–49.
Holcomb HH, Lahti AC, Medoff DR, Weiler M, Tamminga CA (2001). Sequential regional cerebral blood flow brain scans using PET with H2(15)O demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology 25: 165–172.
Homayoun H, Jackson ME, Moghaddam B (2005). Activation of metabotropic glutamate 2/3 (mGlu2/3) receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J Neurophysiol 93: 1989–2001.
Javitt DC, Zukin SR (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308.
Jentsch JD, Redmond Jr DE, Elsworth JD, Taylor JR, Youngren KD, Roth RH (1997). Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 277: 953–955.
Kirkpatrick B, Buchanan RW, McKenney PD, Alphs LD, Carpenter Jr WT (1989). The schedule for the deficit syndrome: an instrument for research in schizophrenia. Psychiatry Res 30: 119–123.
Konradi C, Heckers S (2003). Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther 97: 153–179.
Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R (2003). NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 169: 215–233.
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD et al (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gene Psychiatry 51: 199–214.
Lahti AC, Holcomb HH, Medoff DR, Tamminga CA (1995a). Ketamine activates psychosis and alters limbic blood flow in schizophrenia. NeuroReport 6: 869–872.
Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995b). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13: 9–19.
Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA (2001a). Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25: 455–467.
Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA (2001b). Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25: 455–467.
Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA (2003). Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117: 697–706.
Malhotra AK, Adler CM, Kennison SD, Elman I, Pickar D, Breier A (1997). Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: a study with ketamine. Biol Psychiatry 42: 664–668.
Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D et al (1996). NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology 14: 301–307.
Moghaddam B (2003). Bringing order to the glutamate chaos in schizophrenia. Neuron 40: 881–884.
Moghaddam B, Adams B, Verma A, Daly D (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17: 2921–2927.
Moghaddam B, Adams BW (1998). Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281: 1349–1352.
Moghaddam B, Jackson ME (2003). Glutamatergic animal models of schizophrenia. In: Moghaddam B, Wolf ME (eds). Glutamate and Disorders of Cognition and Motivation. The New York Academy of Sciences: New York. pp 131–137.
Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T et al (1999). Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 20: 106–118.
Olney JW, Newcomer JW, Farber NB (1999). NMDA receptor hy-pofunction model of schizophrenia. J Psychiatr Res 33: 523–533.
Overall JF, Gorham DR (1962). The brief psychiatric rating scale. Psychol Rep 10: 799–812.
Patel AB, De Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL (2003). Coupling of glutamatergic neurotransmission and neuronal glucose oxidation over the entire range of cerebral cortex activity. Ann NY Acad Sci 1003: 452–453.
Pearlson GD (1981). Psychiatric and medical syndromes associated with phencyclidine (PCP) abuse. John Hopkins Med J 148: 25–33.
Rothman DL, Behar KL, Hyder F, Shulman RG (2003). In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu Rev Physiol 65: 401–427.
Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E et al (2005). Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4.0T Proton MRS study. Am J Psychiatry 162: 394–396.
Sharp FR, Tomitaka M, Bernaudin M, Tomitaka S (2001). Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci 24: 330–334.
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF et al (1999). Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci USA 96: 8235–8240.
Shulman RG, Hyder F, Rothman DL (2002). Biophysical basis of brain activity: implications for neuroimaging. Q Rev Biophys 35: 287–325.
Strauss JS, Carpenter Jr WT (1974). The prediction of outcome in schizophrenia. II. Relationships between predictor and outcome variables: a report from the WHO international pilot study of schizophrenia. Arch Gene Psychiatry 31: 37–42.
Takahata R, Moghaddam B (2003). Activation of glutamate neurotransmission in the prefrontal cortex sustains the motoric and dopaminergic effects of phencyclidine. Neuropsychopharmacology 28: 1117–1124.
Tamminga CA (1998). Schizophrenia and glutamatergic transmission. Crit Rev Neurobiol 12: 21–36.
Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J et al (2002). Glutamate and glutamine measured with 4.0T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry 159: 1944–1946.
Tomitaka S, Tomitaka M, Tolliver BK, Sharp FR (2000). Bilateral blockade of NMDA receptors in anterior thalamus by dizocilpine (MK-801) injures pyramidal neurons in rat retrosplenial cortex. Eur J Neurosci 12: 1420–1430.
Vollenweider FX, Leenders KL, Oye I, Hell D, Angst J (1997a). Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET). Eur Neuropsychopharmacol 7: 25–38.
Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J et al (1997b). Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol 7: 9–24.
Zukin SR, Zukin RS (1979). Specific [3H]phencyclidine binding in rat central nervous system. Proc Natl Acad Sci USA 76: 5372–5376.
Acknowledgements
We gratefully acknowledge the expert assistance of Dr Martin Weiler and T Michaelidis for help with ketamine infusions and patient assessments. Dr Robert Dannals, Robert Smoot, David Clough, and Karen Edmonds, of the Johns Hopkins Cyclotron Facility, made the PET work possible. The William K Warren Biomedical Research Foundation and NIH Grant RO1 DA09483-01 supported this study. Drs LM Rowland, R Schwarcz, WT Carpenter, and R McMahon provided exceptionally useful comments and guidance in the manuscript's writing and editing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Holcomb, H., Lahti, A., Medoff, D. et al. Effects of Noncompetitive NMDA Receptor Blockade on Anterior Cingulate Cerebral Blood Flow in Volunteers with Schizophrenia. Neuropsychopharmacol 30, 2275–2282 (2005). https://doi.org/10.1038/sj.npp.1300824
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300824
Keywords
This article is cited by
-
Region- and time- specific effects of ketamine on cerebral blood flow: a randomized controlled trial
Neuropsychopharmacology (2023)
-
The NMDA receptor antagonist ketamine impairs and delays context-dependent decision making in the parietal cortex
Communications Biology (2022)
-
Acute effects of ketamine on the pregenual anterior cingulate: linking spontaneous activation, functional connectivity, and glutamate metabolism
European Archives of Psychiatry and Clinical Neuroscience (2022)
-
Altered functional connectivity of the cingulate subregions in schizophrenia
Translational Psychiatry (2015)
-
Phenomenologically distinct psychotomimetic effects of ketamine are associated with cerebral blood flow changes in functionally relevant cerebral foci: a continuous arterial spin labelling study
Psychopharmacology (2015)
