
Abstract
Backcross breeding with marker-assisted selection was used to construct an intervarietal set of part chromosome substitution lines in Brassica napus, formed from a cross between two winter varieties of oilseed rape: Tapidor and Victor. A total of 22 lines from this substitution library were examined over a 3-year period, in a total of nine field trials, for seed oil fatty acid composition and seed oil content. Trialing of the substitution lines gave evidence for the existence of 13 quantitative trait loci (QTL). All 13 QTL affected fatty acid composition of the seed, and were distributed among linkage groups 1, 3, 6, 7, 8, 11, 13, 14, 18, and 19. Seven of these QTL, on linkage groups 3, 6, 8, 13, 14, 18, and 19, also affected total seed oil content. The positions of these QTL are compared to those in the published literature and with respect to erucic acid QTL previously identified in a backcross population of the same cross. The substitution line approach gives increased precision and sensitivity for QTL mapping compared to other methods.
Similar content being viewed by others


Introduction
Brassica napus is an important crop in parts of Europe, North America, Australia and Asia, because of the oil found in its seed. This oil, which can account for up to 46% of the total dry weight of the seed, is used for both nutritional and industrial purposes, and the value of the crop is determined by the levels of the constituent fatty acids found in the seed oil. Compared to other vegetable oils, it is unusual in containing long-chain monomeric fatty acids, of which the principal five are: oleic (C18:1), linoleic (C18:2), linolenic (C18:3), eicosenoic (C20:1), and erucic acid (C22:1).
Most rapeseed oil is used for human consumption in cooking oils and margarine but if the oil is high in erucic acid it may prove a health risk. However, oils with high levels of erucic acid have commercial benefits for use as high-temperature lubricants and, after minor modifications, as water repellents, plasticizers, waxes, and surface-active agents (Luhs and Friedt, 1993; Ecke et al, 1995). Most modern cultivars of oilseed rape have reduced levels of erucic acid and relatively high linolenic acid content. Oil rich in linolenic acid is unsuitable for human consumption because the unstable, oxidative nature of the fatty acid affects the oil's flavour and quality, and chemical hydrogenation is required to further modify the fatty acid for cooking purposes. On the other hand, low levels of linolenic acid are required as a precursor to producing chemicals essential for normal immunological and vascular system control (Ecke et al, 1995; Jourdren et al, 1996; Thormann et al, 1996). Hence, the value of the crop as a major oil producer has been dependent upon the successful breeding of varieties low in both erucic and linolenic acid, and so the identification of quantitative trait loci (QTL) controlling these traits would be a valuable asset to breeding via marker-assisted selection (MAS).
A number of QTL mapping methods are currently available, relying on statistical techniques to find relationships between molecular markers and QTL in segregating populations (Soller et al, 1976; Lander and Botstein, 1989; Luo and Kearsey, 1989; Haley and Knott, 1992; Jansen and Stam, 1994; Kearsey and Hyne, 1994). However, all of these methods can suffer from lack of precision of QTL mapping owing to a number of factors. These factors include: the heritability of the trait; the total number of QTL governing the trait, the distribution of these QTL over the genome, their interactions with each other, the random variation because of the environment and other unlinked genetic factors, the type and size of the population studied, the genome size, and the number and distribution of marker loci (van Ooijen, 1992; Darvasi et al, 1993).
Computer simulations, used to test the precision and reliability of QTL mapping, have shown that segregating populations often underestimate the number of QTL (Hyne et al, 1995), result in large confidence intervals (CIs) associated with QTL position (van Ooijen, 1992; Darvasi et al, 1993; Hyne et al, 1995; Burns, 1997), and overestimate the size of the QTL effects, particularly at low population sizes and heritability (Burns, 1997).
The reliable scoring of agronomic traits generally requires plot trials involving genetically uniform individuals within each plot. QTL analysis of such traits benefits from the use of ‘immortal’ doubled-haploid or recombinant inbred lines because such populations allow the replication of an individual line almost indefinitely, resulting in greater precision of QTL mapping. However, these specialised sets of lines require a substantial investment in time in their construction, while studies have shown that large population sizes are needed to detect QTL which exhibit only small effects (Soller et al, 1976), the scale of which are typically outside the size of the normal experimental field design when plot trials are used.
An alternative approach is to use MAS to produce substitution libraries (Tanksley, 1993; Ramsay et al, 1996; Rae et al, 1999). In its ideal state, a substitution line will carry a single segment of a donor genotype, in an otherwise pure genetic background of the recurrent genotype. A substitution library consists of a set of substitution lines representing the donor genome, where each substitution line carries a different introgressed region of the donor genome. In an experimental field trial, the performance of a substitution line can be compared to the recurrent parent and any significant differences found between the two lines can be attributed to QTL within the defined introgressed region. Such homozygous substitution lines can be multiplied indefinitely to improve precision in QTL mapping experiments, eliminate the genetic ‘noise’ of segregating, unlinked loci, and require smaller population sizes, with practical implications for traits scored on a field scale.
In this paper, QTL analysis of seed oil content and fatty acid composition in a B. napus substitution library is presented. The library was formed from a cross of an old oilseed rape winter variety ‘Victor’, with a modern homozygous oilseed rape winter variety ‘Tapidor’ (Howell et al, 1996). The original intention was to produce a complete set of 38 half-chromosome substitution lines, but not all were achieved in the time available. These lines were trialed at sites across Europe and scored for oil yield and content. The positions and effects of the QTL identified in this study are compared to those already published and to those identified at the first backcross stage of the same cross.
Materials and methods
Production of substitution library
The substitution library was produced by backcross breeding with MAS, as described in Howell et al (1996). The donor parent was an old, winter-sown variety, ‘Victor’. The recurrent parent, TapDH1, was a doubled-haploid line produced from a modern homozygous winter variety, ‘Tapidor’, via microspore culture. ‘Victor’ and ‘Tapidor’ have high and low levels of seed erucic acid, respectively. A single Victor (DH) plant was used to pollinate a single Tapidor (DH) plant, to produce the F1. The first backcross (B1) population was produced by crossing a Tapidor (DH) plant as seed parent to a single F1 individual, and so inherited the Tapidor cytoplasm. The B1 population consisted of 200 individuals genotyped with 158 restriction fragment length polymorphism (RFLP) markers distributed over the 19 linkage groups. MAS was used to choose six B1 individuals for their potential to produce the substitution library. To reduce the number of independently segregating Victor donor segments, the selected lines were backcrossed again to the B3 and B4 stages. Finally, B3S1 and B4S1 individuals (from selfing individual B3 and B4 plants) were genotyped to identify plants with the homozygous substituted segments. Multiplication of each line was carried out at independent field sites in order to produce enough seed for the field trials.
Field trials
Replicated field trials of the substitution lines were conducted over a period of 3 years at sites in England and France, giving a total of nine trials. Three trials were conducted in each of the seasons 1996/97, 1997/98, and 1998/99. All substitution lines were trialed at least three times during this 3-year period, and were included in the field trials as they became available from the breeding programme. All trials involved a fully randomised block design and followed a common field trial structure. Trials 1 and 2 were conducted at a field site in Banbury in Oxfordshire, UK, trial 3 was conducted at Framlingham in Suffolk, UK, trials 4–6 inclusive were conducted at Chartainvilliers, near to Chartres in France, and trials 7–9 inclusive were conducted at Granchester in Cambridgeshire, UK. Details of trial structures are shown in Table 1. All trials in a given year involved the same lines. Fertiliser was applied to the seed beds where necessary to correct for any nutrient disorders, in accordance with standard agricultural practice.
Herbicides were applied post-emergence and insecticides were applied at the start of flowering. No other pesticide treatments were used. Fungicides were not used owing to possible inhibitory effects on growth. Harvesting was conducted following plot swathing using a Haldrup plot swather. When plants were dry, seed was harvested using a Hege plot combine 140. Post-harvest, the seed samples were cleaned and oven-dried, before undergoing quality analysis by NIR/NMR, as per standard agricultural practice. Total seed oil content was measured for nine trials but fatty acid composition was measured in trials 4, 5 and 6 only.
Measurement of total seed oil content
Nuclear magnetic resonance (NMR) was used to measure total seed oil content in trials 4, 5, and 6, while near-infra-red spectroscopy analysis (NIR) was used in all other trials.
NMR is a standard nondestructive test and used an Oxford 400 NMR machine (Oxford Instruments Industrial Analysis Group, Abingdon, Oxon.). Ten grams of oven-dried seed were weighed in 40 ml sample tubes and calibrated using a sample of pure rapeseed oil. The readings are expressed as the percentage oil content of the whole seed at 9% moisture.
For NIR analysis a NIR Systems 5000 scanning instrument, running ISI software was used, and calibrated to measure 25 gm representative samples from each harvested plot.
Both NMR and NIR are calibrated to measure oil content by reference to wet chemistry methods, and after calibration give directly comparable results.
Measurement of fatty acid composition
Individual fatty acids were only recorded in trials 4, 5 and 6. Lipids were extracted by grinding 5 g of seed in 5 ml of n-heptane, and methylated with 0.5 ml 2 M KOH in methanol (Carlo Erba Mega 2, Chrompack CPWAX 58CB 25 m×0.25 mm, Inj Split 1:100 at 250°C, run isothermal at 230°C, FID at 300°C; Carrier H2). Individual fatty acid results are expressed as a percentage of the total identified fatty acids, as data for absolute yields were not available.
Substitution library data analysis
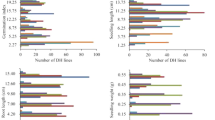
A model-fitting approach was used to analyse the data for the presence of QTL. Figure 1a shows the genotype of the 22 substitution lines used in the analysis, and how these relate to the substituted segments used in the model fitting. Given the structure of the substituted segments, there are a maximum of 25 regions of chromosomes where we might find Victor QTL. These are shown in Figure 1 and can be explained as follows.
-
i)
In the case of chromosomes 8, 14, 15, 18, and 19, the substituted line contains potentially the whole chromosome and hence we can specify just one potential QTL per chromosome (ie QTL 8, 14, 15, 18, 19).
-
ii)
Chromosomes 1, 2, and 11 have single substitutions at the ends of each chromosome so these regions are specified by potential QTL 1, 2, 11. The opposite end of each chromosome is Tapidor, but without a Victor substitution, we cannot specifically identify any QTL that may map there (see (vi)).
-
iii)
Chromosome 6 has two different substitutions, 6-A and 6-B. There are thus potentially two regions containing QTL, QTL 6-i in the top 30 cM and 6-ii in the bottom 50 cM. Line 6-A could have both 6-i and 6-ii, while 6-B would have only 6-ii.
-
iv)
Chromosome 7 is represented by three substitutions and there are potentially three QTL regions: 7-i in the top 3 cM, 7-ii in the next 8 cM, and 7-iii in the bottom 21 cM. Line 7-A could contain the Victor allele of QTL 7-i, 7-B could contain QTL 7-i and 7-ii, while 7-C could contain QTL 7-i, 7-ii and 7-iii.
-
v)
Chromosomes 3 and 13 are represented by several substitutions each, with potential QTL 3-i, to 3-v and 13-i to 13-vii as illustrated in Figure 1.
-
vi)
All chromosome regions not represented by substitutions into Tapidor are combined as a single effect, ‘Victor non-substituted’. This class includes whole chromosomes 4, 5, 9, 10, 12, 16, 17, as well as parts of other chromosomes as explained in (ii) above.
-
vii)
The year effects.

Substitution lines used in analysis. The 22 substitution lines used in the analysis are shown. For each substitution line, the size of the bar on the y-axis represents the estimated size of that linkage group in cM. Black areas represent Victor substituted segments. Grey areas represent regions of recombination between Victor and Tapidor. Single lines represent the Tapidor recurrent background. Alongside each substitution line, the substituted segments are represented by vertical arrows, which were used to calculate CI associated with QTL position used in the model fitting.
For each trait, the data were analysed by ANOVA to test for line (L), trial (T) and interaction (L×T) effects. The mean scores (over trials in a given year where applicable) for Tapidor, Victor, and each substitution line were calculated and the L×T mean square from the ANOVA was used to calculate the variance of each mean. The reciprocals of these variances were used as weights to estimate the genetic effects of the substitutions across years.
The model containing the specified QTL and year effects is as follows:

where μ is the mean of the parental lines; yr(i) is the deviation from the mean due to year i; ΣQ(j) is the deviation of Victor from μ; and ΣQ(kl) is the summed effect because of the ‘k’ Victor alleles in substitution line ‘l’.
The parameters were estimated by weighted least squares. Using the rule of parsimony, the simplest model (ie fewest parameters) which fitted the data was accepted. In such a model, all parameter estimates had to be significant and the χ2 testing for the adequacy of the model had to be nonsignificant (Kearsey and Pooni, 1996).
Results
Production of substitution library
A total of 22 substitution lines were produced from the original cross, each line representing a different single Victor segment, in an otherwise pure Tapidor recurrent parent background. The RFLP linkage map of Brassica napus, based on the B1 segregation data had a total map length of 1238 cM (Kosambi, 1944), as presented in Howell et al, 1996. These 22 substitution lines give a total minimum coverage of the Victor genome of 45% (based on the minimum size of the introgressed segments as defined by the marker genotypes). The maximum coverage of the Victor genome by the substitution library was 54% (inclusive of regions where recombination would have taken place between the introgressed segment and the Tapidor recurrent parent).
Data analysis of substitution library
Appendices 1 and 2 show the mean values of the substitution lines and the parents within each trial, for total seed oil content and fatty acid composition, respectively.
Table 2a shows the full analysis of variance for the character of oil content of the seed, which was conducted across all nine trials for the 22 substitution lines. Both main effects of substitution lines and trials were significantly different at the 0.1% level, as was the interaction term. However, the mean square for the interaction term was a lot smaller than the other two main effects.
Table 2b shows the summary analysis of variance for the fatty acids, and lists the mean squares and probabilities associated with the line, trial, and line-by-trial interaction terms. The analysis was conducted across the three trials of trials 4, 5 and 6 where fatty acids were measured. Except for C14:0, there were significant differences (C18:3 and C22:0, P<0.05; all others P<0.001) between lines detected by ANOVA for all traits scored when tested against the ‘lines-by-trials interaction’ mean squares. Although there are some significant interactions with trials, the interaction mean squares were relatively small compared to the line mean squares.
The location and maximum extent of each region capable of containing a QTL (as defined in Figure 1) is shown in Table 3, together with the sizes of the QTL effects actually detected. It shows, for example, that three Victor-substituted segments produce significantly more oil (3-v, 8, and 18) while four segments produce significantly less oil than Tapidor (6-ii, 13-vii, 14, and 19). Thus, there would appear to be increasing alleles for QTL in Victor at the bottom of N3 and at unspecified positions on N8 and N18. Conversely, decreasing QTL alleles in Victor are located at the bottom of N6 and N13, at an unspecified position on N14 and at the top of N19. For oil content, the model fitting using the weighted least-squares approach fits the data with a χ2 value of 36.17 with 41 degrees of freedom (df), which is nonsignificant (indicating that the model fits the data well). Victor has alleles that both increase and decrease the oil content. Overall, the effect of the Victor nonsubstituted segments is negative, showing that the other nonsubstituted Victor segments have QTL that, overall, decrease the oil content, or that the established effects are nonadditive.
The results of the analysis for fatty acids reveals the following QTL which are summarised in Table 3. The use of the terms positive and negative below refers to the direction of the effect attributable to the Victor allele.
C14:0
The model fits the data well (χ2: 39.71 with 45 df). Only two QTL are identified: a positive effect at the bottom of N6 and a negative effect midway through N13. The nonsubstituted Victor segments have no significant effect.
C16:0
The model fits the data well (χ2: 33.65 with 40 df). Three significant QTL are evident: one large positive effect at an unspecified region of N14 and two smaller, negative effects at an unspecified region of N8 and the top of N11. The nonsubstituted Victor segments have no significant effect.
C18:0
The model fits the data well (χ2: 44.35 with 42 df). Totally, 4 QTL are identified: one large and positive effect at the bottom of N1 and three small and negative effects at the bottom of N6, the top of N7 (which represents a CI of only 9 cM) and at an undefined location on N8. The sum total of the Victor nonsubstituted segments is negative.
C18:1 (Oleic acid)
The best model does not fit the data well; χ2=271.53 with 43 df (P≪0.001). In this model, four QTL are indicated: three large negative effects (at the bottom of N3 and at undefined locations on N8 and N18) and a small positive QTL effect at the top of N11, with a CI of only 9 cM. The Victor nonsubstituted segments have a positive effect. Reanalysing the data by fitting models for each year separately resulted in models which fitted the data well and with the same QTL effects being significant. This implied that the poor fit of the overall model was a consequence of line-by-trial (year) effects and Table 2b shows that there is a highly significant line-by-trial interaction for C18:1.
C18:2 (Linoleic acid)
The model fits the data well (χ2: 54.79 with 43 df) and indicates the presence of five QTL: two positive effects (at the bottom of N13 and an undefined region on N14) and three negative effects (at the bottom on N3, and undefined regions on N8 and N18). The nonsubstituted Victor segments are nonsignificant, indicating that most variation has been accounted for using this set of substitution lines.
C18:3 (Linolenic acid)
The model fits the data well (χ2: 50.67 with 41 df). Totally, five QTL are indicated: three positive effects (in the middle of N6, the middle of N7, and a small effect at a unspecified region of N18) and two negative effects (at the bottom of N7 and a small effect at the top of N11 with a 9 cM CI). The sum total of the Victor nonsubstituted segments is positive. The model fitting approach using substituted segments has allowed the effective differentiation of two antagonistic QTL effects on N7 for linolenic acid.
C20:0
The model fits the data well (χ2: 46.64 with 44 df). A total of 3 QTL are indicated, all with positive effects. There are two minor effects (at the bottom of N3, with a CI of 14 cM and an unspecified region on N18), and a large effect on N8.
C20:1 (Eicosenoic acid)
The model does not fit the data (χ2=501.94 with 43 df, P≪0.001). As with C18:1, splitting up the analysis into its three component years results in simple well-fitting models involving the same significant QTL. This suggests that the overall model does not fit well because of significant genotype by year effects. Table 2b shows that significant line-by-trial interactions were observed for C20:1. Similar QTL effects are evident for both C20:0 and C20:1.
C22:0
The model fits the data (χ2: 53.91 with 43 df). Here, four significant QTL effects are seen: two positive effects at an unspecified region of N8 and the bottom of N13, and two negative effects at the bottom of N3 (with a 14 cM CI) and an unspecified region of N18. The sum total of Victor nonsubstituted segments is positive.
C22:1 (Erucic acid)
The model does not fit the combined data well (χ2: 436.05 with 43 df, P≪0.001). Only three large positive QTL effects are detected, on the bottom of N3 (14 cM CI), an unspecified region on N18, and the largest effect on an unspecified region of N8. The sum total of Victor nonsubstituted segments is negative, implying that a number of smaller QTL, or one large QTL, still exist in the Victor genotype that significantly decreases the C22:1 content. These are either not represented by this set of 22 substitution lines or may be because of the nonadditive effects of the established QTL. Analysing the data for the three component years separately results in the model fitting the data well on each occasion.
The fact that the model fits the data well on a year-by-year basis implies that the major component of the variation which is unaccounted for in the combined mode was line-by-trial interaction. Table 2b shows that there was a significant line-by-trial interaction for erucic acid in the analysis of variance.
Discussion
Averaged over all the trials, Tapidor and Victor have 0.18 and 27.30% of their total seed oil content as erucic acid, respectively. As expected, Victor alleles increased concentrations of erucic acid, C22:1, for all QTL identified (Table 3). However, there were indications that QTL may exist in the Victor genotype that could decrease erucic acid content, as indicated by the negative effect of the combined Victor segments which were not represented in the substitution lines (Figure 1).
The following paragraphs compare QTL found within this study, to what appears to be similar ‘significant’ QTL from other studies. However, uncertainty may arise through these comparisons across studies as significance levels for the identification of QTL may vary, raising questions as to whether QTL identified in the same chromosomal region between studies are exactly the same QTL.
RFLP marker data from 200 B1 individuals of the same cross (Tapidor×Victor), were used to produce a linkage map, as described in Howell et al (1996). The B1S1 families from the original B1 population were scored for seed erucic acid concentration and QTL affecting erucic acid were determined using interval mapping with MAPMAKER/QTL v.1.1 (Lincoln et al, 1992), using a LOD threshold of 3 to infer the presence of QTL (P. Howell, personal communication). Two QTL for erucic acid, eru1 and eru2, were identified on linkage groups 8 and 13 respectively, which significantly increased erucic acid concentrations in the Victor genotype using this approach. eru1 was positioned at the proximal end of linkage group 8, between markers pN168E1b and pC2E2 with a 95% CI of approximately 12 cM. Eru2 mapped to the distal end of linkage group 13 near terminal marker pO87E4 with a 95% CI of approximately 26 cM. LOD scores were used to estimate these QTL positions, but these have been shown to give biased estimates of the CI of QTL position when the QTL effect is unknown (Mangin and Goffinet, 1997). Collectively, eru1 and eru2 accounted for 64% of the variation for erucic acid levels seen in the B1 population but no other regions were implicated (P. Howell, personal communication). In the present study, QTL 8 on linkage group 8 in the substitution lines affected erucic acid concentration and thus may be synonymous with eru1 (Table 3). The substituted segment used to infer the presence of QTL 8 was a whole chromosome substitution on linkage group 8, with a CI of 60 cM. None of the substitution lines representing the distal end of linkage group 13 scored significantly higher than Tapidor in the present study, so we were unable to confirm eru2.
Thormann et al (1996), identified two QTL affecting erucic acid levels, and two QTL affecting linolenic acid levels in a doubled-haploid population formed from a cross between rapeseed varieties ‘Major’ and ‘Stellar’, using MAPMAKER/QTL v1.1 (Lincoln et al, 1992). The QTL affecting erucic acid levels were mapped to LG7 and LG15 of that cross, and accounted for 95% of the phenotypic variation in the population. Their QTL on LG7 may be synonymous with eru1 (QTL 8) on linkage group 8 of the Tapidor by Victor cross, while that on LG15 is probably the same QTL as eru2 on linkage group 13, which was not seen in the substitution lines (P. Howell, personal communication). The QTL for linolenic acid accounted for 60% of the phenotypic variation seen in the ‘Major x Stellar’ doubled-haploid population, and were found on LG12 and LG14. Comparative mapping between the two maps of Thormann et al (1996), and Howell et al (1996) would be necessary to establish how LG12 and LG14 relate to those in the present study.
Ecke et al (1995), identified two QTL affecting erucic acid concentrations, and three QTL affecting seed oil content, in doubled-haploid populations formed from the varieties of ‘Mansholt's Hamburger Raps’ and ‘Samourai’, using MAPMAKER/QTL (Lincoln et al, 1992). It was suggested that the erucic acid QTL may be synonymous with two of the seed oil content QTL, because of the strong positive correlation between the two traits in selected crosses, and the coincidence of positions of the QTL. Summed over trials 4, 5, and 6 in the present study, oil content and erucic acid concentrations have a low correlation (r=0.18). Table 3 also shows that QTL from Victor which increased oil content in the Tapidor background, also increased erucic acid content, supporting the hypothesis of Ecke et al (1995). The strongest correlation that existed with erucic acid was with oleic acid (r=−0.90). This is to be expected, because erucic acid is derived from the precursor of oleic acid by the addition of two C2 units (Ecke et al, 1995).
Although it has been postulated that erucic acid levels are controlled by only two genes with multiple alleles in B. napus (Kondra and Stefansson, 1965), our substitution lines have identified three QTL. One of the QTL (QTL 8) was identified previously using the B1 data. Comparative mapping across the linkage maps by Howell et al (1996), Ecke et al (1995), and Thormann et al (1996), is necessary to establish if the other two erucic acid QTL were unique or had been identified before. The extra QTL identified in the present study may result from the increased precision and sensitivity of mapping QTL afforded by the substitution lines.
The Victor genotype increased the concentration of fatty acids C20:0, C20:1, and C22:1 (Table 3). For the remaining fatty acids, QTL were identified in the Victor genotype that both increase and decrease specific fatty acid concentrations.
Victor did not possess all the alleles for QTL that increased oil content, because a number of substituted segments scored significantly lower than Tapidor (Table 3). Butruille et al (1999) identified a QTL affecting oil content in backcross inbred lines formed from crosses of ‘Ceres’ with ‘Marnoo’ and ‘Westar’, which corresponds to linkage group 1 of the Tapidor by Victor cross. This QTL may not have been identified in the present study because substitution line 1-A represented less than half of linkage group 1.
Precision and sensitivity of QTL mapping
In segregating populations the precision of QTL mapping methods are relatively poor, and rarely fall below a 95% CI of about 20 cM (van Ooijen, 1992; Hyne et al, 1995; Burns, 1997). The CI in the substitution lines is defined precisely as the upper limits of extent of the donor segments as indicated in Figure 1. Many of the potential QTL containing regions can be seen to have CI values less than 10 cM, such as 3-ii, 3-v, 11-i, 13-i, etc. For example, the results presented in this paper show that substituted segment 11 (QTL 11) significantly affects C16:0, C18:1, and C18:3, and has a CI of only 9 cM (Table 3). This CI is dependent upon the accuracy with which introgressed segments are defined. Further backcrossing of the substitution lines could be used to reduce the size of the CI associated with the QTL even further (van Ooijen, 1992; Darvasi et al, 1993; Mackinnon and Weller, 1995; Ramsay et al, 1996). The model-fitting approach using substituted segments has also allowed the identification and separation of two QTL within a 32 cM interval on linkage group 7 effecting linolenic acid (C18:3) which act in opposite directions. As a result of the limited resolution inherent in population-based mapping strategies, it is unlikely that such a precise result could have been achieved.
Other results show that QTL 8 on linkage group 8 affects nine out of the 11 characters scored, indicating this region may possess multiple QTL or, more probably, QTL of pleiotropic effect.
Future uses
The present study has identified QTL affecting seed oil content and fatty acid composition in B. napus using a substitution line approach, the majority of which have not been identified before. Since the QTL mapping was conducted in the commercial varieties Tapidor and Victor, it will allow breeders to use marker-assisted breeding to introduce favourable alleles from either parent into new cultivars. The substitution library can provide a better understanding of the number of genes controlling a trait, their distribution over the genome, and their individual effects. It can also show how these genes interact and how they are affected by the environment. The substitution library can be used as material for commercial trialing, as well as a future resource for rapeseed breeding. The substitution library can be used in further field trials to determine QTL for resistances to common Brassica pests and diseases, such as phoma, stem canker, Cabbage stem beetle, Winter stem weevil, Cabbage root fly, Pollen beetle, and Brassica pod midge.
A maximum of 54% of the Victor genome was represented by the substitution library. Further substitution lines have been developed from the original cross, giving an increased coverage of the Victor genome, and these extra lines could be used to identify additional QTL affecting oil content and fatty acid composition.
Although the present study presents results just for oil content and composition, the field trials were also scored for other traits, including flowering time, yield, seed glucosinolate levels, emergence, establishment, lodging, and height. The analysis of these will be reported elsewhere.
References
Burns MJ (1997). Quantitative Trait Loci Mapping in Arabidopsis – Theory and Practice. PhD Thesis. University of Birmingham.
Butruille DV, Guries RP, Osborn TC (1999). Linkage analysis of molecular markers and quantitative trait loci in populations of inbred backcross lines in Brassica napus L. Genetics 153: 949 964.
Darvasi A, Weinreb A, Minke V, Weller JI, Soller M (1993). Detecting marker-QTL linkage and estimating QTL gene effect and map location using a saturated genetic-map. Genetics 134: 943 951.
Ecke W, Uzunova M, Weissleder K (1995). Mapping the genome of rapeseed (Brassica napus L.) II. Localization of genes controlling erucic acid synthesis and seed oil content. Theor Appl Genet 91: 972 977.
Haley CS, Knott SA (1992). A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315 324.
Howell PM, Marshall DF, Lydiate DJ (1996). Towards developing intervarietal substitution lines in Brassica napus using marker assisted selection. Genome 39: 348 358.
Hyne V, Kearsey MJ, Pike DJ, Snape JW (1995). QTL analysis – unreliability and bias in estimation procedures. Mol Breed, 1: 273 282.
Jansen RC, Stam P (1994). High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447 1455.
Jourdren C, Barret P, Horvais R, Delourme R, Renard M (1996). Identification of RAPD markers linked to linolenic acid genes in rapeseed. Euphytica 90: 351 357.
Kearsey MJ, Hyne V (1994). QTL analysis: a simple marker-regression approach. Theor Appl Genet 89: 698 702.
Kearsey MJ and Pooni HS (1996). The genetical analysis of quantitative traits. London: Chapman and Hall.
Kondra ZP, Stefansson BR (1965). Inheritance of erucic and eicosenoic acid content of rape-seed oil (Brassica napus). Can J Genet Cytol 7: 505 510.
Kosambi DD (1944). The estimation of map distances from recombination values. Ann Eugen 12: 172 175.
Lander ES, Botstein D (1989). Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185 199.
Lincoln S, Daly M, Lander E (1992). Mapping Genes Controlling Quantitative Traits with MAPMAKER/QTL 1.1, 2nd ed. Whitehead Institute Technical Report.
Luhs W, Friedt W (1993). Nonfood uses of vegetable oils and fatty acids. In: Murphy DJ (ed) Designer Oil Crops: Breeding, Processing and Biotechnology, VCH: Cambridge, UK, pp 73 130.
Luo ZW, Kearsey MJ (1989). Maximum likelihood estimation of linkage between a marker gene and a quantitative locus. Heredity 63: 401 408.
Mackinnon MJ, Weller JI (1995). Methodology and accuracy of estimation of quantitative trait loci parameters in a half-sib design using maximum likelihood. Genetics 141: 755 770.
Mangin B, Goffinet B (1997). Comparison of several confidence intervals for QTL location. Heredity 78: 345 353.
Rae AM, Howell EC, Kearsey MJ (1999). More QTL for flowering time revealed by substitution lines in Brassica oleracea. Heredity 83: 586 596.
Ramsay LD, Jennings DE, Bohuon EJR, Arthur AE, Lydiate DJ, Kearsey MJ, Marshall DF (1996). The construction of a substitution library of recombinant backcross lines in Brassica oleracea for the precision mapping of quantitative trait loci. Genome 39: 558 567.
Soller M, Brody T, Genizi A (1976). On the power of experimental designs for the detection of linkage between marker loci and quantitative loci in crosses between inbred lines. Theor Appl Genet 47: 35 39.
Tanksley SD (1993). Mapping polygenes. Ann Rev Genet 27: 205 233.
Thormann CE, Romero J, Mantet J, Osborn TC (1996). Mapping loci controlling the concentrations of erucic and linolenic acids in seed oil of Brassica napus L. Theor Appl Genet 93: 282 286.
van Ooijen JW (1992). Accuracy of mapping quantitative trait loci in autogamous species. Theor Appl Genet 84: 803 811.
Acknowledgements
The results reported in this paper are from a project started in 1992, involving collaboration between the plant breeding companies of Advanta, CPB Twyford Ltd, Monsanto UK Ltd and, Nickerson (UK) Ltd, together with the John Innes Centre (Norwich, UK) and the University of Birmingham (UK). The work was also supported by the United Kingdom (UK) Biotechnology and Biological Sciences Research Council, and the UK. Department of Trade and Industry (GICP Scheme ‘Genetic analysis in oilseed rape’).
The authors would also like to thank Dr's M Boddy, E Arthur, D Lydiate, and P Howell for their help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burns, M., Barnes, S., Bowman, J. et al. QTL analysis of an intervarietal set of substitution lines in Brassica napus: (i) Seed oil content and fatty acid composition. Heredity 90, 39–48 (2003). https://doi.org/10.1038/sj.hdy.6800176
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800176
Keywords
This article is cited by
-
Molecular mapping and candidate gene identification of two major quantitative trait loci associated with silique length in oilseed rape (Brassica napus L.)
Molecular Breeding (2024)
-
Mapping of flowering time, seed quality and clubroot resistance in rutabaga × spring canola populations and their association
Euphytica (2021)
-
GWAS and co-expression network combination uncovers multigenes with close linkage effects on the oleic acid content accumulation in Brassica napus
BMC Genomics (2020)
-
Genome-wide identification of silique-related traits based on high-density genetic linkage map in Brassica napus
Molecular Breeding (2019)
-
Unconditional and conditional QTL analyses of seed fatty acid composition in Brassica napus L.
BMC Plant Biology (2018)
