
Abstract
Previously, we have found that lipid rafts/caveolae were essential for insulin-like growth factor-1 (IGF-1) receptor signaling during 3T3-L1 preadipocytes differentiation induction. However, it was not identified as to which of the membrane lipid-ordered microdomains mediates the receptor signal. Using small double-stranded RNA-mediated interference (RNAi), we successfully suppressed the caveolin-1 protein expression. In cells stably transfected with vector expressing small interfering RNA (siRNA) fragment, no caveolin-1 protein or caveola was detected. On the other hand, removal of caveolin-1 did not affect the caveolinless lipid rafts or the localization of IGF-1 receptor in lipid rafts on plasma membrane. IGF-1 receptor signal transduction and induced cellular differentiation were normal in RNAi cells with only lipid rafts. Furthermore, these IGF-1 receptor signaling events were still sensitive to the cholesterol-binding reagents. Thus, our results suggest that lipid rafts are sufficient for IGF-1 receptor signaling and the recruitment of signal molecules by caveolin-1 is not essential for IGF-1 receptor signaling.
Similar content being viewed by others

Introduction
Lipid rafts are distinct plasma membrane lipid microdomains highly enriched with sphingolipids and cholesterol.1, 2, 3, 4 With integration of structure protein caveolins, the lateral lipid rafts structures will form flask-shaped vesicular invaginations in the plasma membrane, called caveolae.5, 6, 7 Caveolae are considered as a subtype of lipid rafts and only formed in cells expressing caveolin-1. If the cells become deficient in caveolin-1, they lose caveolae.8, 9 The presence of structure proteins (caveolins) makes caveolae morphologically distinct from noncaveolar lipid rafts.
The involvement of plasma membrane microdomains (lipid rafts and its subtype caveolae) in membrane receptor signaling has been extensively studied and is substantiated by the association of many membrane receptors and signal molecules with isolated lipid rafts and caveolae.1, 2, 3, 4, 10, 11, 12, 13 The study for the function of protein components in membrane lipid microdomains has largely been focused on the structural protein of caveolae, that is, caveolins (especially caveolin-1). Besides involved in vesicular trafficking and cholesterol trafficking, caveolin-1 has been indicated to negatively regulate the endothelial nitric oxide synthase (eNOS) activity and to mediate transmembrane signaling by organizing a ‘preassembled signal complex’ at the inner membrane side of caveolae.4, 14, 15, 16, 17 However, the animal model studies provided by caveolin-1 knockout mice suggest that the function of caveolin-1 and caveolae is not absolutely required. The animals lacking caveolin-1 lose all the caveolae on cell membrane, but they are viable with relatively minor defects.8, 9 These animal model results have raised the question whether caveolin play an irreplaceable role in the regulation of cellular activities, for example, transmembrane signal transduction.
The importance of the lipid composition in membrane lipid microdomains has been demonstrated by the studies of the cholesterol trafficking in lipid rafts/caveolae and its role in signaling.4, 14, 15, 16, 18, 19, 20, 21, 22, 23 Depletion of cholesterol from plasma membrane leads to the disruption of lipid rafts and blockade of membrane receptor signal transduction. It blocks signal transduction in both lymphocytes that is rich in caveolin-free rafts as well as endothelial cells and adipocytes that are rich in caveolae.20, 21, 22, 23, 24, 25
In order to differentiate the functional differences between caveolae and noncaveolar lipid rafts in mediating transmembrane signal, it is necessary to disrupt only one type of lipid microdomains. Since caveolin is the key component, which is present in caveolae but not in noncaveolar lipid rafts, by suppressing the expression of caveolin it is possible to generate animals or cells with only noncaveolar lipid rafts. Compared with caveolin-1 knockout animals that provide the model for the studies on the whole animal, caveolin-free cells are good models to analyze the intracellular mechanisms of caveolae and caveolin in the regulation of cellular activities.
In our previous study, we have indicated that cholesterol-enriched membrane microdomains play an important role in IGF-1 receptor transmembrane signaling in inducing 3T3-L1 cell differentiation. However, it is not clear whether caveolin-1 protein by forming caveolae are responsible for the IGF-1 receptor signaling.26 In this study, using RNA interference method, we have generated 3T3-L1 preadipocyte without caveolin and caveolae. Our results clearly demonstrated that the important component in IGF-1 receptor transmembrane signaling is the noncaveolar lipid rafts but not caveolae. Without caveolin and caveolae, preadipocytes could still differentiate into adipocytes under hormonal induction.
Results
Suppression of caveolin-1 protein expression by small double-stranded RNA-mediated interference (RNAi)
Plasmid vectors expressing small interfering RNA (siRNA) fragment for caveolin-1 mRNA were constructed and transfected into 3T3-L1 preadipocyte (for details, see in Materials and Methods). Several 3T3-L1 cell lines transfected with vector expressing siRNA fragment or control vector expressing the shuffle-sequence were selected and analyzed (Figure 1). Both caveolin-1 sequence fragments selected for RNA interference exhibited the RNAi effect. The caveolin-1 protein level in the RNAi vector-transfected cells was significantly reduced (Figure 1a). However, the Sequence-1 fragment (for details, see in Materials and Methods) was more effective than Sequence-2 (Figure 1a). Caveolin-1 protein expression was completely blocked in three independently selected cell lines stably transfected with Sequence-1 RNAi vector (Figure 1a and b). Even after differentiation induction, there was still no detectable caveolin-1 protein in these cells (Figure 1b). These three stable cell lines (1-1, 1-2 and 1-3 in Figure 1a) were selected for further studies and labeled as Ri-1, Ri-2 and Ri-3. In addition, only the transcript of caveolin-1, but not caveolin-2, was reduced by the vector-expressed caveolin-1 siRNA fragment (Figure 1c). Thus, caveolin-1 siRNA fragment specifically induced the degradation of cavolin-1 mRNA, but not caveolin-2 mRNA.

RNAi of caveolin-1. (a) Western blot analysis of caveolin-1 protein in stable cell lines. Equal amount of protein was loaded for each sample. After SDS-PAGE, the proteins were transferred to Immobilon-P membrane and caveolin-1 protein was detected by anti-caveolin-1 antibody. 1-1, 1-2, 1-3 and 1-4, Four independent stable cell lines transfected with Sequence-1 RNAi vector; 2-1 and 2-2, Two independent stable cell lines transfected with Sequence-2 RNAi vector; S1, Shuffle-sequence-1 RNAi vector transfected cell; S2, Shuffle-sequence-2 RNAi vector-transfected cell; C, The wild-type 3T3-L1 cell. The detailed information of sequences and RNAi vectors is described in the ‘Materials and Methods’. (b) caveolin-1 and caveolin-2 expression in caveolin-1 RNAi vector transfected cells. Cell samples were subjected to Western blot by anti-caveolin-1 antibody and anti-caveolin-2 antibody. Ri1, Ri2, Ri3 and S are 1-1, 1-2, 1-3 and S1 in panel A, respectively. C is the wild-type 3T3-L1 cell. D0 preadipocyte, The 2-day post-confluent preadipocytes; D6 adipocyte, Differentiated adipocytes 6 days after the addition of differentiation induction cocktail. (c) RT-PCR analysis of caveolin-1 and caveolin-2 mRNAs from control and transfected cells. Total RNA was isolated from cells as described in the ‘Materials and Methods’. RT-PCR and the primers are described in the ‘Materials and Methods’. C, Ri1, Ri2, S, D0 preadipocyte and D6 adipocyte are the same as in panel B. Cycle No refers to the number of PCR cycles before the termination of the reaction. Actin indicates the housekeeping gene in these RNAi cell lines following the same RT-PCR analysis
Results from caveolin-1 knockout mice studies indicate that protein expression of caveolin-2 depends on the presence of caveolin-1 protein (8). In cav-1−/− mice, the transcript of caveolin-2 is present, but the caveolin-2 protein is absent. In our caveolin-1 RNAi cell lines, the protein level of caveolin-2 was also dramatically reduced, but the mRNA level of caveolin-2 was not significantly altered by the caveolin-1 RNA interference (Figure 1b and c). Thus, the results of the caveolin-1 RNA interference in 3T3-L1 cells were consistent with the findings from the caveolin-1 knockout mice.
Ablation of caveolae but not rafts by caveolin-1 RNA interference
As shown in Figure 1, caveolin-1 siRNA fragment could completely block the caveolin-1 protein expression in cells stably transfected with vector expressing siRNA fragment. Thus, experiments were carried out to determine whether in these transfected cells the caveolae were also ablated. The immunofluorescence staining by caveolin-1 antibody detected no caveolin-1 protein on the plasma membrane of RNAi vector transfected cells. In contrast, 3T3-L1 cells transfected with vector expressing the shuffle-sequence and the wild-type 3T3-L1 cells exhibited strong caveolin-1 antibody staining (Figure 2a). Further analysis with electron microscope confirmed the immunofluorescence staining results. In wild-type 3T3-L1 cells or cells transfected with the shuffle-sequence vector, caveolae on the plasma membrane were clearly visible under electron microscope. However, there were no caveolae on the plasma membrane of cells transfected with the caveolin-1 RNAi vector (Figure 3). Even after extensively searching under the electron microscope, no caveolar structures were found in these caveolinless RNAi vector transfected cells. Only the representative microscopic images were presented in Figure 3. Taken together, these results provided compelling evidence that caveolin-1 siRNA fragment blocked the expression of caveolin-1 protein and lead to the ablation of caveolae on the cell surface.

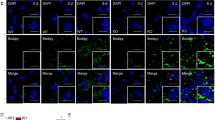
Immunofluorescence staining of caveolin-1, IGF-1 receptor and glycosphingolipids. (a) Cells of immunofluorescence staining were visualized with confocal microscope. Control refers to the wild-type 3T3-L1 preadipocyte. S refers to the cell stably transfected with the shuffle-sequence-1 RNAi vector. Ri-1, Ri-2 and Ri-3 indicate three independent cell lines stably transfected with Sequence-1 RNAi vector. The detailed information of Sequence-1 and shuffle-sequence-1 is described in the ‘Materials and Methods’. Caveolin-1 was detected by rhodamine red fluorescence (Cav-1) and IGF-1 receptor by FITC green fluorescence (IGF-1R). Glycosphingolipids were detected by FITC conjugated cholera toxin subunit B (CTXb). (b) Double immunofluorescence staining of caveolin-1 and glycosphingolipids. Ri-1, Ri-2 and Ri-3 cells are not analyzed by the double immunofluorescence because they have no caveolin-1 protein. Control, S, Cav-1 and CTXb are the same as described in panel A. The microscope images of caveolin-1 (detected by rhodamine red fluorescence (Cav-1)) and glycosphingolipids (detected by FITC green fluorescence (CTXb)) in the same cell were merged into the overlapped image (merged). (c) Double immunofluorescence staining of IGF-1 receptor and glycosphingolipids. C, S, Ri-1, Ri-2 and Ri-3 are the same as described in panel A. IGF-1 receptor is shown by rhodamine red fluorescence (IGF-1R) and glycosphingolipids are shown by FITC green fluorescence (CTXb). The overlapped picture of these two images is shown as merged

Visualization of caveolae by electron microscope: Electron microscope was used to visualize caveolae on the plasma membrane. Control, S, Ri-1, Ri-2 and Ri-3 are the same cell lines as described in Figure 2a. Caveolae are pointed by arrowheads. The enlarged areas of membrane section are shown to reveal the details
Although caveolae were abolished by caveolin-1 RNA interference, caveolin-free lipid rafts appeared to be unaffected in these RNAi cell lines. Immunofluorescence staining with cholera toxin subunit B revealed that the patterns of glycosphingolipid distribution on the cell surface of all these cell lines (the wild-type 3T3-L1 cell, caveolin-1 RNAi vector-transfected cell and control vector-transfected cell) were the same (Figure 2a). This result suggested that the distribution of sphingolipids on plasma membrane (hence lipid rafts) is independent from the presence of caveolin. In addition, the staining pattern of cholera toxin was very similar to that of caveolin-1 antibody in cells having caveolae, indicating the binding of caveolin-1 to most of the membrane lipid microdomains in cells with high level of caveolin-1 protein (Figure 2b). Thus, caveolae are likely the predominant type of membrane lipid microdomains in 3T3-L1 cell. With the wild-type 3T3-L1 cells predominantly having caveolae and caveolin-1 RNAi cells having only noncaveolar lipid rafts, it is possible for us to differentiate the functional differences of caveolae and lipid rafts in IGF-1 receptor signal transduction.
Localization of IGF-1 receptor in membrane lipid ordered microdomains in caveolaeless cells
Our previous study indicates that insulin-like growth factor-1 receptor was located in the lipid rafts/caveolae on the cell surface.26 In the cells transfected with caveolin-1 RNAi vector, caveolae no longer existed on the cell plasma membrane. However, IGF-1 receptor remained to be located in the membrane lipid microdomains in these cells (Figures 2c and 4. Fraction Nos. 1–21 are the fractions from the density gradient and Fraction 1 is the top of the gradient. WB, Western blot; CTXb, Western blot by cholera toxin subunit B; α-IGF-1Rβ, Western blot by antibody against IGF-1 receptor β-subunit; α-Cav-1, Western blot by antibody against caveolin-1 α isoform). By double-immunofluorescence staining with anti-IGF-1 receptor antibody and cholera toxin subunit B, IGF-1 receptor was found to be located in the glycosphingolipid-enriched area (Figure 2c). This colocalization of IGF-1 receptor with glycosphingolipid-enriched area was independent from the presence of caveolin-1 protein. The immunofluorescence staining patterns of IGF-1 receptor antibody or cholera toxin B subunit were very similar between the wild-type cells and cells without caveolae (Figures 2a and c). In addition, the results from the sodium carbonate extraction and sucrose density gradient centrifugation confirmed the observation of immunofluorescence staining. In caveolin-1 RNAi cells, IGF-1 receptor cofractionated with glycosphingolipid-enriched low-density membrane, which was the same as in the wild type 3T3-L1 preadipocyte (Figure 4). It is clear that the localization of IGF-1 receptor in membrane lipid microdomain did not require the mediation of caveolin. The sucrose density gradient centrifugation also showed that IGF-1 receptor was better cofractionated with glycosphingolipid-enriched low-density membrane than caveolin-1-enriched low-density membrane (Figure 4).

Density gradient centrifugation separation of membrane lipid microdomains. Cell membrane extraction, gradient centrifugation and Western blot were conducted as described in the ‘Materials and Methods’. For each cell line, samples from the same density gradient centrifugation were used in all the Western blots. Control, S, Ri-1 and Ri-2 are the same cell lines as described in Figure 2a
IGF-1 receptor activation and its signaling in caveolaeless cells
The downstream signal transduction of IGF-1 receptor requires intact lipid rafts/caveolae.26 However, it is not clear if there are any functional differences between caveolin-free rafts and caveolae in mediating IGF-1 receptor signal. Owing to the lack of caveolin-1 protein, signal molecules recruited to caveolae through the interaction with caveolin-scaffolding domain may not congregate in the lipid rafts. If the recruitment of signal molecules by caveolin into caveolae is functionally important for membrane lipid microdomain-mediated membrane receptor signal transduction, we would expect that IGF-1 receptor signal would be affected in caveolin-1 RNAi 3T3-L1 preadipocyte, which only has caveolin-free lipid rafts. Thus, the IGF-1 receptor activation and its downstream signaling in wild-type 3T3-L1 cell and caveolinless RNAi vector transfected 3T3-L1 cell were compared.
Although IGF-1 receptor activation by ligand is independent from the membrane microdomains26 and caveolin-free lipid rafts were sufficient for the proper localization of IGF-1 receptor in the plasma membrane, the change from predominantly caveolae in wild-type cells to only lipid rafts in caveolin-1 RNAi cells represented a significant environment change for IGF-1 receptor. Therefore, IGF-1 receptor activation in caveolinless cells was analyzed. As shown in Figure 5, there was no difference in IGF-1 receptor autophosphorylation activated by the ligand stimulation between the wild-type 3T3-L1 cells and caveolinless RNAi vector transfected cells.

Activation of IGF-1 receptor, insulin receptor and IRS-1 by ligand in caveolinless cells. IGF-1 receptor, insulin receptor and IRS-1 were immunoprecipitated from cell extracts as described in the ‘Materials and Methods’. Control, S, Ri-1 and Ri-2 are the same cell lines as described in Figure 2a. The time points (0, 10, 30 and 60 min) indicate the time of insulin stimulation. IP, immunoprecipitation; IB, Western immunoblot; α-IGF-1Rβ, antibody against IGF-1 receptor β-subunit; α-IRβ, antibody against insulin receptor β-subunit; α-IRS-1, antibody against insulin receptor substrate-1; α-P-Tyr, antibody against phosphotyrosine
IGF-1 receptor is responsible for inducing the adipocyte differentiation process in 3T3-L1 preadipocytes;27 however, insulin receptor and IGF-1/insulin hybrid receptor are also present in the preadipocytes.28 As shown in Figure 5, although the homodimer receptors were the dominant form, the hybrid IGF-1/insulin receptor was present in both control and RNAi cells. When anti-IGF-1 receptor antibody was used to immunoprecipitate the receptor, insulin receptor was detectable but was much less than IGF-1 receptor, and vice versa (Figure 5). This was also confirmed by the anti-phosphotyrosine antibody Western blot. With IGF-1 receptor antibody immunoprecipitation, the phosphorylated IGF-1 receptor β-subunit was more than phosphorylated insulin receptor β-subunit, whereas with insulin receptor antibody immunoprecipitation the phosphorylated insulin receptor was the dominant form. This is consistent with the results reported by other researchers, which indicate that the IGF-1/insulin hybrid receptor is low in preadipocyte and high in adipocyte.28 It should be noted here that in identical experimental conditions, the ECL exposure of insulin receptor Western blot for samples immunoprecipitated by insulin receptor antibody took much longer than that for IGF-1 receptor (20 min versus 30 s). When used for samples of 3T3-L1 adipocytes, the insulin receptor antibody did not exhibit significantly less affinity than the IGF-1 receptor antibody.26 Thus, it is likely that the number of insulin receptor was less than that of IGF-1 receptor in these cells. These results indicated that IGF-1 receptor, insulin receptor and IGF-1/insulin hybrid receptor, all were induced by the insulin stimulation in RNAi cells. Since IGF-1 receptor is the major receptor in preadipocytes while insulin receptor and IGF-1/insulin hybrid receptor are most abundant in adipocytes not preadipocytes, the downstream signaling observed here was mostly attributed to IGF-1 receptor. Currently, we are using the differentiated RNAi cells to analyze the insulin receptor downstream signaling.
The cholesterol depletion study indicates that the intact membrane lipid microdomain is an essential precondition for IGF-1 receptor downstream signal transduction.26 If the receptor downstream signal transduction is unaffected by removing caveolin-1, it is the lipid rafts not caveolae essential for the receptor downstream signal transduction. Insulin receptor substrate-1 (IRS-1) is the immediate downstream substrate of IGF-1 receptor tyrosine kinase and is phosphorylated on tyrosine when the receptor is activated by ligand.29, 30 As shown in Figure 5, IRS-1 was phosphorylated on tyrosine by IGF-1 receptor kinase in both caveolinless RNAi cells and the wild-type 3T3-L1 cells after the insulin stimulation. The lack of caveolin had no effect on IRS-1 tyrosine phosphorylation by IGF-1 receptor kinase. It appeared that caveolinless lipid rafts alone were sufficient to support the IGF-1 receptor signal transduction.
To provide further evidence that IGF-1 receptor signal transduction does not require caveolin and caveolae, signal molecules further downstream in IGF-1 receptor signal cascade were analyzed. PKB/Akt and ERK, two intracellular kinases involved in the regulation of many cellular activities, are activated by IGF-1 receptor signaling.26, 31, 32, 33, 34, 35 The activation of PKB/Akt by IGF-1 receptor is reported to be mediated by IRS-1 and phosphatidylinositol 3-kinase, and the activation of ERK by the receptor is through Ras and MEK. Since these two kinases are activated by IGF-1 receptor through the mediation of several signal molecules, to analyze their activation by the receptor signal in caveolinless RNAi cells will help to understand the effect of the lack of caveolin-1 on the intracellular signal molecules associated with membrane lipid microdomains. As shown in Figure 6a and b, PKB/Akt and ERK were activated by IGF-1 receptor signal in both wild-type 3T3-L1 cells and caveolinless RNAi cells. As expected, the activation of PKB/Akt by IGF-1 receptor signaling in caveolinless transfected cells could be inhibited by LY294002, which inhibits the generation of phosphatidylinositol phosphate by blocking the phosphatidylinositol 3-kinase activity,36 while the activation of ERK by the receptor signal was specifically inhibited by PD98059, an MEK-1 inhibitor (Figure 6a and b).37 These results suggested that the intermediate signal molecules were able to mediate the IGF-1 receptor signal in cells completely lacking caveolin-1 protein and caveolae. Their sensitivity to the inhibitors implied the activation of PKB/Akt and ERK by IGF-1 receptor through the normal activation pathways in these caveolinless cells. Importantly, in cells without caveolin-1 and caveolae the activation of PKB/Akt or ERK still required the integrity of lipid rafts, because the treatment with β-methylcyclodextrin, which disrupts the plasma membrane lipid microdomains by extracting cholesterol from cell membrane,15, 23, 38, 39, 40 blocked their activation by the receptor signal (Figure 6). These results suggested that the function of lipid rafts in IGF-1 receptor signaling is irreplaceable, while caveolae play nonessential role in the receptor signaling. In addition, cavelin-1 protein did not seem to be required for the recruitment of signal molecules to the membrane lipid microdomains.

Activation of PKB/Akt and ERK by IGF-1 receptor signal. Cells were treated with inhibitors and insulin as described in the ‘Materials and Methods’, and then subjected to Western blot analysis. (a) Activation of PKB/Akt by IGF-1 receptor signaling. Control, S, Ri-1, Ri-2 and Ri-3 are the same cell lines as described in Figure 2a. None, Cells without inhibitor pretreatment; +β-MCD, Cells pretreated with 10 mM β-methylcyclodextrin for 60 min; +LY, Cells pretreated with 20 μ M LY294002 for 30 min; +PD, Cells pretreated with 20 μ M PD98059 for 60 min. +insulin and the time points (0, 2, 5, 10, 30 and 60 min) indicate the insulin stimulation times. WB, Western blot; α-pAkt, Western blot by antibody against Ser473 phosphorylated PKB/Akt; α-Akt, Western blot by antibody against PKB/Akt protein. (b) Activation of ERK by IGF-1 receptor signal. The labelings are the same as in panel A. α-pERK, Western blot by antibody against Tyr204 phosphorylated ERK; α-ERK, Western blot by antibody against ERK protein
Adipocyte differentiation induction and mitotic clonal expansion in caveolinless 3T3-L1 cells
Since the lack of caveolin-1 and caveolae did not interfere with IGF-1 receptor signal transduction, we would like to know if the receptor signal-induced cellular responses were also normal in caveolin-1 RNAi cells. In wild-type 3T3-L1 cells, IGF-1 receptor signaling induces adipocyte differentiation and mitotic clonal expansion.27, 41 Thus, in caveolin-1 RNAi cells these two cellular responses to IGF-1 receptor signaling induction were analyzed. As shown in Figure 7, the adipocyte differentiation of the caveolinless RNAi cells was not affected by the absence of caveolin-1 protein. Although some clonal variations existed between the RNAi vector transfected cell lines, the adipocyte differentiation was evident. In addition, the cells with greatly decreased caveolin-1 protein but not selected for further analysis (Figure 1a) also differentiated into adipocytes (results not shown).

Adipocyte differentiation induction in caveolaeless cells. Cells were treated with inhibitors and induced to differentiation as described in the ‘Materials and Methods’. The differentiated adipocyte monolayers were stained with Oil-Red-O and photographed. Control, S, Ri-1, Ri-2 and Ri-3 refer to the cell lines as described in Figure 2a. None, Cells with standard differentiation induction; +Xyla, Differentiation induced in the presence of 50 μg/ml xylazine; +β-MCD, Differentiation induced in the presence of 3.5 mM β-methylcyclodextrin; +Filip, Differentiation induced in the presence of 1.4 μg/ml filipin
Our previous studies indicate that 3T3-L1 cell differentiation induction is blocked by cholesterol-binding reagents, such as β-methylcyclodextrin or filipin.26 Thus, the effect of cholesterol depletion on the adipocyte differentiation in caveolinless RNAi cells was also investigated. The results clearly showed that the adipocyte differentiation of caveolinless cells was cholesterol sensitive (Figure 7). Xylazine as expected had no inhibitory effect on the induced adipocyte differentiation. In every aspect we analyzed (the appearance of triglyceride droplets, the morphology of differentiated adipocytes etc.), there was no significant difference between caveolinless RNAi vector transfected cells and wild-type 3T3-L1 cells or control vector transfected cells (results not shown).
Mitotic clonal expansion is another cellular response induced by IGF-1 receptor signaling during the differentiation induction process and sensitive to the cellular cholesterol level.26 Again, caveolinless RNAi cells exhibited the same phenotype as wild-type 3T3-L1 cells in mitotic clonal expansion (Figure 8). Taken together, our results suggested that in IGF-1 receptor signal transduction and its induced adipocyte differentiation process, caveolin-free lipid rafts is sufficient and the functions of caveolin or caveolae in the differentiation induction (if they had any function) can be fully complemented by caveolin-free lipid rafts.

Analysis of mitotic clonal expansion during differentiation induction. Cells were induced to differentiate as described in the ‘Materials and Methods’. Cell numbers at different stages after induction were determined and normalized against that on day 0. The cell numbers of 3 culture plates were averaged. Control, S, Ri-1, Ri-2 and Ri-3 refer to the cell lines as described in Figure 2a. The fold-increases of cell numbers during differentiation induction over that on day 0 were shown, and the five bars in each group represent the five cell lines, wild type cell, shuffle sequence vector transfected cell and three RNAi cells. Mitotic clonal expansion of control and RNAi cells during standard differentiation induction (+MDI), in the presence of β-methylcyclodextrin (+β-MCD), in the presence of Filipin (+Filipin) and in the presence of xylazine (+Xylazine) were shown in panels a, b, c and d respectively
Discussion
Our previous study indicates that membrane lipid microdomains are essential for IGF-1 receptor signaling in inducing 3T3-L1 adipocyte differentiation.26 However, it was not identified which type of the membrane lipid microdomains (caveolae or noncaveolar lipid rafts) is essential. Although caveolin-free lipid rafts and its subtype caveolae have similar lipid composition,1, 2, 3, 4, 5 the major difference between these two structures is the protein component of caveolae–caveolins, which give caveolae the invaginated shape. So far, it is not clear whether caveolae and lipid rafts are dynamically interconvertible in the cell. The role of caveolin and caveolae in membrane signal transduction is controversial. Most studies including our previous work26 on the role of membrane lipid microdomains in transmembrane signal transduction involve the cofractionation of receptor or signal molecules with low-density membranes in density-gradient centrifugation or the depletion of cholesterol with cholesterol-binding reagents, such as β-methylcyclodextrin and filipin. However, these methods cannot distinguish the caveolae and caveolin-free lipid rafts from each other. The low-density membranes isolated in density-gradient centrifugation contain both caveolae and noncaveolar lipid rafts. In the cells with abundant caveolin-1 protein, such as 3T3-L1 preadipocyte, caveolae appear to be the predominant type of lipid microdomains on the cell membrane because most glycosphingolipids-enriched areas were covered by caveolin-1 (Figures 2b and 3). IGF-1 receptor is likely to be identified into caveolar membrane fraction. However, improved analysis indicated that IGF-1 receptor was better co-localized with glycosphingolipid-enriched low-density membranes than caveolin-enriched low-density membranes (Figure 4). In addition, this colocalization of IGF-1 receptor with glycosphingolipid-enriched low-density membrane did not require the mediation of caveolin-1 protein since in cells without caveolin-1 protein the colocalization of IGF-1 receptor with glycosphingolipid-enriched membranes was unaffected (Figure 4). These results strongly suggested that the localization of IGF-1 receptor in membrane lipid microdomains is not a caveolin-mediated process.
Since cholesterol-binding reagents disrupt both caveolae and noncaveolar lipid rafts, by targeting caveolin-1 with caveolin-1 RNA interference we successfully disrupted the caveolae without affecting the non-caveolar lipid rafts (Figures 2 and 3). It is evident that without caveolin-1 3T3-L1 cells grow and differentiate normally (Figures 7 and 8). Furthermore, the signal transduction of IGF-1 receptor in cells without caveolin-1 and caveolae was also undisrupted. There were no difference in the receptor activation by ligand and its downstream signaling between wild-type 3T3-L1 cells and 3T3-L1 cells with only noncaveolar lipid rafts (Figures 5 and 6). Interestingly, the IGF-1 receptor signal transduction and its induced 3T3-L1 adipocyte differentiation and mitotic clonal expansion were still sensitive to the cholesterol-binding reagent in caveolin-1 RNAi cells (Figures 5, 6, 7 and 8). Thus, lipid rafts appeared to be still required for IGF-1 receptor signaling events. Taken together, it is clear that IGF-1 receptor signal transduction and its induced adipocyte differentiation require the membrane lipid microdomains but not caveolin-1 protein per se. Although our current investigation is focusing on a specific case of IGF-1 receptor signaling in 3T3-L1 cells differentiation induction, given the importance of IGF-1 receptor in regulating cell activities and its representative position in receptor tyrosine kinase, the results could also have implication on the signal transduction of other receptor tyrosine kinases systems. In addition, the study on the cell line could allow analyzing the detailed intracellular signal mechanisms, which could not be easily done by the knockout animal studies.
Caveolin-1 has been suggested as an organizer to recruit signal molecules into caveolae to form ‘preassembled signal complexes’ for mediating signal transduction.11, 12, 13 However, the sufficiency of lipid rafts in mediating IGF-1 receptor signal in caveolin-1 RNAi 3T3-L1 cells has questioned whether caveolin-1 protein functions as an organizer in the formation of signal molecule congregation in membrane lipid microdomains. Our current study indicated that after removing caveolin-1 protein, lipid rafts could still assemble sufficient signal molecules for IGF-1 receptor signaling. Removal of caveolin-1 protein from 3T3-L1 preadipocytes and adipocytes created a dramatic change for the local environment of IGF-1 receptor and the plasma membrane microstructures, since these are caveolae and caveolin-1-enriched cells. However, it did not affect the cell signal transduction, growth and differentiation. Taken together with the normal cellular development in caveolin-1 knockout mice,8, 9 these studies suggest that caveolin-1 protein per se does not play an irreplaceable role in membrane lipid microdomain mediated signal transduction.
Materials and Methods
Materials
Anti-IGF-1 receptor β-subunit antibody was purchased from Oncogene Research Products. Anti-caveolin-2 antibody was from BD Transduction Laboratories. Anti-phospho-tyrosine antibody was from UBI (Upstate Biotechnology). Anti-caveolin-1 α isoform, anti-insulin receptor-substrate-1, anti-insulin receptor β-subunit, anti-ERK, anti-p-ERK (against the critical Tyr residue phosphorylated peptide), anti-PKB/Akt, and anti-pPKB/Akt (against Ser-473 phosphorylated form) antibodies were from Santa Cruz Biotechnology. Horseradish peroxidase (HRP)-conjugated, fluorescein isothiocyanate (FITC)-conjugated or rhodamine-conjugated secondary antibodies, HRP- or FITC-conjugated cholera toxin subunit B, β-methylcyclodextrin, filipin, xylazine, dexamethasone, 1-methyl-3-isobutylxanthine and insulin were from Sigma. PD98059 and LY294002 were from Calbiochem. Dulbecco's modified Eagle's medium (DMEM), protein A-agarose, transfection reagent Lipofectamine and G418 were from Invitrogen.
Cell culture, differentiation induction of 3T3-L1 preadipocytes and Oil-Red-O staining
3T3-L1 preadipocytes were cultured and induced to differentiate following the protocol described previously.42, 43, 44, 45 The differentiated 3T3-L1 adipocytes were stained with Oil-Red-O to show triglyceride droplets.45
RNAi
The RNA interference experiment was carried out by using plasmid expressing siRNA fragment.46 Two sequences for RNAi were selected from caveolin-1 cDNA coding region started with GGG and were analyzed by BLAST search to ensure that they did not have significant sequence homology with other genes. Sequence-1, 5′-GGGACACACAGTTTCGACGGC, was corresponding to nucleotides 229-249 (translation initiation site ATG as nucleotide 1) and Sequence-2, 5′-GGGCATTTACTTTGCCATTCT, corresponding to nucleotides 345–365. The reverse and complement strands were designed as described by Sui et al.46 For Sequence-1, two pairs of oligonucleotides were synthesized: pair-S1-1 5′-GGACACACAGTTTCGACGGCA (forward) and 5′-AGCTTGCCGTCGAAACTGTGTGTCC (complement), and pair-S1-2 5′-AGCTTGCCGTCGAAACTGTGTGTCCCTTTTTG (forward) and 5′-AATTCAAAAAGGGACACACAGTTTCGACGGCA (complement). The annealed pair-S1-1 double-strand oligonucleotide was inserted into the U6 promoter Bluescript plasmid between Apa I (nuclease blunted end) and Hind III site to make the intermediate plasmid.46 The annealed pair-S1-2 oligonucleotide was then inserted into the Hind III and EcoR I site of the intermediate plasmid to make the caveolin-1 RNAi plasmid. Thus, the constructed caveolin-1 RNAi plasmid can express an RNA fragment, which will form a 21-bp double-strand RNA hairpin with a 6-bp loop. The sequence of 21-bp double-strand RNA hairpin is corresponding to the Sequence-1. For Sequence-2, the same strategy was used to make the RNAi plasmid: pair-S2-1 5′GGCATTTACTTTGCCATTCTA (forward) and 5′-AGCTTAGAATGGCAAAGTAAATGCC (complement), and pair-S2-2 5′-AGCTTAGAATGGCAAAGTAAATGCCCTTTTTG (forward) and 5′-AATTCAAAAAGGGCATTTACTTTGCCATTCTA (complement).
As control to these two RNAi plasmids for caveolin-1, corresponding shuffle sequences with same nucleotide composition but different sequences were designed and the plasmids were constructed to express the hairpin RNA fragments. The shuffle sequence for Sequence-1 is 5′-GGGACACCAGACTTTCAGCGG, and the two pairs of oligonucleotide for the construction of plasmid were: pair-Sh1-1 5'-GGACACCAGACTTTCAGCGGA (forward) and 5′-AGCTTCCGCTGAAAGTCTGGTGTCC (complement), and pair-Sh1-2 5′-AGCTTCCGCTGAAAGTCTGGTGTCCCTTTTTG (forward) and 5′-AATTCAAAAAGGAACACCAGACTTTCAGCGGA (complement). For Sequence-2, the shuffle sequence was 5′-GGGATACCTTTCGTTTCTAC and the two pairs of oligonucleotides were: pair-Sh2-1 5′-GGATATCCTTTCGTTTCTACA (forward) and 5′-AGCTTGTAGAAACGAAAGGATATCC (complement), and pair-Sh2-2 5′-AGCTTGTAGAAACGAAAGGATATCCCTTTTTG (forward) and 5′-AATTCAAAAAGGGATATCCTTTCGTTTCTACA (complement). The plasmids were constructed in the same way as the caveolin-1 RNAi plasmids.
In order to make stable cell lines expressing the siRNA fragment, the cassette of U6 promoter with RNAi sequence insertion was digested from the constructed plasmids, and inserted into pBCMGneo plasmid between Bam HI and Xba I site.47, 48 3T3-L1 preadipocyte was transfected with pBCMGneo caveolin-1 RNAi plasmids and the stable cell lines were selected by the resistance to G418. In total, four plasmids (pBCMGneo-Sequence-1, pBCMGneo-Sequence-2, pBCMGneo-Shuffle-1 or pBCMGneo-Shuffle-2) were transfected into 3T3-L1 preadipocytes respectively, and independent foci were selected for each plasmid.
Cell transfection
A total of 1 × 105 exponentially growing low passage 3T3-L1 preadipocytes were plated into 60 mm culture dish and cultured in DMEM with 10% calf serum at 37°C CO2 incubator for 24 h. The cells were then transfected with the desired plasmid by LipofectamineRT following the protocol provided by Invitrogen. A measure of 400 μg/ml G418 was added to the medium 24 h after the transfection. G418 resistant foci were generally formed within 2–3 weeks. Individual focus was selected and propagated for further analysis.
RNA preparation and reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA in two 100 mm 3T3-L1 preadipocyte or adipocyte monolayers were extracted with TRIzolRT reagent (Invitrogen) following the protocol provided by the manufacturer. The extracted RNA was dissolved in 200 μl diethyl pyrocarbonate (DEPC)-treated water and the concentration was determined by UV absorption at 260 nm. An amount of 1.5 μg total RNA for each sample was reverse transcribed with Oligo dT primer by Superscript II (Invitrogen) following the protocol provided by the manufacturer. One-twentieth reverse transcription mixture was used as the template for PCR reaction. The primers of PCR for caveolin-1 were 5′-ATGTCTGGGGGCAAATACGTGG-3′ and 5′-AGCACGCAGAAAGAGATATGAG-3′, and for caveolin-2 were 5′-TGTGCAGCTCTTCATGGCCGAT-3′ and 5′-ATGCAACTGAGCCACGACTGAC-3′. The PCR product of caveolin-1 was 538 base-pairs (bp) and caveolin-2 464 bp. The PCR amplification condition was 94°C 30 s, 56°C 30 s and 72°C 1 min with Taq polymerase in total 50 μl reaction mixture. PCR reaction was terminated at 20, 25 and 30 cycles, respectively. The PCR-produced DNA fragment was analyzed with 1.2% agarose gel electrophoresis and revealed by ethidium bromide staining. Control of housekeeping gene, actin, was conducted in the same condition with the same reverse transcribed samples. Two primers for actin were 5′-AATGGGTCAGAAGGACTC-3′ and 5′-AGAGCAACATAGCACAGC-3′.
Western immunoblot
3T3-L1 cell monolayers (35 mm) were lysed directly in 1 × boiling Laemmli SDS sample buffer with 20 mM dithiothreitol. Samples were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Immobilon-P membrane (Millipore). After blocking with 2% nonfat-dried milk in 1 × Tween-Tris-buffered saline (TTBS) (0.05% Tween, 25 mM Tris-HCl, pH 7.5 and 150 mM NaCl) for 2 h at room temperature, membranes were incubated with primary antibody for 2 h at room temperature, followed by HRP-conjugated secondary antibody for 45 min. The targeted protein was revealed by enhanced chemiluminescence (ECL).
Immunofluorescence staining
Caveolin-1 or IGF-1 receptor β-subunit immunofluorescence staining was carried out following the protocol described previously.26 Caveolin-1 and IGF-1 receptor β-subunit were revealed with rhodamine- or FITC-conjugated secondary antibody. The cells were visualized by confocal microscope (Leica).
For the staining of glycosphingolipid on the 3T3-L1 cell membrane by FITC conjugated cholera toxin B subunit (FITC-CTXb), 3T3-L1 preadipocytes cultured on coverslips were rinsed with ice-cold phosphate-buffered saline (PBS) three times and incubated with FITC-CTXb in blocking buffer (1% bovine serum albumin in TTBS) on ice for 10 min. After washing with ice-cold PBS, the cells were immediately fixed with 100% methanol for 5 min at –20°C and visualized under confocal microscope.
For glycosphingolipid and caveolin-1 (or IGF-1 receptor β-subunit) double immunofluorescence staining, cells on coverslips were stained with FITC-CTXb as described in the previous paragraph. After the methanol fixation, the cells were washed with PBS and incubated in blocking buffer for 30 min at room temperature. The cells were then incubated with anti-caveolin-1 or anti- IGF-1 receptor β-subunit antibody in blocking buffer for 1 h at room temperature. After washing with PBS, the coverslips were incubated with rhodamine-conjugated secondary antibody.
Electron microscopy
Confluent 3T3-L1 cells were washed with PBS, scraped from the culture dishes and collected in 1.5 ml. Eppendorf tube by centrifugation at 1200 r.p.m. for 5 min. The cell pellets were fixed in 2.5% glutaraldehyde, washed with PBS and incubated in 1% OsO4. After gradual dehydration in ethanol, the fixed cells were embedded in epoxy matrix. The samples were observed in electron microscope.
Sodium carbonate extraction and sucrose density gradient fractionation of lipid rafts/caveolae
The experiments were carried out following the detergent-free protocol developed by Song et al.49 Fractions from the sucrose density gradient centrifugation were collected and analyzed by Western immunoblot using anti-caveolin-1 or anti-IGF-1 receptor β-subunit antibody as described previously.26 For analysis of glycosphingolipid, 2 μl sample of each fraction from the density gradient centrifugation were dot-blotted onto Hybond C membrane (Amersham Biosciences). After blocking in 2% nonfat-dried milk TTBS buffer for 1 h, the membrane was incubated with HRP-conjugate cholera toxin subunit B (HRP-CTXb) for 45 min at room temperature. ECL was carried out to reveal the samples enriched with glycosphingolipids.
Analysis of IGF-1 receptor and insulin receptor autophosphorylation and IRS-1 phosphorylation
The 2-day postconfluent 3T3-L1 cells or cells transfected with RNAi vector were fed with DMEM containing 10% FBS for 48 h to minimize the serum effect. A measure of 1 μ M insulin was then added to the cells. At indicated time point as described in Figure 5, cells were washed with ice-cold PBS and lysed in 1% Triton X-100 buffer (1% Triton X-100, 50 mM HEPES, pH 7.4, 2.5 mM EDTA, 150 mM NaCl, 30 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM PMSF and 2 μl/ml protease inhibitor cocktail 1 and 2). The cell lysate was homogenized and extracted at 4°C for 1 h. After centrifugation at 12,000 × g for 15 min, the supernatant (500 μg protein) was mixed with 1 μg anti-IGF-1 receptor β-subunit antibody, anti-insulin receptor β-subunit antibody or 1 μg anti-IRS-1 antibody for 2 h at 4°C, and then with protein A–agarose overnight at 4°C. The protein A–agarose was collected by centrifugation at 1000 × g for 5 min, washed with Triton lysis buffer four times and resuspended in 1 × Laemmli SDS sample buffer for Western blot analysis.
Analysis of PKB/Akt and ERK activation by IGF-1 receptor signal
The 2-day post confluent cells were fed with DMEM containing 10% FBS for 48 h to minimizing the serum effect. Cells were pretreated with or without inhibitor (10 mM β-methylcyclodextrin for 1 h, 20 μ M LY294002 for 30 min or 20 μ M PD98059 for 1 h respectively), and then treated with 1 μ M insulin. At indicated time points as described in Figure 6, the cells were lysed in 1 × Laemmli SDS sample buffer with 20 mM dithiothreitol for Western blot analysis.
Adipocyte differentiation induction and mitotic clonal expansion in caveolinless 3T3-L1 cell
Postconfluent 3T3-L1 preadipocytes and RNAi vector-transfected cells were pretreated with cholesterol binding reagents (3.5 mM β-methylcyclodextrin or 1.4 μg/ml filipin) or nonspecific lipid binding reagent (50 μg/ml xylazine) for 24 h, and then were induced to differentiate as described previously.26 During differentiation induction, cell numbers were counted to analyze the mitotic clonal expansion. By day 8, the cells were stained with Oil-Red-O.
Abbreviations
- IGF-1:
-
insulin-like growth factor-1
- IRS-1:
-
insulin receptor substrate-1
- RNAi:
-
small double-stranded RNA-mediated interference
- siRNA:
-
small interfering RNA
- PKB/Akt:
-
protein kinase B
- ERK:
-
extracellular signal-regulated kinase
- MEK:
-
MAP kinase kinase
- DMEM:
-
Dulbecco's modified Eagle's medium
- SDS-PAGE:
-
SDS polyacrylamide gel electrophoresis
References
Galbiati F, Razani B and Lisanti M (2001) Emerging themes in lipid rafts and caveolae. Cell 106: 403–411
Kurzchalia TV and Parton RG (1999) Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 11: 424–431
Brown DA and London E (1998) Function of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14: 111–136
Fielding CJ and Fielding PE (2003) Relationship between cholesterol trafficking and signaling in rafts and caveolae. Biochim Biophys Acta. 1610: 219–228
Anderson RGW (1998) The caveolae membrane system. Annu. Rev. Biochem. 67: 199–225
Parton RG (2001) Life without caveolae. Science 293: 2404–2405
Parton RG (1996) Caveolae and caveolins. Curr. Opin. Cell Biol. 8: 542–548
Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H and Kurzchalia TV (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293: 2449–2452
Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou Jr H, Kneitz B, Lagaud G, Christ GJ, Edelmann W and Lisanti MP (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276: 38121–38138
Simons K and Toomre D (2000) Lipid rafts and signal transduction. Nature Rev. Mol. Cell. Biol. 1: 31–39
Liu P, Rudick M and Anderson RGW (2002) Multiple functions of caveolin-1. J. Biol. Chem. 277: 41285–41298
Okamoto T, Schlegel A, Scherer PE and Lisanti MP (1998) Caveolins, a family of scaffolding proteins for organizing ‘preassembled signaling complexes’ at the plasma membrane. J. Biol. Chem. 273: 5419–5422
Couet J, Li S, Okamoto T, Ikezu T and Lisanti MP (1997) Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272: 6525–6533
Ikonen E and Parton RG (2000) Caveolins and cellular cholesterol balance. Traffic. 1: 212–217
Fielding CJ and Fielding PE (2000) Cholesterol and caveolae: structural and functional relationships. Biochim. Biophys. Acta. 1529: 210–222
Schlegel A and Lisanti MP (2001) The caveolin triad: caveolae biogenesis, cholesterol trafficking, and signal transduction. Cytokine Growth Factor Rev. 12: 41–51
Sowa G, Pypaert M and Sessa W (2001) Distinction between signaling mechanisms in lipid rafts vs. caveolae. Proc. Natl. Acad. Sci. USA. 98: 14072–14077
Incardona JP and Eaton S (2000) Cholesterol in signal transduction. Curr. Opin. Cell Biol. 12: 193–203
Kranenburg O, Verlaan I and Moolenaar WH (2001) Regulating c-Ras function. Cholesterol depletion affects caveolin association, GTP loading, and signaling. Curr. Biol. 11: 1880–1884
Muller G, Hanekop N, Wied S and Frick W (2002) Cholesterol depletion blocks redistribution of lipid raft components and insulin-mimetic signaling by glimepiride and phosphoinositolglycans in rat adipocytes. Mol. Med. 8: 120–136
Zhuang L, Lin J, Lu ML, Solomon KR and Freeman MR (2002) Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 62: 2227–2231
Ushio-Fukai M, Hilenski L, Santanam N, Becker PL, Ma Y, Griendling KK and Alexander RW (2001) Cholesterol depletion inhibits epidermal growth factor receptor transactivation by angiotensin II in vascular smooth muscle cells: role of cholesterol-rich microdomains and focal adhesions in angiotensin II signaling. J. Biol. Chem. 276: 48269–48275
Parpal S, Karlsson M, Thorn H and Stralfors P (2001) Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J. Biol. Chem. 276: 9670–9678
Vereb G, Matko J, Vamosi G, Ibrahim SM, Magyar E, Varga S, Szollosi J, Jenei A, Gaspar Jr R, Waldmann TA and Damjanovich S (2000) Cholesterol-dependent clustering of IL-2Ralpha and its colocalization with HLA and CD48 on T lymphoma cells suggest their functional association with lipid rafts. Proc. Natl. Acad. Sci. USA 97: 6013–6018
Kabouridis PS, Janzen J, Magee AL and Ley SC (2000) Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur. J. Immunol. 30: 954–963
Huo H, Guo X, Hong S, Jiang M, Liu X and Liao K (2003) Lipid rafts/caveolae are essential for insulin-like growth factor-1 receptor signaling during 3T3-L1 preadipocyte differentiation induction. J. Biol. Chem. 278: 11561–11569
Smith PJ, Wise LS, Berkowitz R, Wan C and Rubin CS (1988) Insulin-like growth factor-I is an essential regulator of the differentiation of 3T3-L1 adipocytes. J. Biol. Chem. 263: 9402–9408
Modan-Moses D, Janicot M, McLenithan JC, Lane MD and Casella SJ (1998) Expression and function of insulin/insulin-like growth factor 1 hybrid receptors during differentiation of 3T3-L1 preadipocytes. Biochem. J. 333: 825–831
Kadowaki T, Tobe K, Honda-Yamamoto R, Tamemoto H, Kaburagi Y, Momomura K, Ueki K, Takahashi Y, Yamauchi T, Akanuma Y and Yazaki Y (1996) Signal transduction mechanism of insulin and insulin-like growth factor-1. Endocr. J. 43: S33–41
Myers Jr MG, Grammer TC, Wang LM, Sun XJ, Pierce JH, Blenis J and White MF (1994) Insulin receptor substrate-1 mediates phosphatidylinositol 3'-kinase and p70S6k signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. J. Biol. Chem. 269: 28783–28789
Zhang D and Brodt P (2003) Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene 22: 974–982
Banno Y, Takuwa Y, Yamada M, Takuwa N, Ohguchi K, Hara A and Nozawa Y (2003) Involvement of phospholipase D in insulin-like growth factor-I-induced activation of extracellular signal-regulated kinase, but not phosphoinositide 3-kinase or Akt, in Chinese hamster ovary cells. Biochem. J. 369: 363–368
Dunn SE, Torres JV, Oh JS, Cykert DM and Barrett JC (2001) Up-regulation of urokinase-type plasminogen activator by insulin-like growth factor-I depends upon phosphatidylinositol-3 kinase and mitogen-activated protein kinase kinase. Cancer Res. 61: 1367–1374
Kulik G, Klippel A and Weber MJ (1997) Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell Biol. 17: 1595–1606
Webster J, Prager D and Melmed S (1994) Insulin-like growth factor-1 activation of extracellular signal-related kinase-1 and -2 in growth hormone-secreting cells. Mol. Endocrinol. 8: 539–544
Vlahos CJ, Matter WF, Hui KY and Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269: 5241–5248
Dudley DT, Pang L, Decker SJ, Bridges AJ and Saltiel AR (1995) A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 92: 7686–7689
Chang WJ, Rothberg KG, Kamen BA and Anderson RGW (1992) Lowering the cholesterol content of MA104 cells inhibits receptor-mediated transport of folate. J. Cell Biol. 118: 63–69
Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR and Anderson RGW (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68: 673–682
Hailstones D, Sleer LS, Parton RG and Stanley KK (1998) Regulation of caveolin and caveolae by cholesterol in MDCK cells. J. Lipid Res. 39: 369–379
Qiu Z, Wei Y, Chen N, Jiang M, Wu J and Liao K (2001) DNA synthesis and mitotic clonal expansion is not a required step for 3T3-L1 preadipocyte differentiation into adipocytes. J. Biol. Chem. 276: 11988–11995
Mackall JC, Student AK, Polakis SE and Lane MD (1976) Induction of lipogenesis during differentiation in a ‘preadipocyte’ cell line. J. Biol. Chem. 251: 6462–6464
Rosen OM, Smith CJ, Hirsch A, Lai E and Rubin CS (1979) Recent studies of the 3T3-L1 adipocyte-like cell line. Recent Prog. Horm. Res. 35: 477–499
Student AK, Hsu RY and Lane MD (1980) Induction of fatty acid synthetase synthesis in differentiating 3T3-L1 preadipocytes. J. Biol. Chem. 255: 4745–4750
Jin S, Zhai B, Qiu Z, Wu J, Lane MD and Liao K (2000) c-Crk, a substrate of the insulin-like growth factor-1 receptor tyrosine kinase, functions as an early signal mediator in the adipocyte differentiation process. J. Biol. Chem. 275: 34344–34352
Sui G, Soohoo C, Affar EB, Gay F, Shi Y, Forrester WC and Shi Y (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA 99: 5515–5520
Lin FT and Lane MD (1992) Antisense CCAAT/enhancer-binding protein RNA suppresses coordinate gene expression and triglyceride accumulation during differentiation of 3T3-L1 preadipocytes. Genes Dev. 6: 533–544
Liao K and Lane MD (1995) The blockade of preadipocyte differentiation by protein-tyrosine phosphatase HA2 is reversed by vanadate. J. Biol. Chem. 270: 12123–12132
Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M and Lisanti MP (1996) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J. Biol. Chem. 271: 9690–9697
Acknowledgements
This work was supported by Chinese National Nature Sciences Foundation Research Grants 30270336 and 90208007, Chinese Academy of Sciences Grant KSCX2-SW-203, and the Ministry of Science and Technology Grants 2002CB513000. We thank Ms. Shan Hongying of Institute of Biochemistry and Cell Biology and Ms Jin Feng of Institute of Bio-product for assistance in electron microscope.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Dr G Melino
Rights and permissions
About this article
Cite this article
Hong, S., Huo, H., Xu, J. et al. Insulin-like growth factor-1 receptor signaling in 3T3-L1 adipocyte differentiation requires lipid rafts but not caveolae. Cell Death Differ 11, 714–723 (2004). https://doi.org/10.1038/sj.cdd.4401405
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401405
Keywords
This article is cited by
-
Unraveling the rationale and conducting a comprehensive assessment of KD025 (Belumosudil) as a candidate drug for inhibiting adipogenic differentiation—a systematic review
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
The Cross-Talk Between Sphingolipids and Insulin-Like Growth Factor Signaling: Significance for Aging and Neurodegeneration
Molecular Neurobiology (2019)
-
Intake of Xylooligosaccharides Alters the Structural Organization of Liver Plasma Membrane Bilayer
Food Biophysics (2014)
-
Neutral sphingomyelinase 2 modulates cytotoxic effects of protopanaxadiol on different human cancer cells
BMC Complementary and Alternative Medicine (2013)
-
Cocoa polyphenols suppress adipogenesis in vitro and obesity in vivo by targeting insulin receptor
International Journal of Obesity (2013)
