
Abstract
The Fusarium wilt disease caused by Fusarium oxysporum f. sp. batatas (Fob) is one of the devastating diseases of sweetpotato. However, the molecular mechanisms of sweetpotato response to Fob is poorly understood. In the present study, comparative quantitative proteomic analysis was conducted to investigate the defense mechanisms involved. Two sweetpotato cultivars with differential Fob infection responses were inoculated with Fob spore suspensions and quantitatively analyzed by Tandem Mass Tags (TMT). 2267 proteins were identified and 1897 of them were quantified. There were 817 proteins with quantitative ratios of 1.2-fold change between Fob-inoculated and mock-treated samples. Further, nine differentially expressed proteins were validated by Parallel Reaction Monitoring (PRM). According to Gene Ontology (GO) annotation information, the proteins functioned in molecular metabolism, cellular component formation, and biological processes. Interestingly, the results showed that sweetpotato resistant response to Fob infection included many proteins associated with signaling transduction, plant resistance, chitinase and subtilisin-like protease. The functions and possible roles of those proteins were discussed. The results provides first insight into molecular mechanisms involved in sweetpotato defense responses to Fob.
Similar content being viewed by others


Introduction
Sweetpotato (Ipomoea batatas L.) is one of the most important food crops in the world and has made great contributions to our food security. The latest statistics showed that in 2017, 9.20 Mha of Sweetpotato were planted in the world with a total production of 12.84 M tons. Currently, sweetpotato is mainly grown in Asia and Africa, In Africa, 4.72 Mha is cultivated with Sweetpotato which surpasses that of Asia 3.97 Mha1 .The production of sweetpotato is constrained by numerous biotic and abiotic stresses among which Fusarium wilt is one of the severest. The disease is caused by Fusarium oxysporum f. sp. batatas (Fob). It is one of the major factors that lead to the general sweetpotato yield loss of 10–50% in southern China2. One of the most effective measures of preventing Fusarium wilt is by growing cultivars that are resistant to the disease. Understanding the mechanisms of interaction and co-evolution between the sweetpotato and Fusarium wilt will be advantageous in improving the efficiency of breeding new Fusarium wilt resistant cultivars.
Interactions between crops and pathogens are complex. The gene-for-gene hypothesis proposed by Flor3 explains the relationships between crops and pathogenic microorganisms. According to Jones4, PAMP-triggered immunity (PTI) and effector-triggered immunity (ETI) are two main mechanisms of crop response to pathogenic microorganisms. The R protein can recognize diverse pathogen effectors directly or indirectly and then deliver the signal downstream by signal transducers such as kinases, transcription factors, phosphatases, and then induce pathogenesis-related proteins, these therefore, enhances serious defense reactions5. Many resistance (R) genes were identified and the mechanism of resistance in crops were elucidated by this mode6,7.
Recently, the rapid development of molecular biology and bioinformatics, the application of proteomic with high throughput sequencing technologies have enhanced research on protein–protein interaction between crops and pathogens, improving our understanding of the immune system of many crops. Comparative proteomic analysis was conducted to investigate the mechanisms of response to infectious pathogens in a range of crops such as rice, alfalfa, and peanut8,9,10. In sweetpotato, proteomics comparisons were performed to understand the differences between pencil and storage roots, light orange- and purple-fleshed storage roots, contrasting ecotypes, and how the crop responds to drought stress and root-knot nematode11,12,13,14,15. However, the differential proteomics of sweetpotato with different resistance response to Fob resistance remains unclear.
In this study, quantitative proteomics were conducted for a comprehensive understanding of the response of sweetpotato to Fob. Tandem mass tags (TMT) combined with UPLC-MS/MS were used with nine interested proteins. The results of this study widens our understanding of the molecular mechanisms of sweetpotato resistance to Fob.
Results
Quantitative analysis of sweetpotato defense response to fob
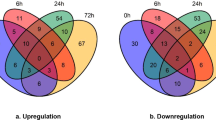
The integrated approach involving TMT labeling and LC–MS/MS was used to quantify the dynamic changes of the whole proteomes in three biological replicates of two sweetpotato cultivars JS57 and XZH. They were exposed to challenged by the Fusarium oxysporum f. sp. batatas pathogenic strain Fob-07. A total of 2,267 proteins were identified in the three replicates identified and 1,897 were quantified. In order to exploit the potential proteins with special functions related to disease resistance, the fold-change cutoff was set. The proteins with quantitative ratios above 1.2 or below 0.833 are deemed significant. Among the quantified proteins, the amounts of up- or down-regulated proteins in the comparison groups of JS57-07/JS57-CK and XZH-07/XZH-CK in different change folds were similar. Comparisons were also made the comparison between the two comparison groups (JS57-07/JS57-CK) and (XZH-07/XZH-CK) to find out the difference in resistant and susceptible cultivars. The numbers of up- or down-regulated proteins filtered with the threshold value of expression fold change and p value < 0.05 were displayed (Table 1).
To further understand the functions and features of the identified and quantified proteins, the function or feature of proteins from several different categories, including Gene Ontology, Domain, Pathway, and Subcellular Localizations were annotated. 2267 proteins were identified, quantifiable and annotated. GO annotation information of identified proteins was used to sum up the amount of the differentially expressed proteins in each GO term of level 2. The results showed that the identified proteins covered the range of molecular functions, cellular components and the biological processes. In the molecular functions, the major functional categories identified are binding (1562), catalytic activity (1478), structural molecule activity (173), transporter activity (136) and antioxidant activity (54). In the cellular components, the major categories are cell (1885), organelle (1435), membrane (811), macromolecular complex (514) and symplast (178). In the biological process, the major functional categories identified are cellular process (1607), metabolic process (1600), single-organism process (1289), response to stimulus (564), localization (298), biological regulation (293) and cellular component organization or biogenesis (228).
Analysis of differential expressed of proteins in sweetpotato response to fob
JS57-07 vs JS57-CK
Based on the GO annotation information of identified proteins (Table 2), the amount of the differentially expressed proteins in each GO term of level 2 were summed up. The results showed that a total of 1,086 proteins were up-regulated in the resistant cultivar JS57 in response to Fob07, which belonged to 22 GO terms with the lowest p value for 9 biological processes, 9 cellular components and 4 molecular functions. The most enriched GO terms among the 22 GO terms were the catalytic activity (132 proteins), followed by metabolic process (131 proteins), cell (118 proteins), binding (118 proteins), single-organism process (111 proteins) and cellular process (104 proteins). There were 1,563 proteins that were down-regulated and they belonged to 24 GO terms with the lowest p value for 10 biological processes, 8 cellular components and 6 molecular functions. The most enriched GO terms among the 24 GO terms were the cell (192 proteins), followed by cellular process (178 proteins), metabolic process (171 proteins), binding (160 proteins), and catalytic activity (154 proteins).
KEGG pathway-based enrichment analysis16 revealed that 11 pathways with the lowest p value were up-regulated in JS57 in response to Fob07, and 5 pathways were down-regulated. The most enriched up-regulated pathways were the Valine, leucine and isoleucine degradation, followed by 2-oxocarboxylic acid metabolism and glycine, serine, and threonine metabolism. There were 5 down-regulated pathways among which amino sugar and nucleotide sugar metabolism were the most enriched ones, followed by photosynthesis and fructose and mannose metabolism.
XZH-07 vs XZH-CK
From the GO annotation information of the identified proteins (Table 2), a total of 1091 proteins were found to be up-regulated in the susceptible cultivar XZH in response to Fob07 which belonged to 23 GO terms with the lowest p value for 8 biological processes, 9 cellular components and 6 molecular functions. The most enriched GO terms among the 23 GO terms were the catalytic activity (142 proteins), followed by metabolic process (134 proteins), cell (121 proteins) and binding (113 proteins) (Table 2). There were 1,223 proteins down-regulated which belonged to 23 GO terms with the lowest p value for 10 biological processes, 8 cellular components and 5 molecular functions. The most enriched GO terms among the 23 GO terms were cell (156 proteins), followed by cellular process (150 proteins), metabolic process (135 proteins), binding (128 proteins), and catalytic activity (115 proteins) (Table 2).
KEGG pathway-based enrichment analysis16 revealed that 7 pathways with the lowest p value were up-regulated in XZH in response to Fob07, and 5 were down-regulated. The most enriched up-regulated ones were valine, leucine and isoleucine degradation, followed by 2-oxocarboxylic acid metabolism and citrate cycle (TCA cycle). There were 5 down-regulated pathways observed among which porphyrin and chlorophyll metabolism were the most enriched ones, followed by amino sugar and nucleotide sugar metabolism and photosynthesis.
JS57-07/JS57-CK vs XZH-07/XZH-CK
In order to find out the differences in response to Fob07 between the two different resistant cultivars JS57 and XZH, the two different proteomics of A (JS57-07 vs JS57-CK) and B (XZH-07 vs XZH-CK) were compared (Table 2). It was found that in the comparisons of A vs B, there were totally 183 up-regulated proteins in total that belonged to 21 GO terms with the lowest p value for 10 biological processes, 7 cellular components and 4 molecular functions. The most enriched GO terms among them were catalytic activity (22 proteins) and metabolic process (22 proteins), followed by cell (21 proteins), cellular process (20 proteins), binding (18 proteins), and single-organism process (17 proteins). It was also observed that there were 356 down-regulated proteins which belonged to 21 GO terms with the lowest p value of 10 biological processes, 7 cellular components and 4 molecular functions. The most enriched GO terms among the 21 GO terms were catalytic activity (46 proteins), followed by metabolic process (43 proteins), single-organism process (40 proteins), cell (38 proteins) and cellular process (38 proteins) (Table 2).
KEGG pathway-based enrichment analysis16 revealed that 3 pathways with the lowest p value were up-regulated in A when compared to B, and 5 were down-regulated. The 3 enriched up-regulated pathways were phenylpropanoid biosynthesis, alanine, aspartate and glutamate metabolism, arginine biosynthesis, respectively. The 5 enriched down-regulated pathways were amino sugar and nucleotide sugar metabolism, alanine, aspartate and glutamate metabolism, glycine, serine and threonine metabolism, valine, leucine and isoleucine degradation, ascorbate and aldarate metabolism, respectively.
Validation by parallel reaction monitoring (PRM) of interested proteins
In order to validated the accuracy of the proteomes using TMT label-based quantitative analysis technique, nine proteins were selected for analysis by PRM (Table 3), Q56YA5 (Serine-glyoxylate aminotransferase), Q6SYB9 (S-adenosylmethionine synthase), P24805 (Stem-specific protein), Q0JF58 (Argonaute 4B), Q9SRH6 (Hypersensitive-induced response protein 3), Q9FYV1 (Inositol-3-phosphate synthase), P13046 (Pathogenesis-related protein R), Q96518 (Peroxidase 16) and O04887 (Pectinesterase 2). The correlation of protein expression levels quantified by TMT and by PRM were compared to examine the accuracy of the data obtained by these two methods. Bivariate correlation analysis was used to calculate the Pearson correlation coefficient among the proteins. The results showed that all of the nine proteins were significantly correlated when both the TMT and by PRM were used to validate the data (r2 > 0.8, p < 0.01) (Table 3).
The results obtained (Table 4) showed that the mean expression levels of these proteins were all higher in the cultivars of JS57 and XZH when challenged with Fob-07 than those in the CKs, and the trends were the same when the proteins were quantified by TMT and PRM. However, the range of up-regulated proteins varied for differentially expressed proteins (DEPs), especially for the proteins P13046 and O04887, the ranges quantified by PRM were much higher than those quantified by TMT.
Identification of the proteins involved in the defense system of sweetpotato response to fob
Sets of proteins that are differentially expressed and directly involved in the immune systems of sweetpotato were differentially expressed was identified (Table 5). Among the eight proteins associated with signaling transduction, three proteins, i.e. Linoleate 9S-lipoxygenase, Auxin-induced protein, Aldo–keto reductase were up-regulated and one protein, i.e. Inositol-3-phosphate synthase was down-regulated in both in the two cultivars. One calcium-binding protein CML13 was down-regulated in the resistant cultivar JS57, while Phospholipase A1 and another Linoleate 9S-lipoxygenase 1 were highly up-regulated extremely. Intriguingly, we found the Aldo–keto reductase 4 was found to be up-regulated in JS57 while it was down-regulated in XZH, suggesting that it may play an important role in the defense response of sweetpotato to Fob.
A number of defense-related proteins differentially regulated were screened, they include the PR, chitinase, Subtilisin-like protease, Peroxidase, Pectinesterase and so on. Most of them were found to be up-regulated in both the two cultivars. Five were found to be up- or down-regulated in JS57 only. Interestingly, two subtilisin-like proteases were found not to be up- or down-regulated after exposed to Fob, but the levels were higher in JS57 than those of XZH which implies the subtilisin-like protease may contribute to the defense system acting as PTI.
Discussion
One of the most effective ways to control plant diseases is by breeding resistant cultivars for use in production and cultivation. Understanding the mechanisms of plant’s resistance to diseases and identifying resistance genes helps to promote the breeding process. There exists a complicated interactions between crops and pathogens. The crops recognize invading pathogens by receptors and trigger defense responses by activating signaling transduction through MAP kinases (MAPKs), oxidative burst, ion influx which induces expression of defense-related genes17.
In this study, the quantitative proteomics was used and a number of defense-related proteins were identified in sweetpotato and how they respond to Fob. Some proteins were found to be associated with signal transduction and were differentially expressed between the resistant cultivar JS57 and the susceptible cultivar XZH, which may contribute to their defense response. It was also discovered that there were two aldo–keto reductases which may have different roles in the response of sweetpotato to Fob infection:the aldo–keto reductases 1 was up-regulated both in JS57 and XZH, and the aldo–keto reductases 4, was up-regulated in JS57 while down-regulated in XZH. The aldo–keto reductases belong to a superfamily that contains more than 190 members associated with carbonyl substrates reduction in all phyla18. In plants the aldo–keto reductase may enhance tolerance to various abiotic stresses. One of the aldo–keto reductase genes from sweetpotato was found to transfer high tolerance of cadmium stress in tobacco19. In the current study, it was discovered that aldo–keto reductases also contributed to Sweetpotato tolerance to biotic stress (Fob) and the aldo–keto reductases 4 may have special roles since it was differentially regulated in JS57 and XZH.
Mitogen-activated protein kinases (MAPKs) are important signal transducing enzymes that are involved in many aspects of cellular regulations20. Phospholipases may be activated in response to various cellular and environmental cues and affect various cellular processes through their roles in signal transduction. In this study, Phospholipase A1 was fond to be highly up-regulated (31.18 times) in the susceptible cultivar XZH’s response to Fob only. This explains its important role in conferring resistance. Thus, the Phospholipase A1 needs to be better understood than other members of the family such as Phospholipase A2, D, C and B20.
Pectins are the major components of plant’s cell walls and play a key role in forming the first defense barrier against pathogen colonization. The degree of esterification of pectin, determined by Pectinesterase, affects the structure and properties of the cell wall which is involved in resistance to pathogens21. The abundance of fully de-esterified, low and partially methylesterified HGs Pectinesterase is related to pathogen susceptibility22. Overexpression of pectin methyl esterases inhibitor proteins reduces the activity or the expression level of pectin mechyl esterification resulting in higher resistance to pathogens23. Pectinesterase 1 was found to be down-regulated in resistant cultivar JS57’s response to Fob, which may have resulted from the structural stability of pectin in the sweetpotato’s cell walls. Intriguingly, we found another Pectinesterase 2 located in the vacuolar membrane was found to be up-regulated by 10.43 times in JS57. The levels in expressions of both Pectinesterases were unchanged in the susceptible cultivar XZH in its response to Fob. These results demonstrated that pectinesterases plays important roles in plant-pathogen interactions but the way each member of the family works is different.
It was reported that S-adenosylmethionine synthase (SAMs) catalyzes the biosynthesis of S-adenosylmethionine which serves as the methyl donor for pectin methylesterase24 and the precursor for the biosynthesis of ethylene25. This shows that SAMs is very essential to the biotic stress response of plants, as the pectin is one of the components of cell wall and ethylene is a key molecule in signal transduction.
Subtilisin-like proteases are a large family of enzymes consisting of many diverse members in plants with various biological functions including protein turnover, plant development and interactions with the environment. Recently, subtilases were found to be involved in pathogen resistance and plant immunity26. In this study, two subtilisin-like proteases of the subfamily 3, SBT3.11 and SBT3.9, with higher expressions in JS57 than in XZH were identified. However, protease was not found to increase in the response to Fob, and did not show any difference in the two cultivars. These indicated that they may play a significant role in the PTI.
From the current research, sets of proteins that are involved in the defense systems of sweetpotato to Fob were identified, their functions of which need to be further exploited. Previously, the same cultivars (JS57 than XZH) and pathogens (Fob) were used to identify disease resistance genes in sweetpotato by performing de novo transcriptome assembly and digital gene expression analysis27, but there were few identical genes/proteins found in the study.
Materials and methods
Plant and pathogen materials, treatment
Two sweetpotato cultivars, JS57, with high resistance, and XZH, highly susceptible to Fusarium wilt, were used as materials. These cultivars were the ones used to investigate the digital gene expression response against Fusarium oxysporum f. sp. batatas 27.
The seedlings of the two cultivars were grown in the field for 4 weeks and then freshly excised into a length of about 15 cm and prepared for subsequent treatment.
The pathogen strain F07 of Fusarium oxysporum f. sp. batatas was activated by growing on potato-saccharose-agar (PSA) medium in a Petri dish at 28 °C for about 1 weeks, and then moved into a new Petri dish with fresh PSA until the mycelia covered the surface of the medium. The spores were collected and prepared into conidia solutions of 1 × 107 conidia/mL in glass bottles, as described by Yang20. The sweetpotato seedlings were inoculated into the bottles with conidia solutions and water as a control and kept in growth chambers at 28 °C for 24 h. Then the basic stems with a length of 2 cm were excised as samples and immediately kept frozen in liquid nitrogen. The experiment was composed of three independent biological replicates with four treatments as follows: JS57 cultured in the Fob solution (JS57-07), JS57 cultured in water (JS57-CK), XZH cultured in the Fob solution (XZH-07), and XZH cultured in water (XZH-CK).
Protein isolation, digestion and TMT labeling
The seedling samples were grounded for 30 min in liquid nitrogen and then sonicated three times using a high intensity ultrasonic processor (Scientz) on ice in lysis buffer (8 M urea, 2 mM EDTA, 10 mM DTT and 1% Protease Inhibitor Cocktail). After the samples were centrifuged at 20,000×g at 4 °C for 10 min, the remaining debris was removed, and the protein was precipitated with exposure to 15% TCA for 4 h at − 20 °C. The supernatant was discarded after centrifugation at 4 °C for 3 min. The proteins were rinsed three times with cold acetone and then re-dissolved in a buffer and the concentration determined with a 2-D Quant kit following the manufacturer’s guide28.
For the digestion, the proteins solution were reduced with a 5 mM DTT for 30 min at 56 °C and alkylated with 11 mM IAA for 15 min at room temperature in darkness. For trypsin digestion, the proteins samples were diluted by adding 200 mM TEAB to urea concentration (less than 2 M). The trypsin was added at a ratio of 1:50 trypsin-to-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratio for the second 4 h-digestion. Approximately 100 μg protein from each of the samples was digested with trypsin for the subsequent experiments28.
After trypsin digestion, peptide was desalted by Strata X C18 SPE column (Phenomenex) and vacuum-dried. It was reconstituted in 1 M TEAB and processed according to the manufacturer’s protocol for 6-plex TMT kit. Briefly, one unit of TMT reagent (defined as the amount of reagent required to label 100 μg of protein) was thawed and reconstituted in 24 μl ACN. The peptide mixtures were then incubated for 2 h at room temperature and pooled, desalted and dried by vacuum centrifugation28.
HPLC fractionation and LC–MS/MS analysis
The samples were then fractionated into fractions by high pH reverse-phase HPLC using Agilent 300Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length). Peptides were first separated briefly with a gradient of 2% to 60% acetonitrile in 10 mM ammonium bicarbonate pH 9.0 over 80 min into 80 fractions. Then, the peptides were combined into 18 fractions and dried by vacuum centrifugation28.
The Peptides were dissolved in 0.1% FA and directly loaded onto a reversed-phase pre-column (Acclaim PepMap 100, Thermo Fisher Scientific). Peptide separation was performed using a reversed-phase analytical column (Acclaim PepMap RSLC, Thermo Fisher Scientific). The gradient was composed of an increase from 5 to 25% solvent B (0.1% FA in 98% ACN) over 26 min, 25% to 40% in 8 min and climbing to 80% in 3 min then holding at 80% for the last 3 min, all at a constant flow rate of 350 nL/min on an EASY-nLC 1000 UPLC system28.
The peptides were subjected to NSI source followed by a tandem mass spectrometry (MS/MS) in Q Exactive (Thermo Fisher Scientific) coupled online to the UPLC. The intact peptides were detected in the orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using NCE setting as 28; ion fragments were detected in the orbitrap at a resolution of 17,500. A data-dependent procedure that alternates between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 1E4 in the MS survey scan with 30.0 s dynamic exclusion. The electrospray voltage applied was 2.0 kV. Automatic gain control (AGC) was used to prevent overfilling of the orbitrap; 5E4 ions were accumulated for generation of MS/MS spectra. For MS scans, the m/z scan range was 350 to 1800. Fixed first mass was set at 100 m/z28.
Database search
The resulting MS/MS data were processed using MaxQuant with integrated Andromeda search engine (v.1.5.2.8). Tandem mass spectra were searched against SwissProt Green Plant database. Trypsin/P was specified as cleavage enzyme allowing up to 2 missing cleavages. Mass error was set at10 ppm for precursor ions and 0.02 Da for fragment ions. Carbamidomethyl on Cys, was specified as fixed modification and oxidation on Met and acetylation on protein N-term was specified as variable modifications. For the protein quantification method, TMT 6-plex was selected in Mascot. FDR was adjusted to < 1% at protein, peptide and PSM level29.
Validation by parallel reaction monitoring (PRM)
Nine proteins Q56YA5 (serine-glyoxylate aminotransferase), Q6SYB9 (S-adenosylmethionine synthase), P24805 (stem-specific protein), Q0JF58 (argonaute 4B), Q9SRH6 (hypersensitive-induced response protein 3), Q9FYV1 (inositol-3-phosphate synthase), P13046 (pathogenesis-related protein R), Q96518 (peroxidase 16), O04887 (pectinesterase 2) were selected for validation by Parallel Reaction Monitoring (PRM). The proteins of the sample were extracted and then digested by trypsin. The tryptic peptides were dissolved in 0.1% formic acid (solvent A), directly loaded onto a home-made reversed-phase analytical column (15-cm length, 75 μm i.d.). The gradient was composed of an increase from 6 to 23% solvent B (0.1% formic acid in 98% acetonitrile) over 38 min, 23% to 35% in 14 min and climbing to 80% in 4 min then holding at 80% for the last 4 min, all at a constant flow rate of 400 nL/min on an EASY-nLC 1000 UPLC system29.
The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q Exactive Plus (Thermo Fisher Scientific) coupled online to the UPLC. The electrospray voltage applied was 2.0 kV. The m/z scan range was 350 to 1000 for full scan, and intact peptides were detected in the Orbitrap at a resolution of 35,000. Peptides were then selected for MS/MS using NCE settings at 27 and the fragments were detected in the Orbitrap at a resolution of 17,500. A data-independent procedure that alternated between one MS scan followed by 20 MS/MS scans. Automatic gain control (AGC) was fixed at 3E6 for full MS and 1E5 for MS/MS. The maximum IT was set at 20 ms for full MS and auto for MS/MS. The isolation window for MS/MS was set at 2.0 m/z29.
The generated MS data were processed using Skyline (v.3.6). Peptide settings: enzyme was set as Trypsin [KR/P], Max missed cleavage set at 2. The peptide length was set at 8–25, Variable modification was set as Carbamidomethyl on Cys and oxidation on Met, and max variable modifications were set at 3. Transition settings: precursor charges were set as 2, 3, ion charges were set at 1, 2 and ion types were set as b, y, p. The productions were set as from ion 3 to last ion, the ion match tolerance was set at 0.02 Da29.
References
FAO. http://www.fao.org/statistics/en (2017).
Fang, S. A preliminary study on inheritance of fusarium wilt resistance of sweet potato cultivars. J. Fujian Agric. Univ. 26(4), 446–448 (1997).
Stahl, E. A. & Bishop, J. G. Plant–pathogen arms races at the molecular level. Curr. Opin. Plant Biol. 3(4), 299–304 (2000).
Jones, J. D. & Dangl, J. L. The plant immune system. Nature 444(7117), 323–329 (2006).
Gassmann, W. & Bhattacharjee, S. Effector-triggered immunity signaling: from gene-for-gene pathways to protein-protein interaction networks. Mol. Plant-Microbe Interact. 25(7), 862 (2012).
Takken, F. & Rep, M. The arms race between tomato and Fusarium oxysporum. Mol. Plant Pathol. 11(2), 309–314 (2010).
Imam, J. et al. Molecular screening for identification of blast resistance genes in North East and Eastern Indian rice germplasm (Oryza sativa L.) with PCR based makers. Euphytica 196(2), 199–211 (2014).
Yan, D. et al. Comparative proteomic analysis of susceptible and resistant rice plants during early infestation by small brown planthopper. Front. Plant Sci. 8, 1744 (2017).
Tian, D. et al. Proteomic analysis of the defense response to Magnaporthe oryzae in rice harboring the blast resistance gene Piz-t. Rice 11(1), 47 (2018).
Wang, Z. et al. Proteomic analysis reveals an aflatoxin-triggered immune response in cotyledons of Arachis hypogaea infected with Aspergillus flavus. J. Proteome Res. 11(5), 2739–2753 (2012).
Lee, J. J. et al. A comparative study of proteomic differences between pencil and storage roots of sweetpotato [Ipomoea batatas (L.) Lam]. Plant Physiol. Biochem. 87, 92–101 (2015).
Lee, J. J. et al. Comparative proteomic study between tuberous roots of light orange- and purple-fleshed sweetpotato cultivars. Plant Sci. 193–194(1), 120–129 (2012).
Shekhar, S. et al. Comparison of proteomic and metabolomic profiles of two contrasting ecotypes of sweetpotato (Ipomoea batata L.). J. Proteom. 143, 306–317 (2016).
Wang, B. et al. Proteomic analysis of roots in seedling stage of two sweetpotato varieties with different drought tolerance. J. Nuclear Agric. Sci. 31(2), 232–240 (2017).
Ha, J. et al. Comparative proteomic analysis of the response of fibrous roots of nematode-resistant and -sensitive sweet potato cultivars to root-knot nematode Meloidogyne incognita. Acta Physiol. Plant. 39(12), 262 (2017).
Kanehisa, M. et al. KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Wu, L. et al. Comparative proteomic analysis of the maize responses to early leaf senescence induced by preventing pollination. J. Proteom. 177, 75–87 (2018).
Penning, T. M. The aldo-keto reductases (AKRs): overview. Chem. Biol. Interact. 234, 236–246 (2015).
Huo, J. et al. A novel aldo-keto reductase gene, IbAKR, from sweet potato confers higher tolerance to cadmium stress in tobacco. Front. Agric. Sci. Eng. 5(2) (2018).
Yang, Z. et al. A rapid seedling assay for determining sweetpotato resistance to fusarium wilt. Crop Sci. 58, 1558–1565 (2018).
Lionetti, V., Cervone, F. & Bellincampi, D. Methyl esterification of pectin plays a role during plant–pathogen interactions and affects plant resistance to diseases. J. Plant Physiol. 169(16), 1623–1630 (2012).
Ma, L. et al. Wound-induced pectin methylesterases enhance banana (Musa spp. AAA) susceptibility to Fusarium oxysporum f. sp. cubense. J. Exp. Bot. 64(8), 2219–2229 (2013).
Volpi, C. et al. The ectopic expression of a pectin methyl esterase inhibitor increases pectin methyl esterification and limits fungal diseases in wheat. MPMI 24(9), 1012 (2011).
Lamblin, F. et al. Overexpression of a heterologous sam gene encoding S-adenosylmethioninesynthetase in flax (Linum usitatissimum) cells: consequences onmethylation of lignin precursors and pectins. Physiol. Plant. 112(2), 223–232 (2010).
Yang, S. & Hoffman, N. Ethylene biosynthesis and its regulation in higher plants. Annu. Rev. Plant Physiol 35(1), 155–189 (2003).
Figueiredo, J., Silva, M. S. & Figueiredo, A. Subtilisin-like proteases in plant defence: The past, the present and beyond. Mol. Plant Pathol. 19(4), 12567 (2017).
Lin, Y. et al. Transcriptome profiling and digital gene expression analysis of sweet potato for the identification of putative genes involved in the defense response against Fusarium oxysporum f. sp. batatas. PLoS ONE 12(11), e0187838 (2017).
Wang, H. et al. Quantitative analysis of the global proteome in peripheral blood mononuclear cells from patients with new-onset psoriasis. Proteomics 1, 1800003 (2018).
Wu, Q. et al. Ultra-deep lysine crotonylome reveals the crotonylation enhancement on both histones and non-histone proteins by SAHA treatment. J. Proteom. Res. 7, 380 (2017).
Acknowledgements
This work was financially sponsored by grants from the Fujian Provincial Department of Science & Technology (CN)(2017NZ0002-2).
Author information
Authors and Affiliations
Contributions
X.C., S.L., G.C. and N.N., contributed in condducting the experiment. S.L., Z.Y., B.H., C.B. and X.H. validated the data and did the analysis. X.C. administrated the project. All contributed in the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, S., Yang, Z., Huang, B. et al. Comparative proteomic analysis of the sweetpotato provides insights into response mechanisms to Fusarium oxysporum f. sp. batatas. Sci Rep 10, 21368 (2020). https://doi.org/10.1038/s41598-020-78557-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78557-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
