
Abstract
Sclerotinia stem rot (Sclerotinia sclerotiorum) is a major disease of Brassica oilseeds. As suitable donors to develop resistant cultivars are not available in crop Brassicas, we introgressed resistance from a wild Brassicaceae species, B. fruticulosa. We produced 206 B. juncea-B. fruticulosa introgression lines (ILs). These were assessed for pollen grain fertility, genome size variations and resistance responses to Sclerotinia following stem inoculations under disease-conducive conditions. Of these, 115 ILs showing normal fertility and genome size were selected for cytogenetic characterization using florescent genomic in situ hybridization (Fl-GISH). B. fruticulosa segment substitutions were indicated in 28 ILs. These were predominantly terminal and located on B-genome chromosomes. A final set of 93 highly fertile and euploid (2n = 36) ILs were repeat-evaluated for their resistance responses during 2014–15. They were also genotyped with 202 transferable and 60 candidate gene SSRs. Association mapping allowed detection of ten significant marker trait associations (MTAs) after Bonferroni correction. These were: CNU-m157-2, RA2G05, CNU-m353-3, CNU-m442-5, ACMP00454-2, ACMP00454-3, EIN2-3-1, M641-1, Na10D09-1 and Na10D11-1. This is the first time such a molecular mapping technique has been deployed with introgression lines carrying genomic segments from B. fruticulosa, and the first to show that they possess high levels of resistance against S. sclerotiorum.
Similar content being viewed by others


Introduction
Indian mustard (Brassica juncea) is an important winter season oilseed crop in India. It contributes nearly 28% of edible oil supplies there1. B. juncea is also cultivated in China and eastern Europe as oilseed, vegetable or condiment crop(s)2. With the development of canola quality B. juncea, possibilities exist for its further expansion to drier regions in Australia and Canada3.While enhancing yield remains a major breeding objective, protecting yield potential against disease threats is equally important. Such threats to B. juncea include diseases such as sclerotinia stem rot, alternaria blight, white rust and downy mildew. Of these, sclerotinia stem rot, caused by Sclerotinia sclerotiorum 4, is now a major threat to the continued sustainability of this crop across the mustard growing belt of India5,6,7. In severe cases, incidence of sclerotinia stem rot can be up to 92%8,9,10. Sclerotinia stem rot is also now recognized as a major yield limiting factor for B. napus in Canada, Europe, Australia and China11,12,13,14,15.
S. sclerotiorum has a vast host range worldwide16, 17. While no commercial crop Brassica varieties have been bred specifically for resistance to sclerotinia stem rot, some B. napus varieties with increased tolerance to this disease have been released in China18, 19. However, available level of resistance within the crop Brassicas is insufficient as a source to meet the level of resistance needed to effectively combat this pathogen in commercial varieties. Identifying sources of resistance in Brassica has been challenging due to variations in plant screening assays utilized and varied aggressiveness of S. sclerotiorum isolates20,21,22.
Studies on genetics of resistance show a complex picture of being either monogenic and/or polygenic depending on the plant species and material under investigation23,24,25,26. Quantitative trait loci (QTL) mapping method for polygenic resistance is helping to identify loci related to S. sclerotiorum resistance in several crop species like soybean, common bean, sunflower and B. napus 27,28,29,30,31,32,33. Several of the resistance-related QTLs were found to be associated with A (A02, A03, A09) or C (C02, C04, C06, C07, C09) genomes of B. napus. A, B and C genomes are associated with B. rapa, B. nigra and B. oleracea species, respectively34,35,36. Comparative mapping analysis with Arabidopsis thaliana further showed a candidate gene, BnaC.IGMT5, was concomitant with a major QTL SRC6 in B. napus. Co-localization of four QTLs for field resistance with three flowering time QTLs within an 8.1-cM region on the linkage group C02 was also reported37. However, the phenotypic variation explained by such QTLs is generally very small like in B. napus, ranging between 4–10% and 6–22%. Furthermore, Wei et al.37 identified 6 and 5 QTLs for Sclerotinia resistance in field and controlled conditions, respectively, and 17 QTLs for flowering time in B. napus 38. Identification of QTLs with such a low level of phenotypic variation can be attributed to a low-moderate level of resistance in the populations/parents used for mapping. In QTL mapping studies, there can be issues related to limited recombination events, low allele number and the need for longer research time. Association mapping is now rated highly in terms of mapping resolution, allele number and time-saving in establishing marker trait associations39, 40. However, there are very few reports of association mapping in B. juncea 41, despite its extensive usage for characterizing resistance to blackleg stem canker caused by Leptosphaeria maculans and for establishing genetic diversity and LD in 192 inbred lines by using 451 single locus SSRs42, 43. Separate QTLs for leaf and stem resistance against S. sclerotiorum have been reported in B. oleracea44. In other crops, like soybean, Iquira et al. genotyped and mapped 3 QTLs against Sclerotinia stem rot by GBS approach45.
Although, the taxonomic family Brassicaceae (Cruciferae) represents a huge pool of genetic diversity, only a few species such as Erucastrum gallicum, Capsella bursa-pastoris, E. cardaminoides, D. tenuisiliqua and B. fruticulosa and B. oleracea have been reported to carry high levels of resistance against this pathogen46,47,48,49,50. While there is only one report regarding introgression of genomic segments responsible for resistance from the wild species E. cardaminoides or D. tenuisiliqua into the cultivated B. juncea, subsequently it was also possible to introgress B. fruticulosa resistance into B. juncea (S. S. Banga, unpublished). In this communication, we report molecular characterization of B. juncea-B. fruticulosa introgression lines (ILs) using molecular cytogenetic studies and a set of transferable and candidate gene based SSR markers. We also report resistance-associated QTLs and the underlying candidate genes against S. sclerotiorum. This is the first time such a molecular mapping technique has been deployed with ILs carrying genomic segments from wild Brassicaceae species possessing high levels of resistance against S. sclerotiorum.
Results
Pollen grain fertility and genome size variations
Pollen grain fertility in the introgression set (AD-4), comprising 206 ILs, varied from 40% (IL-251) to 90% (IL-152, IL-215, IL-332) with a relatively greater number of genotypes clustering around 75% pollen fertility (Fig. 1). Twenty four lines showed intermediate pollen grain fertility. The estimation of DNA content in cell nuclei of B. juncea-B. fruticulosa ILs showed significant genome size variations (SE 2.08, CV 2.66). Generally, the 1C DNA content in the ILs varied from 870.42–1085.58 Mbp (Fig. 2). Natural B. juncea genotype, RLC-1 has an average genome size of 1045.482 Mbp. The largest genome size was found in IL-244 (1085.58 Mbp) followed by IL-100 (1060.152 Mbp). IL-217 (870.42 Mbp) had the smallest genome size.

Pollen grain fertility of introgression lines, showing variation from 40% to 90%.

Variation in genome size between introgression lines. Genome size varied from 870.42 to 1085.58 Mbp. Genotype RLC-1 had an average genome size of 1045.482 Mbp.
In situ–hybridization
One hundred and fifteen B. juncea-B. fruticulosa ILs with high pollen fertility and near normal genome size were selected for intensive molecular cytogenetic investigations. These were checked for chromosome number and also explored for the presence of B. fruticulosa introgression(s) using B. fruticulosa specific probes. We also used B. nigra probes to differentiate between A- or B-genomes of B. juncea, found to be harbouring fruticulosa introgressions. Out of 115 ILs investigated, 108 had normal euploid chromosome number (2n = 36). Of these, 28 ILs revealed large B. fruticulosa chromosome fragment substitutions. Segment substitutions were recorded on 2–6 recipient chromosomes in 28 ILs (Fig. 3A–F). Number and size of such alien chromosome fragment substitutions varied. Simultaneous staining with B. nigra specific probes revealed that B-genome chromosomes of B. juncea harboured 83.84% of the identified B. fruticulosa chromosome segment substitutions. Alien fragment substitutions were primarily terminal. Only a small fraction of introgressed fragments were located on intercalary positions. These showed normal chromosome pairing during meiosis.

Genomic in situ hybridization on mitotic spreads of introgression lines (ILs) of B. juncea (AABB). B genome is painted in red while B. fruticulosa introgressions are shown in green colour. (A) B. juncea with no introgression, (B) IL with two fruticulosa segment substitutions in the A-genome, (C) IL with three fruticulosa segment substitutions in the B-genome, (D) IL with three fruticulosa segment substitutions in the B-genome and one in A-genome, (E) IL with five fruticulosa segment substitutions in the B-genome, (F) IL with six fruticulosa segment substitutions in the A-genome.
Disease Expression
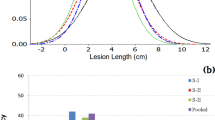
We first evaluated an extended set of 206 ILs along with resistant donor (B. fruticulosa) and susceptible recipient (B. juncea cv. RLC 1) parents for their responses to stem inoculation during 2011–12. Of these, a smaller set of 93 ILs were selected on the basis of high fertility and euploid chromosome number. These were repeat evaluated during 2014–15 for their responses to stem inoculation. Significant (P < 0.05) variations were indicated among the ILs in terms of expression of resistance responses to stem inoculation. Test genotypes were classified between highly susceptible to highly resistant classes (Fig. 4). Results were largely consistent over two crop seasons (Fig. 5). The LS mean value for stem lesion length in season I (SI) was 5.72 cm and for season II (SII) it was 3.66 cm. The corresponding values for the susceptible parent RLC-1 and resistant B. fruticulosa were 11.5, 11.75 and 0.62, 0.32 cm, during seasons I and II, respectively. Of the ILs evaluated, 38 and 65 ILs fell under the resistant category during two crop seasons. Among these, 13 genotypes showed a highly resistant response in season I and 29 genotypes in season II, with a mean lesion length less than 2.5 cm. Dunnet test established them to be significantly superior than the susceptible recipient, RLC 1. Frequency histogram revealed a marginal overrepresentation of the resistant class (Fig. 5). The analysis of variance revealed significant genotypic differences between the test ILs for their resistance responses to stem inoculation.

Resistance expression of introgression lines against Sclerotinia sclerotiorum. Introgression lines were grouped into five resistance categories. (A) Highly susceptible, (B) susceptible, (C) moderately resistant, (D) resistant, (E and F) hypersensitive resistance response; with stem lesion lengths ranging from >10.0; 7.5 to 10.0; 5.0 to <7.5; 2.5 to <5.0; and <2.5 cm, respectively.

The logarithmic values (% proportion) for 93 B. juncea-B. fruticulosa introgression lines in season I (2012–2013), season II (2013–2014), and pooled across both seasons I and II, as against K values. Histogram shows frequency of ILs in season I and season II against Sclerotinia sclerotiorum.
Genetic diversity, population structure, linkage disequilibrium
ILs along with two parents were genotyped with a set of 262 SSRs. These included 202 transferable SSRs, obtained from A- or B-genomes of crop Brassicas. Remaining 60 SSRs were developed on the basis of sequence information from 14 candidate genes, reportedly associated with sclerotinia resistance. These markers amplified 509 alleles. DNA polymorphism data were then used to establish genetic diversity in the given set of ILs. Diversity tree based on the SSR markers formed four clusters on the basis of Gower’s similarity coefficient (Fig. 6A). These four clusters included 29, 25, 27 and 12 genotypes, respectively. Wild donor species B. fruticulosa was included in cluster one, while the recipient B. juncea parent RLC 1 was present in cluster four. In terms of resistant genotypes, clusters three and four included maximum proportion(s) of resistant genotypes (Fig. 6B).

(A) Dendrogram based on Gower’s similarity index showing patterns of genetic variation in introgression lines. (B) Box plots showing average performance of introgression lines grouped in a given cluster.
As the genetic structure of the mapping panel is important for association mapping, we used STRUCTURE software to deduce the groups or clusters based on the average LnP(D) values. ∆K appeared maximum at K = 4, dividing the population into four distinct clusters, epitomized by different colors (Fig. 7). A total of 34 ILs was included in the cluster I, whereas clusters II, III and IV comprised of 26, 14 and 19 ILs, respectively. Most of the resistant and highly resistant introgression lines were included in population groups I and II (<4.0 LS-mean stem lesion length in pooled data). These groups are represented by green and blue colors respectively. In contrast groups III and IV constituted a mixture of moderately resistant, susceptible and highly susceptible lines.

Population structure of introgression lines based on 262 markers. The K is at 4 as the introgression lines clearly differentiated into three distinct clusters.
Association mapping
LS-means of phenotypic data were used for association studies. Of the four different models considered, GLM (P3) was the best fitted model, having observed P-values closest to the expected P values. Linkage disequilibrium (LD) was evaluated for global markers as well as significant markers in SI, SII and data pooled over two crop seasons. The R2 values of SSR markers were plotted against genetic distances. The R2 values of global SSRs in SI and SII were 0.013406 and 0.015688, respectively. For significant marker loci, R2 values were 0.1054 and 0.1241, respectively. Significant (P < 0.0001) LD blocks were observed for SSR markers EIN2-2, CNUm157, CNUm442,CNU_m353, NA10011, NA10009, RA2G05 (Supplementary Fig. S1). The markers with −log10 P value > 3.0 after Bonferroni correction were considered significantly associated with S. sclerotiorum resistance.
A total of 10 significant marker trait associations were detected across two seasons (Table 1). Of these, 5 marker loci were A or B-genome specific, 5 were with unknown marker positions. One marker loci, RAG2G05, exhibited association in season I as well as on the basis of data pooled over two seasons, having −log10P values varying from 3.01 to 3.45. This marker explained 10.0 to 13.06% of phenotypic variation, respectively. Other significantly associated markers were CNU_m353, ACMP00454 and M6341. These had −log10P values varying from 3.01 to 4.08 as observed during season II as well as for data pooled over two seasons. Phenotypic variation explained by these significant marker loci ranged from 10.51 to 15.28%. CNU_m157, Cnu_m442, EIN2-3, m6341, Na10D09 and Na10D11 were also involved in significant markers-trait-associations. Gene prediction analyses established the biological relevance of these trait markers associations as follows: CNU_m353 appeared associated with major intrinsic protein Aquaporine like (NIP3-1) and methyl transferase activity; Na10D11was co-related with cysteine type peptidase activity (Ulp-1protease family); and ACMP00454 seemed concomitant with the defensive AT hook motif nuclear localized protein.
Discussion
Introgressive breeding is an important technique to introduce novel traits into the cultivated forms. It is especially consequential, if the genetic variation for the desired trait is not available in the primary germplasm. B. juncea is generally highly susceptible to S. sclerotiorum. There is only one previous report demonstrating introgression of genomic segments responsible for resistance against S. sclerotiorum from the wild species E. cardaminoides or D. tenuisiliqua into the cultivated B. juncea 49 and a single unpublished study demonstrating successful introgression of B. fruticulosa resistance into B. juncea (S. S. Banga, unpublished). Advanced generation BC1S5 ILs, selected for the present studies were fully fertile and had euploid chromosome number. These were developed in the genetic background of a recipient B. juncea cv. RLC 1. The presence of a uniform background in ILs with different and homozygous introgressed donor segments allowed us to establish differences among the introgressed and native alleles. Molecular markers have been used in the past for mapping populations derived from distantly related crosses and many QTLs were identified51. IL analysis uses lines fixed for each QTL in the background of an elite breeding line, and can produce acceptable results. Molecular cytogenetic (Fl-GISH) coupled with molecular mapping technique helped to identify ILs carrying genomic segments from B. fruticulosa. These possessed high levels of resistance against S. sclerotiorum. Resistance-associated QTLs and the underlying candidates in introgression lines were successfully aligned with their resistance responses to stem inoculation with S. sclerotiorum. Ten significant marker trait associations were detected, of which five involved A-genome marker loci. The present study not only provides important new insights into the resistance mechanisms within B. juncea-B. fruticulosa introgression lines against S. sclerotiorum, but opens the way for novel engineering of new B. juncea varieties that express resistance that is critical to enabling better management of this worldwide devastating pathogen of rapeseed-mustard crops.
GISH studies involving B. juncea-B. fruticulosa introgression lines confirmed large B. fruticulosa introgression or segment substitution in 28 lines. These were predominantly terminal and ranged from 2–6 chromosome fragment substitution per IL. However, it was not possible to estimate exact size of introgressed fragments as Brassica chromosomes are very small and there were cell to cell variations for chromosome condensation patterns. Allosyndetic pairing, chromosome breakages and reunions are known to precipitate multiple translocations leading to introgression of genomic fragments from wild into crop genomes. Most of such chromosome fragment substitutions are expected to be terminal as these require only one break while intercalary fragment exchanges necessitates two breaks. Their location on B-genome chromosomes of B. juncea was probably due to strong homoeology that exist between B. nigra and B. fruticulosa; both belong to the same Sinapis lineage52,53,54. Random introgressions of uncharacterized DNA segments from unadapted wild germplasm are also likely to have structural and/or functional consequences for the recipient genome. This was apparent from low pollen grain fertility and genome size variations in many B. juncea-B. fruticulosa ILs carrying alien chromosomal fragments. Such variations primarily result from loss or gain of chromatin during initial generations following wide hybridization54, 55. The observed DNA changes were not simply unidirectional as upward as well as the downward shift in the genome size was indicated in the present study. However, average 1C genome size (1008.561 Mbp) of the ILs was very close to the genome size of natural B. juncea (1045.482 Mbp).
As expected in an advanced generation of backcross selfs, the ILs were phenotypically homogeneous, as introgressed genomic variation is expected to become stabilized and also Mendelized after many generations of selfing. While variation in pollen fertility observed in ILs may result from the genomic rearrangements expected during inter-specific hybridization, male sterility was the likely outcome of nucleo-cytoplasmic interaction between B. juncea and B. fruticulosa cytoplasm56, 57. Resistance responses of a subset of 91 ILs to S. sclerotiorum infestations revealed excellent variation for the trait investigated. These also suggested that several B. fruticulosa chromosome segments may affect resistance responses, and imply that gene(s) for resistance could be characterized and associated to specific marker alleles. Near normal distribution of resistance responses pointed towards a quantitative inheritance. Group memberships varied between clustering and population structure patterns. Such variations are expected because clustering is based on empirical groupings by similarity values and bootstrapping constancy whereas structure is model based, assuming the loci are at Hardy-Weinberg equilibrium and linkage equilibrium within populations.
Association mapping studies revealed that significant MTAs could explain up to 30% of the phenotypic variation in our introgression set, which possibly explained only a part of the available variation for resistance. In terms of biological relevance of identified MTAs: CNU_m353 could be linked to major intrinsic protein Aquaporine like (NIP3-1) and methyl transferase activity. This confirms a previous report suggesting its association with methyl-transferase activity58. ACMP00454 was related with the defensive AT hook motif nuclear localized protein. The AT hook motif nuclear localized protein is known to be involved in resistance against bacterial and oomycete pathogens59. AT hook motif containing transcription factor (CaAtl1) is associated with the defense response in transgenic tomato plants60. MTA involving EIN2-2 was detected only during the season II. The EIN-like proteins are known to be involved in regulation of biotic, abiotic and oxidative stresses, disease resistances and in regulation of ethylene responsive genes61. Combined use of genomic SSRs and candidate gene based SSRs can be a useful strategy, especially in situations where genomic resources from wild donor species are not available. While genome-wide association mapping surveys the genetic variation in the whole genome and locates signals of association for complex traits62, the candidate gene-based association mapping (CG-AM) allows a more focused approach and requires prior knowledge about gene associations involving a target phenotype63, 64. Extent of associations with identified markers underlined their role in defining introgressed resistance to the stem rot.
Summarizing, we believe that Sclerotinia resistance can be influenced by many uncharacterized genes contributing to overall phenotypic expression, as is commonly observed for quantitative traits. However, results from association mapping studies clearly demonstrated that B. juncea-B. fruticulosa ILs have significant potential breeding applications. This offers mustard breeders a powerful tool to optimize use of the genetic variation available within wild Brassicaceae by bringing together in one genotype the different alleles that maximize resistance to S. sclerotiorum for future mustard cultivars. Such novel engineering of new B. juncea varieties that express these critical resistances to S. sclerotiorum will enable more effective management of this worldwide devastating pathogen of mustard crops.
Methods
Plant materials
B. juncea-B. fruticulosa introgression set was developed by first hybridizing wild crucifer B. fruticulosa (2n = 16) with B. rapa (2n = 20). The synthetic amphiploid (FFAA; 2n = 36) was subsequently backcrossed twice with B. juncea (RLC-1) and advanced by following single seed descent method to develop a BC1S5–6 populations.
Pollen grain fertility
Percent pollen fertility was determined by staining pollen grains from freshly dehisced anther(s) in acetocarmine (2%) solution. For each line, >100 pollen grains were scored in different microscopic fields. Intensely stained and normal shaped pollen grains were scored as fertile while those unstained and/or collapsed were scored as sterile.
Nuclear DNA content
The DNA content was estimated by using Flow cytometry to determine the genome size variations. A reagent kit, PartecCyStain UV precise P, was used for nuclei extraction and DNA staining of nuclear DNA from plant tissues65. The method involves preparation of aqueous suspensions of intact nuclei by placing about 0.5 cm2 of young leaf tissue in a Petri dish and adding 400 µl of extraction buffer. The sample was finely chopped and filtered through a Partec 50 µm Cell Trics disposable filter into the flow cuvette. Staining solution (1.6 ml) was added to this supernatant and allowed to incubate at room temperature for 60 sec. The resultant sample was then analyzed with PartecCyFlowPloidy Analyzer with UV-laser excitation. A suspension of nuclei of CRBCs (chicken red blood cells) was used for calibration with PC-5 (B. carinata) as an internal reference standard. The DNA content of reference sample was calculated based on tomato Lycopersicon esculentum genotype “Stupickepolnityckoverane”, as an external reference standard. It had a 2C content of 1.96 pg. Two replications per each genotype were sampled for ploidy screening. The absolute DNA content of a sample was calculated based on the given formula where 1 pg corresponded to 0.978*109 bp66, 67. Reference genome size of Brassica juncea (http://www.Brassica.info/info/reference/genomesize.php) was used for comparison.
Chromosome preparation
To prepare mitotic slides, roots were first harvested from two days old germinating seeds raised on moist filter paper at 20–25 °C. Harvested roots (=30 mm) were treated with saturated aqueous solution of α-bromonaphthalene for 4 h, followed by fixation in alcohol: acetic acid (3: l) for at least 2 h at 4 °C. To prepare roots for in-situ hybridizations, these were first washed in a citrate buffer for 30 min to remove the fixative and subsequently incubated in the enzyme mixture containing 2% (w/v) cellulase and 20% (v/v) pectinase in 4 mmol citrate buffer (Citric acid monohydrate and trisodium citrate dehydrate), pH 4.8 for about 1 h at 37 °C. Treated roots were agitated with a micropipette tip to discharge mitotic cells in the micro-centrifuge tubes. These were centrifuged for 3 min at 600–800 g, followed by 45 min treatment in KCl (150 mmol) for 20 min. The cells were washed thrice at 800 g for 3 min in freshly prepared fixative to clear the cytoplasm. One drop of 7 µl of suspension was released on acid cleaned chilled slide from a height of 50 mm to spread the cells on the slide. Slides were air-dried in a desiccator before further use.
Probe preparation and in-situ hybridization
Purified DNA of B. fruticulosa, B. nigra and B. juncea were extracted using the DNeasy plant mini kit (Qiagen) according to manufacturer’s instructions. To prepare the probes, genomic DNA of B. fruticulosa was labelled with fluorescein-12-dUTPandgenomic and DNA of B. nigra was labelled with tetramethyl rhodamine-5-dUTP, using a nick translation kit (Roche, Germany) according to manufacturer instruction. GISH was performed in two step hybridization process. For first in situ hybridization, 40 μl of hybridization mixture containing 50% formamide, 2x SSC, 10% dextran sulphate, 0.025 μg salmon sperm DNA, 1.25 mM EDTA, 0.125% SDS, 200 ng of labelled B. fruticulosa probe and 100 fold B. juncea blocking DNA was applied on slides with good chromosome spreads. This was followed by incubation at 80 °C for 4 min in a thermocycler. These slides were kept in the hybridization chamber overnight at 37 °C. Slides were then washed at 42 °C for 2 min in 2x SSC and 5 min in 0.1x SSC (three times), respectively, and cooled to room temperature in 2x SSC for 5 min. After that, slides were incubated in 4x SSC in 1% tween-20 for 5 min. The chromosomes were stained in Vectashield mounting medium containing DAPI. The chromosome preparations were reused for the second GISH detection with B. nigra whole genomic probe without blocking DNA to identify genome specific introgression. Visualization was performed with Zeiss fluorescent microscope (ImagerZ2 AX10). Digital images were captured using Isis® software. Images were cropped and optimized using only functions affecting the whole image with Image J.
DNA extraction and SSR genotyping
Genomic DNA was harvested from young leaves using standard procedures for DNA extraction68 93 ILs, along with parents. Parental polymorphism assays were carried out using over 800 SSR primers, representing all the 18 B. juncea chromosomes. Of these, only 202 SSRs [164 markers for developed from A-genome69 and 38 from B-genome (sequences obtained on MTA from Isobel Parkin, Canada)] were polymorphic. PCR amplification of these markers was positive for B. fruticulosa but negative for recipient B. juncea parent. Fourteen candidate genes for S. sclerotiorum resistance in Arabidopsis thaliana 70, 71 were also used in this study. These included: SAH (S-adenosyl-L homocysteinase), CYP450 (Cytochrome P450), OMT (O-methyltransferase), Photosystem II polypeptide, MYB (Myeloblastosis), COI1 (Coronatine Insensitive 1), IGMT-5 (Indole glucosinolate methyl-transferase 5), NPR1 (Nonexpresser of Pr genes 1), EIN2 (Ethylene Insensitive 2), ABI1 (ABA Insensitive 1), ABI2 (ABA Insensitive 2), DET3 (De-etiolated 3), PAD3 (Phytoalexin Deficient 3) and LACS2 (long-Chain Acyl-CoA Synthetase 2)72, 73. The nucleotide sequences of identified defensive genes were retrieved from the TAIR (Arabidopsis Information Resource) database. The syntenic regions from A. thaliana genome were used to search for similar regions in B. rapa using nucleotide–nucleotide BLAST (blastn) algorithm74. Overlapping primers were designed for identified genes using BLAST tool provided by National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primer3 web software was used to design primers and perform BLASTN search in the selected database to check the specificity of the primers. The primers were designed with optimum melting temperature between 50–60 °C with GC content ranging from 30–60%. Product size varied from 250 to 500 bp. Designed primers were tested for specificity, amplification and optimum annealing temperature on the genomic DNA of both parents, RLC-1 and B. fruticulosa. The list of candidate genes, TAIR accession number along with gene functions and primers developed are presented in Tables S2 and S3. The amplification for total 262 polymorphic makers was carried out in a final volume of 10.33 µl, containing 25 ng of template DNA, primers; 1 µM each forward and reverse, dNTPs- 400 µm, 1 X PCR buffer with MgCl2 and Taq DNA polymerase 1 unit. The PCR reactions were performed in a 384 well Applied Biosystems thermocycler (Model no. EN61328). The amplified DNA product was fractioned using an automated high-throughput electrophoresis system (Caliper Lab Chip GX version 3.0.618.0). Allelic polymorphism of all the markers was recorded and mapping position was inferred from published data75. Markers with more than 25% of missing data were not used for further analysis.
Disease assessment
A set of 206 BC1S5 B. juncea genotypes were initially evaluated for their resistance response against S. sclerotiorum. The test genotypes were sown in a randomized complete block design with two replications during the 2011–12 winter growing season. Of these, a set of 93 genotypes (BC1S6) were selected on the basis of high pollen grain stainability, euploid chromosome number, normal meiosis and disease response. These were repeat-evaluated during 2014–15 winter growing season, in a randomized complete block design with two replications. Each test genotype was raised in 3 m long paired rows with a row-row spacing of 30 cm. High humidity was maintained by using adequately placed foggers (1 fogger/9 m2). Foggers are operated 2–3 times a day for 15 min or more, depending upon the prevailing weather conditions.
Sclerotinia sclerotiorum inoculum
Sclerotia of S. sclerotiorum isolate PAU-4, collected from infested B. juncea fields, were surface sterilized in 50% (v/v) sodium hypochlorite and 70% ethanol, each for 4 min, followed by two washes in sterile distilled water for 1 min76. The isolate was cultured on potato dextrose agar medium at 20 °C with 12/12 h light/dark. The daughter sclerotia were harvested from the incubated plates after four weeks and maintained at 4 °C for subsequent inoculations. The relative resistance responses across ILs were evaluated using a modified stem inoculation method77, 78. Ten random plants in the middle of each row were selected for inoculation when 50% of the plants in the row had achieved flowering. Each selected plant was inoculated by placing a single S. sclerotiorum mycelial plug disc (5–8 mm diameter), cut from the actively growing margin of a 48 h culture grown on a glucose-rich medium containing peptone (peptone 10 g, glucose 20 g, agar 18 g, KH2PO4 0.5 g, H2O 1 L, pH 6.0 before autoclaving), onto the stem immediately above the first node and fixing with Parafilm®. Stem lesion lengths were measured with a linear ruler after three weeks of inoculation as described previously79, 80. ILs were grouped into five resistance categories, viz. highly resistant (HR), resistant (R), moderately resistant (MR), susceptible (S) and highly susceptible (HS) with stem lesion lengths ranging from 0 to <2.5; 2.5 to <5.0; 5.0 to <7.5; 7.5 to 10.0 and >10.0 cm, respectively54. RLC-1 and B. fruticulosa were used as susceptible and resistant checks, respectively. LS-means of lesion length were calculated by PB Tools (http://bbi.irri.org/) for two seasons. Values were also pooled over seasons to determine appropriately adjusted means for the other effects in the model. Boxplot and Dunnett’s test method were used in ANOVA to create confidence intervals in pooled values for differences between the adjusted mean of each introgression line and highly susceptible genotype RLC-1 as a control. The test was carried out with family error rate (0.05) for all comparisons. Dunnett’s method determines the confidence levels for each individual comparison accordingly.
Genetic diversity, population structure and marker-trait-association
Paleontological statistics software package (PAST) was used to establish genetic diversity81 in the introgression set. We estimated the proportion of donor genome introgression in the ILs from the number of SSR markers. We calculated the marker-based value by dividing the total number of SSR markers for the donor genotype that were present in the ILs by the total number of SSR markers used for the SSR genotyping. The Bayesian model based program STRUCTURE 2.2 was used to infer the population structure and K inferred as number of populations82. Real number of clusters was inferred through an ad-hoc measure of K based on relative change in LnP (D) between successive K83. The software STRUCTURE Harvester was used to estimate the actual number of K. For association mapping84, resistance responses in terms of lesion length were standardized by representing lesion length of the test genotype as a proportion of the lesion length of susceptible check. Adjusted mean value lesion length data were used for downstream analysis, using four different models [GLM + K3, MLM + K3), GLM + PC3 and MLM + PC3] as implemented in TASSEL V 2.185. Q and PC were used as model covariates to reduce false associations. The Bonferroni correction threshold was calculated for 262 markers at 5% level of significance. As the probabilities were too small for analysis, they are converted into −log10P values. MEGANTE (https://megante.dna.affrc.go.jp/) was used for gene prediction analyses to establish biological relevance of identified MTAs.
References
Kumar, A. Production barriers and technological options for sustainable production of rapeseed-mustard in India. J. Oilseed Brassica 3, 67–77 (2012).
Rakow, G. Species Origin and Economic Importance of Brassica, Biotechnology in Agriculture and Forestry, Vol. 54, Brassica. In: (eds) E. C. Pua, and C. J. Douglas, Springer-Verlag Berlin Heidelberg (2004).
Oram, R. N. et al. Breeding Indian mustard [Brassica juncea (L.) Czern.] for cold-pressed edible oil production – a review. Aust. J. Agri. Res. 56, 581–596 (2005).
De Bary, A Comparative morphology and biology of the fungi, mycetozoa and bacteria. p. 525, The Clarendon Press, Oxford (1884).
Purdy, L. H. Sclerotinia sclerotiorum: history, diseases and symptomatology, host range, geographic distribution and impact. Phytopathol. 69, 887–890 (1979).
Bolton, M. D., Thomma, B. P. H. J. & Nelson, B. D. Sclerotinia sclerotiorum (Lib.) de Bary: biology and molecular traits of a cosmopolitan pathogen. Mol. Pl.Pathol. 7, 1–16 (2006).
Saharan, G. S. & Mehta, N. Sclerotinia diseases of crop plants biology, ecology and disease Management. Springer Science Business Media BV, Netherlands (2008).
Shivpuri, A., Sharma, K. B. & Chippa, H. P. Some studies on the stem rot diseases Sclerotinia sclerotiorum of rapeseed/mustard in Rajasthan India. J. Mycol. Pl. Pathol. 27, 29–31 (2000).
Sharma, S. K., Yadav, J. L. & Sharma, G. R. Effect of various agronomic practices on the incidence of white rot of Indian mustard caused by Sclerotinia sclerotiorum. J. Mycol. Pl. Pathol. 31, 83–84 (2001).
Yadav, M. S., Yadava, D. K., Nasim, A., Saroj, S. & Bambawale, M. Sclerotinia rot: A threat to rapeseed-mustard and virulence assessment of released varieties against Sclerotinia sclerotiorum. Proc. Nat. Symposium on Strategic Issues in Plant Pathological Research. pp. 202, Dept. Plant Pathology, Chaudhary Sarwan Kumar Himachal Pradesh Krishi Vishvavidyalaya, Palampur, Himachal Pradesh, India (2011).
McCartney, H. A., Lacey, M. E., Li, Q. & Heran, A. Airborne ascospore concentration and the infection of oilseed rape and sunflower by Sclerotinia sclerotiorum. In:Wratten N Salisbury P A (Eds.) Proc. 10thInt. Rapeseed Congress, Canberra, Australia.p.132 (1999).
Barbetti, M. J. & Khangura, R. Fungal diseases of canola in Western Australia. Bull. Agric. West. Aust. pp. 15 (2000).
Hind, T. L., Ash, G. J. & Murray, G. M. Prevalence of Sclerotinia stem rot of canola in New South Wales. Aust. J. Exp. Agric. 43, 163–168 (2003).
Sprague, S. & Stewart, W. S. Sclerotinia in canola results from petal and disease surveys across Victoria in 2001. In: Grains Research and Development Corporation Research Update Southern Region Australia, Grains Research and Development Corporation, Victoria, Australia (2002).
Zhao, J. et al. Quantitative trait loci for resistance to Sclerotinia sclerotiorum and its association with a homeologous non-reciprocal transposition in Brassica napus L. Theor. Appl. Genet. 112, 509–516 (2006).
Willets, H. J. & Wong, J. A. L. The biology of Sclerotinia sclerotiorum, Sclerotinia trifoliorum and Sclerotinia minor with emphasis on specific nomenclature. Bot. Rev. 46, 101–165 (1980).
Boland, G. J. & Hall, R. Index of plant hosts of Sclerotinia sclerotiorum. Can. J. Pl. Pathol. 16, 93–108 (1994).
Li, Y. et al. Breeding, inheritance, and biochemical studies on Brassica napus cv. Zhongyou 821: tolerance to Sclerotinia sclerotiorum (stem rot). In: Proc 10th Int. Rapeseed Congress, Canberra, Australia (1999).
Wang, H., Liu, G., Zheng, Y., Wang, X. & Yang, Q. Breeding of the Brassica napus cultivar Zhongshuang 9 with high-resistance to Sclerotinia sclerotiorum and dynamics of its important defense enzyme activity. Sci. Agric. Sin. 37, 23–28 (2004).
Barbetti, M. J. et al. Host resistance in oilseed Brassicas against Sclerotinia - renewed hope for managing a recalcitrant pathogen. In: Proc. 13th Int. Rapeseed Cong. June 5–9, 2011, Prague, Czech Republic, pp. 713–715 (2011).
Barbetti, M. J., Banga, S. S. & Salisbury, P. A. Challenges for crop production and management from pathogen biodiversity and diseases under current and future climate scenarios – case study with oilseed Brassicas. Field. Crops. Res. 127, 225–240 (2012).
Uloth, M. et al. New sources of resistance to Sclerotinia sclerotiorum for crucifer crops. Field. Crops. Res. 154, 40–52 (2013).
Abawi, G. S., Provvidenti, R., Crosier, D. C. & Hunter, J. E. Inheritance of resistance to white mold disease in Phaseolus coccineus. J. Hered. 69, 200–202 (1978).
Baswana, K. S., Rastogi, K. B. & Sharma, P. P. Inheritance of stalk rot resistance in cauliflower (Brassica oleraceavar. botrytis L.)Euphytica 57, 93–96 (1991).
Zhao, J. & Meng, J. Genetic analysis of loci associated with partial resistance to Sclerotinia sclerotiorum in rapeseed (Brassica napus L.). Theor. Appl. Genet. 106, 759–764 (2003).
Zhao, J., Peltier, A. J., Meng, J., Osborn, T. C. & Grau, C. R. Evaluation of Sclerotinia stem rot resistance in oilseed Brassica napus using a petiole inoculation technique under greenhouse conditions. Pl. Dis. 88, 1033–1039 (2004).
Hartman, G. L., Gardner, M. E., Hymowitz, T. & Naidoo, G. C. Evaluation of perennial Glycine species for resistance to soybean fungal pathogens that cause Sclerotinia stem rot and sudden death syndrome. Crop Sci. 40, 545–549 (2000).
Kim, H. S. & Diers, B. W. Inheritance of partial resistance to Sclerotinia stem rot in soybean. Crop Sci. 40, 55–61 (2000).
Gilmore, B., Myers, J. R. & Kean, D. Completion of testing of Phaseolus coccineus plant introductions for white mold Sclerotinia sclerotiorum resistance. Ann Rept Bean Improv.Coop. pp. 45 (2002).
Godoy, M., Castano, F., Re, J. & Rodriguez, R. Sclerotinia resistance in sunflower genotypic variations of hybrids in three environment of Argentina. Euphytica 145, 147–154 (2005).
Chen, W. et al. Detection of QTLs for six yield related traits in oilseed rape (Brassica napus) using DH and immortalized F2 populations. Theor. Appl. Genet. 115, 849–858 (2007).
Cheng, X. et al. Development and genetic mapping of micro satellite markers from genome survey sequences in Brassica napus. Theor. Appl. Genet. 118, 1121–1131 (2009).
Zhao, J. et al. Association mapping of leaf traits, flowering time, and phytate content in Brassica rapa. Genom. 50, 963–973 (2007).
Wu, J. et al. Identification of QTLs for resistance to Sclerotinia stem rot and BnaC.IGMTa as a candidate gene of the major resistant QTL SRC6 in Brassica napus. PLoS One 8, e67740 (2013).
Yin, X. et al. Mapping of QTLs detected in a Brassica napus DH population for resistance to Sclerotinia sclerotiorum in multiple environments. Euphytica 173, 25–35 (2010).
U, N. Genomic analysis in Brassica with special reference to the experimental formation of B. napus and its peculiar mode of fertilization. Jap. J. of Genet. 7, 784–794 (1935).
Wei, D. et al. Quantitative trait loci analyses for resistance to Sclerotinia sclerotiorum and flowering time in Brassica napus. Mol. Breed. 34, 1797–1804 (2014).
Arahana, V. S., Graef, G. L., Specht, J. E., Steadman, J. R. & Eskridge, K. M. Identification of QTLs for resistance to Sclerotinia sclerotiorum in soybean. Crop Sci. 41, 180–188 (2001).
Yu, J. & Buckler, E. S. Genetic association mapping and genome organization of maize. Curr.Opinion inBiotech. 17, 155–60 (2006).
Flint-Garcia, S. A., Thornsberry, J. M. & Buckler, E. S. Structure of linkage disequilibrium in plants. Ann Rev Pl. Biol. 54, 357–374 (2003).
Akhatar, J. & Banga, S. S. Genome wide association mapping for yield and root traits in Brassica juncea L. Czern and Coss. Mol. Breed. 35, 48–61 (2015).
Jestin, C. et al. Association mapping of quantitative resistance for Leptosphaeria maculans in oilseed rape (Brassica napus L.). Mol. Breed 27, 271–287 (2011).
Xiao, Y. et al. Genetic structure and linkage disequilibrium pattern of a rapeseed (Brassica napus L.) association mapping panel revealed by microsatellites. Theor. Appl. Genet. 125, 437–447 (2012).
Mei, J. et al. Identification of genomic regions involved in resistance against Sclerotinia sclerotiorum from wild Brassica oleracea. Theor.Appl Genet. 126, 549–56 (2013).
Iquira, E., Humira, S. & François, B. Association mapping of QTLs for Sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing approach. BMC Pl. Biol. 15, 5–16 (2015).
Lefol, E., Seguin-Swartz, G. & Downey, R. K. Sexual hybridisation in crosses of cultivated Brassicaspecies with the crucifers Erucastrumgallicum and Raphanus raphanistrum potential for gene introgression. Euphytica 95, 127–39 (1997).
Seguin-Swartz, G. & Lefol, C. Sclerotinia stem rot resistance in crucifers. In: (Eds) Wratten, N. Salisbury, P. A. Proc. 10th Int. Rapeseed Congress, Canberra, Australia. pp. 153 (1999).
Chen, H. F., Wang, H. & Li, Z. Y. Production and genetic analysis of partial hybrids in intertribal crosses between Brassica species (B. rapa, B. napus) and Capsellabursapastoris. Pl. Cell Res. 26, 1791–1800 (2007).
Garg, H. et al. High level of resistance to Sclerotinia sclerotiorum in introgression lines derived from hybridization between wild crucifers and the crop Brassica species Brassica napus and B. juncea. Field Crops Res. 117, 51–58 (2010).
Mei, J. et al. Identification of resistant sources against Sclerotinia sclerotiorum in Brassica species with emphasis on B. oleracea. Euphytica 177, 393–399 (2011).
Eshed, Y. & Zamir, D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141, 1147–1162 (1995).
Chandra, A., Gupta, M. L., Banga, S. S. & Banga, S. K. Production of an interspecific hybrid between B. fruticulosa and B. rapa. Pl. Breed. 123, 497–98 (2004).
Song, K. M., Osborn, T. C. & Williams, P. H. Brassica taxonomy based on nuclear restriction fragment length polymorphism (RFLPs) 3. Genome relationship in Brassica and related genera and the origin of B. oleracea and B. rapa (syn. campestris). Theor. Appl. Genet. 79, 497–506 (1990).
Warwick, S. I. & Black, L. D. Molecular systematics of Brassica and allied genera (Subtribe Brassicinae, Brassiceae): chloroplast genome and cytodeme congruence. Theor. Appl. Genet. 82, 81–92 (1991).
Lukens, L. N. et al. Patterns of sequence loss and cytosine methylation within a population of newly resynthesized Brassica napusallopolyploids. Pl. Physiol. 140, 336–348 (2006).
Chen, Z. J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Ann. Rev. Pl. Biol. 58, 377–406 (2007).
Gupta, M., Atri, C. & Banga, S. S. Cytogenetic stability and genome size variations in newly developed derived Brassica juncea allopolyploid lines. J. Oilseed Brassica 5, 18–127 (2014).
Prakash, S., Bhat, S. R., Quiros, C. R., Kirti, P. B. & Chopra, V. L. Brassica and its close allies: cytogenetics and evolution. Pl. Breed. Rev. 31, 21–161 (2009).
Ahuja, I., Ralph, K. & Bones, A. M. Phtyoalexins in defense against pathogens. Trends Pl. Sci 17, 73–90 (2012).
Kim, S. Y. et al. The chili pepper CaATL1: an AT-hook motif-containing transcription factor implicated in defence responses against pathogens. Mol. Pl. Pathol. 8, 761–71 (2007).
Cao, S. et al. The Arabidopsis Ethylene-Insensitive 2 gene is required for lead resistance. Pl Physiol Biochem. 47, 308–312 (2009).
Risch, N. & Merikangas, K. The future of genetic studies of complex human diseases. Science 273, 1516–1517 (1996).
Pflieger, S., Lefebvre, V. & Causse, M. The candidate gene approach in plant genetics: a review. Mol. Breed. 7, 275–291 (2001).
Hall, D., Tegstrom, C. & Ingvarsson, P. K. Using association mapping to dissect the genetic basis of complex traits in plants. Briefings in Funct Genomics 9, 157–165 (2010).
Dolezel, J. & Bartos, J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 95, 99–110 (2005).
Dolezel, J., Bartos, J., Voglmayr, H. & Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytometry 51, 127–128 (2003).
Suda, J., Kyncl, T. & Freiova, R. Nuclear DNA amounts in macaronesian angiosperms. Ann. Bot. 92, 153–164 (2003).
Doyle, J. J. & Doyle, J. L. Isolation of plant DNA from fresh tissue. Focus 12, 13–15 (1990).
Lowe, A., Moule, C., Trick, M. & Edwards, K. Efficient large scale development of microsatellites for marker and mapping applications in Brassica crop species. Theor. Appl. Genet. 108, 1103–1112 (2004).
Kim, H., Choi, S. R. & Bae, J. Sequenced BAC anchored reference genetic map that reconciles the ten individual chromosomes of Brassica rapa. BMC Genom. 10, 432 (2009).
Liu, R., Zhao, J., Xiao, Y. & Meng, J. Identification of prior candidate genes for Sclerotinia local resistance in Brassica napus using Arabidopsisc DNA microarray and Brassica-Arabidopsis comparative mapping. Sci in China Ser C: Life Sci. 48, 460–470 (2005).
Talukder, I. Z. et al. Candidate gene association mapping of Sclerotinia stalk rot resistance in sunflower (Helianthus annuus L.) uncovers the importance of COI1 homologs. Theor. Appl. Genet. 127, 193–209 (2014).
Guimaraes, R. L. & Stotz, H. U. Oxalate Production by Sclerotinia sclerotiorum deregulates guard cells during infection. Pl. Physiol. 136, 3703–3711 (2004).
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J. & Zhang, Z. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic. Acids. Res. 25, 3389–3402 (1997).
Lowe, A. J. et al. Transferability and genome specificity of a new set of microsatellite primers among Brassica species of the U triangle. Mol. Ecol. Notes 2, 7–11 (2002).
Clarkson, J. P., Staveley, J., Phelps, K., Young, C. S. & Whipps, J. M. Ascospore release and survival in Sclerotinia sclerotiorum. Mycol. Res. 107, 213–222 (2003).
Li, C. et al. Expression of field resistance under Western Australian conditions to Sclerotinia sclerotiorum in Chinese and Australian Brassica napus and Brassica juncea germplasm and its relation with stem diameter. Aust. J. Agric. Res. 57, 1131–1135 (2006).
Buchwaldt, L., Hegedus, D. D. & Rimmer, S. Pathogenesis of Sclerotinia sclerotiorum in relation to screening for resistance. In: Proc. 13th Int. Sclerotinia Workshop Monterey, CA, USA pp. 22 (2005).
Bradley, C. A., Henson, R. A., Porter, P. M., LeGare, D. G. & Khot, S. D. Response of canola cultivars to Sclerotinia sclerotiorum in controlled and field environments. Pl. Dis. 90, 215–219 (2006).
Li, C. et al. The importance of the type and time of inoculation and assessment in the determination of resistance in Brassica napus and B. juncea to Sclerotinia sclerotiorum. Aust. J. Agric. Res. 58, 1198–1203 (2007).
Hammer, Ø., Harper, D.A.T. & Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4(1): 9pp. http://palaeo-electronica.org/2001_/past/issue1_01 (2001).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genet. 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goud, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, A. D. & Vonholdt, B. N. Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conser. Genet. Res. 4, 359–361 (2012).
Bradbury, P. J. et al. Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Acknowledgements
Researches were carried out with the financial assistance from Department of Biotechnology, Government of India in form of Centre of Excellence and Innovation in Biotechnology “Germplasm enhancement for crop architecture and defensive traits in Brassica”. Initial pre-breeding material was developed with financial support from National Professor Project entitled “Broadening the genetic base of Indian mustard (Brassica juncea) through alien introgressions and germplasm enhancement” funded by Indian Council of Agricultural Research. Kusum Rana was supported by Department of Science and Technology (DST) in the form of an INSPIRE fellowship. Authors are also grateful to anonymous reviewers for helpful suggestions.
Author information
Authors and Affiliations
Contributions
S.S.B., K.R. and M.J.B. designed research. S.S.B. and C.A. constructed the genetic population. K.R., P.S.S. and R.J. performed stem inoculations and recorded the disease score. M.G. carried out cytogenetic studies. C.A. and K.R. performed genome size determinations. K.R. performed S.S.R. genotyping. S.S.B. and C.A. provided plant material. K.R., J.A. and N.K. conducted statistical analysis. K.R., S.S.B. and M.J.B. interpreted the data. K.R. and S.S.B. wrote the paper. M.J.B. edited the manuscript, S.S.B. supervised the whole study. All authors have read and approved the version of manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rana, K., Atri, C., Gupta, M. et al. Mapping resistance responses to Sclerotinia infestation in introgression lines of Brassica juncea carrying genomic segments from wild Brassicaceae B. fruticulosa . Sci Rep 7, 5904 (2017). https://doi.org/10.1038/s41598-017-05992-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05992-9
This article is cited by
-
Deciphering the genetic architecture of plant height and main shoot length in backcross introgression lines of Sinapis alba + Brassica juncea allohexaploids
Euphytica (2024)
-
Dissection of QTLs conferring drought tolerance in B. carinata derived B. juncea introgression lines
BMC Plant Biology (2023)
-
Identification of new resistance source for Sclerotinia stem rot in backcross population of B. juncea + S. alba allohexaploids: key to manage disease through host resistance
Euphytica (2023)
-
Assessment of Sclerotinia Stem and Leaf Rot Resistance and its Association with Physical Strength Attributes in Brassicaceae with Special Emphasis on Brassica Juncea
Journal of Plant Growth Regulation (2023)
-
Phenotypic characterization and resistance response to Sclerotinia sclerotiorum of backcross lines developed from stable allohexaploids of Sinapis alba + Brassica juncea
Euphytica (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
