
Abstract
The properties of electrons in matter are of fundamental importance. They give rise to virtually all material properties and determine the physics at play in objects ranging from semiconductor devices to the interior of giant gas planets. Modeling and simulation of such diverse applications rely primarily on density functional theory (DFT), which has become the principal method for predicting the electronic structure of matter. While DFT calculations have proven to be very useful, their computational scaling limits them to small systems. We have developed a machine learning framework for predicting the electronic structure on any length scale. It shows up to three orders of magnitude speedup on systems where DFT is tractable and, more importantly, enables predictions on scales where DFT calculations are infeasible. Our work demonstrates how machine learning circumvents a long-standing computational bottleneck and advances materials science to frontiers intractable with any current solutions.
Similar content being viewed by others

Introduction
Electrons are elementary particles of fundamental importance. Their quantum mechanical interactions with each other and with atomic nuclei give rise to the plethora of phenomena we observe in chemistry and materials science. Knowing the probability distribution of electrons in molecules and materials—their electronic structure—provides insights into the reactivity of molecules, the structure and the energy transport inside planets, and how materials break. Hence, both an understanding and the ability to manipulate the electronic structure in a material propels technologies impacting both industry and society. In light of the global challenges related to climate change, green energy, and energy efficiency, the most notable applications that require an explicit insight into the electronic structure of matter include the search for better batteries1,2 and the identification of more efficient catalysts3,4. The electronic structure is furthermore of great interest to fundamental physics as it determines the Hamiltonian of an interacting many-body quantum system5 and is observable using experimental techniques6.
The quest for predicting the electronic structure of matter dates back to Thomas7, Fermi8, and Dirac9 who formulated the very first theory in terms of electron density distributions. While computationally cheap, their theory was not useful for chemistry or materials science due to its lack of accuracy, as pointed out by Teller10. Subsequently, based on a mathematical existence proof5, the seminal work of Kohn and Sham11 provided a smart reformulation of the electronic structure problem in terms of modern density functional theory (DFT) that has led to a paradigm shift. Due to the balance of accuracy and computational cost it offers, DFT has revolutionized chemistry—with the Nobel Prize in 1998 to Kohn12 and Pople13 marking its breakthrough. It is the reason DFT remains by far the most widely used method for computing the electronic structure of matter. With the advent of exascale high-performance computing systems, DFT continues reshaping computational materials science at an even bigger scale14,15. However, even with an exascale system, the scale one could achieve with DFT is limited due its cubic scaling on system size. We address this limitation and demonstrate that an approach based on machine learning can predict electronic structures at any length scale.
In principle, DFT is an exact method, even though in practice the exchange-correlation functional needs to be approximated16. Sufficiently accurate approximations do exist for useful applications, and the search for ever more accurate functionals that extend the scope of DFT is an active area of research17 where methods of artificial intelligence and machine learning (ML) have led to great advances in accuracy18,19 without addressing the scaling limitation.
Despite these initial successes, DFT calculations are hampered inherently due to their computational cost. The standard algorithm scales as the cube of system size, limiting routine calculations to problems comprised of only a few hundred atoms. This is a fundamental limitation that has impeded large-scale computational studies in chemistry and materials science so far.
Lifting the curse of cubic scaling has been a long-standing challenge. Prior works have attempted to overcome this challenge in terms of either an orbital-free formulation of DFT20 or algorithmic development known as linear-scaling DFT21,22. Neither of these paths has led to a general solution to this problem. In recent years, researchers have delved into the potential of ML techniques to overcome the limitations of the DFT algorithm23. ML has proven to be a valuable tool in learning property mappings of both chemical systems24 and materials25. For example, it has been successfully employed to develop chemical descriptors enabling high-throughput screening of catalytic reactions26, investigate the relationship between the structure and properties of crystals27, and even discover stable materials28. While learning property mappings is computationally less expensive, it typically concentrates on specific properties relevant to particular applications, rather than providing a deep understanding of electronic structures. Another established area where ML has proven useful in predicting the properties of chemical and material systems is the development of interatomic potentials for classical molecular dynamics (MD) simulations. In this approach, ML effectively parametrizes the Born-Oppenheimer potential energy surface of molecules and solids by learning the mapping between the descriptor representation of atomic positions and the corresponding total energy and atomic forces. This enables large-scale MD simulations at an accuracy close to first-principles methods, such as DFT, while the computational overhead is still moderate. Gaussian approximation potentials29, spectral neighborhood analysis potentials30, moment tensor potentials31, and deep neural network potentials32,33 are among the most commonly used ML interatomic potentials. Lastly, ML has been utilized to learn the electronic structure itself, which is more computationally demanding than other approaches but provides the highest fidelity. Pioneering work in this area has focused on learning the electronic charge density using kernel-ridge regression34 or neural networks35,36. However, these works remain at the conceptual level and are only applicable to model systems, small molecules, and low-dimensional solids.
Despite all these efforts, computing the electronic structure of matter at large scales while maintaining first-principles accuracy has remained an elusive goal so far. We provide a solution to this long-standing challenge in the form of a linear-scaling ML surrogate for DFT. Our algorithm enables accurate predictions of the electronic structure of materials, in principle, at any length scale.
Results
Ultra-large scale electronic structure predictions with neural networks
In this work, we circumvent the computational bottleneck of DFT calculations by utilizing neural networks in local atomic environments to predict the local electronic structure. Thereby, we achieve the ability to compute the electronic structure of matter at any length scale with minimal computational effort and at the first-principles accuracy of DFT.
To this end, we train a feed-forward neural network M that performs a simple mapping
where the bispectrum coefficients B of order J serve as descriptors that encode the positions of atoms relative to every point in real space r, while \(\tilde{d}\) approximates the local density of states (LDOS) d at energy ϵ. The LDOS encodes the local electronic structure at each point in real space and energy. More specifically, the LDOS can be used to calculate the electronic density n and density of states D, two important quantities which enable access to a range of observables such as the total free energy A itself37, i.e.,
The key point is that the neural network is trained locally on a given point in real space and therefore has no awareness of the system size. Our underlying working assumption relies on the nearsightedness of the electronic structure38. It sets a characteristic length scale beyond which effects on the electronic structure decay rapidly with distance. Since the mapping defined in Eq. (1) is purely local, i.e., performed individually for each point in real space, the resulting workflow is scalable across the real-space grid, highly parallel, and transferable to different system sizes. Non-locality is factored into the model via the bispectrum descriptors, which are calculated by including information from adjacent points in space up to a specified cutoff radius consistent with the aforementioned length scale.
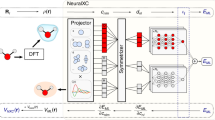
The individual steps of our computational workflow are visualized in Fig. 1. They include combining the calculation of bispectrum descriptors to encode the atomic density, training and evaluation of neural networks to predict the LDOS, and finally, the post-processing of the LDOS to physical observables. The entire workflow is implemented end-to-end as a software package called Materials Learning Algorithms (MALA)39, where we employ interfaces to popular open-source software packages, namely LAMMPS40 (descriptor calculation), PyTorch41 (neural network training and inference), and Quantum ESPRESSO42 (post-processing of the electronic structure data to observables).

ML models created via this workflow can be trained on data from popular first-principles simulation codes such as Quantum ESPRESSO42. The pictograms below the individual workflow steps show, from left to right, the calculation of local descriptors at an arbitrary grid point (green) based on information at adjunct grid points (gray) within a certain cutoff radius (orange), with an atom shown in red; a neural network; the electronic structure, exemplified here as a contour plot of the electronic density for a cell containing Aluminum atoms (red).
We demonstrate the effectiveness of our workflow by computing the electronic structure of a material sample comprising over 100,000 atoms. We selected a disordered system of Beryllium atoms at room temperature as our model system, owing to its suitability for accurately describing the system using DFT calculations. This enables us to evaluate the performance of our ML-based electronic structure prediction model. Despite the inherent accuracy of DFT for such systems, Beryllium’s electronic structure is still complex enough to be amenable to ML techniques. The employed ML model is a feed-forward neural network that is trained on simulation cells containing 256 Beryllium atoms. In Fig. 2, we showcase how our framework predicts multiple observables at previously unattainable scales. Here, we show an atomic snapshot containing 131,072 Beryllium atoms at room temperature into which a stacking fault has been introduced, i.e., three atomic layers have been shifted laterally, changing the local crystal structure from hcp to fcc. Our ML model is then used to predict both the electronic densities and energies of this simulation cell with and without the stacking fault. As expected, our ML predictions reflect the changes in the electronic density due to the changes in the atomic geometry. The energetic differences associated with such a stacking fault are expected to follow a behavior \(\sim {N}^{-\frac{1}{3}}\), where N is the number of atoms. By calculating the energy of progressively larger systems with and without a stacking fault, we find that this expected behavior is indeed obeyed quite closely by our model (Fig. 2b).

a Beryllium simulation cell of 131,072 atoms with a stacking fault, generated by shifting three layers along the y-axis creating a local fcc geometry, as opposed to the hcp crystal structure of Beryllium. The colors in the upper half correspond to the centrosymmetry parameter calculated by OVITO81, where blue corresponds to fcc and red-to-light-green to hcp local geometries. The lower half of the image, generated with VMD82, shows the difference in the electronic density for 131,072 Beryllium atoms with and without a stacking fault. b Energy differences due to introducing a stacking fault into Beryllium cells of differing sizes.
Our findings enable the development of models tailored to specific applications on scales previously unattainable with conventional DFT methods. Our ML predictions for the 131,072-atom system require just 48 min on 150 standard CPUs. The computational cost of our ML workflow is orders of magnitude lower than conventional DFT calculations, which scale as ~ N3. This renders simulations like the one presented here entirely infeasible using conventional DFT (see Fig. 3).

a Comparison of CPU time between conventional DFT (QuantumESPRESSO) and the MALA framework as a function of the number of atoms. Note that for consistency, slightly different computational parameters have been used for the DFT calculations here compared to the DFT reference calculations in Fig. 4. Additional details on the timings can be found in Table 2. b General workflow of size transferability in MALA.
Common research directions for utilizing ML in the realm of electronic structure theory either focus on predicting energies and forces of extended systems (ML interatomic potentials43) or directly predicting observables of interest such as polarizabilities44. MALA models are not limited to singular observables and even give insight into the electronic structure itself, from which a range of relevant observables including the total free energy, the density of states, the electronic density, and atomic forces follow.
The utility of our ML framework for chemistry and materials science relies on two key aspects. It needs to scale well with system size up to the 100,000 atom scale and beyond. Furthermore, it also needs to maintain accuracy as we run inferences on increasingly large systems. Both issues are addressed in the following.
Computational scaling
The computational cost of conventional DFT calculations scales as N3. However, improved algorithms can enable an effective N2 scaling in certain cases and over specific size ranges45. Regardless, one encounters an increasingly formidable computational burden for systems with more than a few thousand atoms. As depicted in Fig. 3a, conventional DFT calculations (here using the Quantum ESPRESSO42 software package) exhibit this scaling behavior.
Contrarily, the computational cost of using MALA models for size extrapolation (as shown in Fig. 3b) grows linearly with the number of atoms and has a significantly smaller computational overhead. We observe speed-ups of up to three orders of magnitude for atom counts up to which DFT calculations are computationally tractable.
MALA model inference consists of three steps. First, the descriptor vectors are calculated on a real-space grid, then the LDOS is computed using a pre-trained neural network for given input descriptors, and finally, the LDOS is post-processed to compute electronic densities, total energies, and other observables. The first two parts of this workflow trivially scale with N, since they strictly perform operations per grid point, and the real space simulation grid grows linearly with N.
Obtaining linear scaling for the last part of the workflow, which includes processing the electronic density to the total free energy, is less trivial since it requires both the evaluation of the ion-ion energy as well as the exchange-correlation energy, which for the pseudopotentials we employ includes the calculation of non-linear core corrections. While both of these terms can be shown to scale linearly with system size in principle, in practice this requires the addition of a few custom routines, as is further outlined in the methods section.
To facilitate a transparent comparison of computational timings, we present Table 2, which details the number of atoms, wall time, number of CPUs for each calculation, and CPU time. CPU time is defined as the product of the number of CPUs and wall time, providing a suitable measure for the total computational cost of different methods with varying levels of parallelization. Our MALA model demonstrates a CPU time of 121 hours for our largest calculation, which includes 131,072 atoms.
To contextualize our model’s timings, we compare them with linear-scaling DFT methods that enable large-scale calculations46, albeit relying on approximations in terms of the density matrix. For this comparison, we consider linear-scaling DFT calculations on bulk silicon46,47, which amount to a CPU time of approximately 500 hours for a system of comparable size. This comparison underscores the competitiveness of our model, which is significantly faster than linear-scaling DFT codes. Furthermore, there is considerable potential for optimizing our ML model, particularly in calculating bispectrum descriptors, where we anticipate achieving substantial additional speed-ups.
Benchmarks at DFT scales (~103 atoms)
We tackle the first part of this problem by investigating a system of Beryllium atoms at room temperature and ambient mass density (1.896 g cm−1). Neural networks are trained on LDOS data generated for 256 atoms. After training, inference was performed for an increasing number of atoms, namely 256, 512, 1024, and 2048 atoms.
The total free energy and the electronic density were used to assess the accuracy of MALA predictions for a total of 10 atomic configurations per system size. In Fig. 4a we report the absolute error of the energy and the mean absolute percentage error (MAPE) of the density across system sizes. It is evident that the errors stay roughly constant across system size and are well within both chemical accuracy (below 43 meV atom−1). Furthermore, the error of the energy is within the 10 meV atom−1 threshold which is considered the gold standard for ML interatomic potentials. Similarly, the error in the electronic density follows the trend observed in the total energy. The MAPE of the electronic density is consistently below 1%. To provide context for this value, we can compare it with the variation in electronic densities across different atomic configurations. Taking into account five distinct atomic configurations per number of atoms and system sizes up to 2048 atoms, the average percentage change in electronic density within this set of atomic configurations surpasses 7%.

a Prediction errors when using MALA to calculate total free energies and electronic densities compared to DFT data. b Correlation between DFT and predicted total energies for MALA and an EAM type interatomic potential for Beryllium (across all system sizes).
The accuracy of absolute total free energy predictions does not suffice to assess model performance, since one is usually interested in energy differences. Therefore, we relate the predicted total free energy to the DFT reference data set in Fig. 4b. The data points are drawn across all system sizes but are given relative to the respective means per system size for the sake of readability. Ideally, the resulting distribution would lie along a straight line. In practice, both a certain spread around this line (unsystematic errors) and a tilt of the line (systematic errors) can be expected. We quantify our results by comparing MALA (red circles) with an embedded-atom-method (EAM) interatomic potential (blue squares)48,49 which is commonly used in molecular dynamics simulations. It can clearly be seen that MALA outperforms the EAM model in both unsystematic as well as systematic errors, and, therefore, delivers physically correct energies beyond the system sizes it was trained on.
Accuracy at ultra-large scales (~105 atoms)
Finally, we tackle the second step of providing evidence that MALA predictions on the ultra-large scale are expected to be as accurate as conventional DFT calculations. This analysis is grounded in the local nature of our workflow. Given that the local environments are similar to those observed in training, predictions for arbitrarily large cells boil down to interpolation, a task at which neural networks excel. Accordingly, our ML model performs a perceived size extrapolation by actually performing local interpolations.
To verify the similarity of the atomic configurations in the training set with those used for inference at ultra-large scales, we employ the radial distribution function. It is a useful quantity that distinguishes between different phases of a material, by giving insight into how likely it is to find an atom at a given distance from a reference point. Since the input to our workflow, B, is calculated based on atomic densities drawn from a certain cutoff radius, a matching radial distribution function up to this point indicates that the individual input vectors B should on average be similar between simulation cells. This comparison is shown in Fig. 5 where the radial distribution functions g(r) of the training (256 atoms, green), inference test (2048 atoms, blue), and ultra-large prediction (131,072 atoms, red and orange) data sets are illustrated. Figure 5a illustrates the absolute values, whereas Fig. 5b shows the difference of the radial distribution function to the training data set. It should be noted that for the sake of comparability, the radial distribution functions for 256 atoms and 2048 were averaged over 30 atomic configurations and 10 atomic configurations, respectively. Furthermore, we do not show the full radial distribution function for radii below 1.5 Å, since it is zero irrespective of the number of atoms, due to the average interatomic distance for this system. Note also that we have shifted these curves along the y axis by a constant value of 0.2 from each other to better illustrate how similar they are.

a Radial distribution functions for Beryllium simulation cells of differing sizes, within the radius in which information is incorporated into bispectrum descriptors. For technical details on the radial distribution function, see the methods section. Note that the curves of the inference test (blue) and prediction (red, orange) data sets have been shifted along the y-axis by a constant value of 0.2 to better illustrate how similar they are. b Absolute difference Δg(r) of the radial distribution functions with respect to the training data set.
In Fig. 5, slight deviations between the radial distribution functions of different system sizes can be seen, most notably for the cells containing a stacking fault. Overall, these deviations are small in magnitude, especially for the unperturbed cells, and generally, all radial distribution functions agree very well up to the cutoff radius (dotted black) from which information is incorporated into the bispectrum descriptors.
This analysis hence provides evidence that training, inference test, and the ultra-large simulation cells possess, on average, the same local environments. It indicates that our MALA predictions of the electronic structure and energy are based on interpolations on observed data. Therefore our models can be expected to be accurate at ultra-large scales far exceeding those for which reference data exists. By comparing the radial distribution functions for 2048 and 131,072 atoms, we can deduce that errors similar to those reported in Fig. 4 can be assumed for these ultra-large scales.
Discussion
We have introduced an ML model that avoids the computational bottleneck of DFT calculations. It scales linearly with system size as opposed to conventional DFT that follows a cubic scaling. Our ML model enables efficient electronic structure predictions at scales far beyond what is tractable with conventional DFT, in fact at any length scale. In contrast to existing ML approaches, our workflow provides direct access to the electronic structure and is not limited to specific observables. Any physical quantity that can be expressed as a functional of the electronic density, the fundamental quantity in DFT, can be predicted using the ML models trained with the workflow presented here.
At system sizes where DFT benchmarks are still available, we demonstrate that our ML model is capable of reproducing energies and electronic densities of extended systems at virtually no loss in accuracy while outperforming other ML models that are based solely on energy. Furthermore, we demonstrate that our ML workflow enables predicting the electronic structure for systems with more than 100,000 atoms at a very low computational cost. We underpin its accuracy at these ultra-large scales by analyzing the radial distribution function and find that our ML models can be expected to deliver accurate results even at such length scales.
We expect our ML model to set standards in a number of ways. Using our ML model either directly or in conjunction with other ML workflows, such as ML interatomic potentials for pre-sampling of atomic configurations, will enable first-principles modeling of materials without finite-size errors. Combined with Monte-Carlo sampling and atomic forces from automatic differentiation, our ML model can replace ML interatomic potentials and yield thermodynamic observables at much higher accuracy. Another application our ML model enables is the prediction of electronic densities in semiconductor devices, for which an accurate modeling capability at the device scale has been notoriously lacking.
Methods
Density functional theory
DFT is the most widely used method for computing (thermodynamic) materials properties in chemistry and materials science because it strikes a balance between computational cost and accuracy. Within DFT, one commonly seeks to describe a coupled system of N ions of charge Zα at collective positions \(\underline{{{{\bf{R}}}}}=({{{{\bf{R}}}}}_{1},{{{{\bf{R}}}}}_{2},...,{{{{\bf{R}}}}}_{N})\) and L electrons at collective positions \(\underline{{{{\bf{r}}}}}=({{{{\bf{r}}}}}_{1},{{{{\bf{r}}}}}_{2},...,{{{{\bf{r}}}}}_{L})\) on a quantum statistical mechanical level5,50. Within the commonly assumed Born–Oppenheimer approximation51, the Hamiltonian
represents a system of interacting electrons in the external field of the ions that are simplified to classical point particles. Here, \(\hat{T}=\mathop{\sum }\nolimits_{j}^{L}-{\nabla }_{j}^{2}/2\) denotes the kinetic energy operator of the electrons, \({\hat{V}}_{{{{\rm{ee}}}}}=\mathop{\sum }\nolimits_{i}^{L}\mathop{\sum }\nolimits_{j\ne i}^{L}1/(2| {{{{\bf{r}}}}}_{i}-{{{{\bf{r}}}}}_{j}| )\) the electron–electron interaction, and \({\hat{V}}_{{{{\rm{ei}}}}}=-\mathop{\sum }\nolimits_{j}^{L}\mathop{\sum }\nolimits_{\alpha }^{N}{Z}_{\alpha }/| {{{{\bf{r}}}}}_{j}-{{{{\bf{R}}}}}_{\alpha }|\) the external potential, i.e., the electron–ion interaction. The Born–Oppenheimer Hamiltonian separates the electronic and ionic problems into a quantum mechanical and classical mechanical problem. Such an assumption is feasible since ionic masses far exceed the electronic mass, leading to vastly different time scales for movement and equilibration.
At finite temperatures τ > 0, the theoretical description is extended to the grand canonical operator52
where \(\hat{S}=-{k}_{{{{\rm{B}}}}}\ln \hat{\Gamma }\) denotes the entropy operator, \(\hat{N}\) the particle-number operator, and μ the chemical potential. Here, we introduced the statistical density operator \(\hat{\Gamma }={\sum }_{L,m}{w}_{L,m}\left\vert {{{\Psi }}}_{L,m}\right\rangle \left\langle {{{\Psi }}}_{L,m}\right\vert\) with the L-electron eigenstates ΨL,m of the Hamiltonian \(\hat{H}\) and wL,m as the normalized statical weights that obey ∑L,mwL,m = 1. Any observable O is then computed as an average
Most importantly, finding the grand potential
amounts to finding a \(\hat{\Gamma }\) that minimizes this expression. The exact solution to this problem evades numerical treatment even with modern hardware and software due to the electron-electron interaction in the Born-Oppenheimer Hamiltonian of Eq. (3). It dictates an exponential growth of complexity with the number of electrons L, i.e., eL.
Based on the theorems of Hohenberg and Kohn5 and of Mermin50, DFT makes solving this problem computationally tractable by employing the electronic density n as the central quantity. The formal scaling reduces to L3 due to the Kohn-Sham approach11. Within DFT, all quantities of interest are formally defined as functionals of the electronic density via a one-to-one correspondence with the external (here, electron-ion) potential. In conjunction with the Kohn-Sham scheme, which introduces an auxiliary system of non-interacting fermions restricted to reproduce the density of the interacting system, practical calculations become feasible. Rather than evaluating Eq. (6) using many-body wave functions ΨL, the grand potential is evaluated as a functional of density n as
with the kinetic energy of the Kohn–Sham system TS, the entropy of the Kohn–Sham system SS, the classical electrostatic interaction energy EH (Hartree energy), the electrostatic interaction energy of the electronic density with the ions Vei, and the exchange-correlation (free) energy EXC. The Kohn–Sham system serves as an auxiliary system that is used to calculate the kinetic energy and entropy terms in Eq. (7). The Kohn–Sham equations are defined as a system of one-electron Schrödinger-like equations
with an effective potential, the Kohn–Sham potential vS(r), that yields the electronic density of the interacting system via
where ϕj denotes the Kohn–Sham orbitals, ϵj the Kohn–Sham eigenalues, and fτ(ϵj) the Fermi-Dirac distribution at temperature τ. The Kohn–Sham potential is a single-particle potential defined as vS(r) = vei(r) + vH(r) + vXC(r), where \({v}_{{{{\rm{ei}}}}}({{{\bf{r}}}})=-\mathop{\sum }\nolimits_{\alpha }^{N}{Z}_{\alpha }/| {{{\bf{r}}}}-{{{{\bf{R}}}}}_{\alpha }|\) denotes the electron–ion interaction potential, \({v}_{{{{\rm{H}}}}}[n]({{{\bf{r}}}})=\delta {E}_{{{{\rm{H}}}}}[n]/\delta n({{{\bf{r}}}})=\int\,d{{{{\bf{r}}}}}^{{\prime} }\,n({{{{\bf{r}}}}}^{{\prime} })/| {{{\bf{r}}}}-{{{{\bf{r}}}}}^{{\prime} }|\) the Hartree potential, and vXC[n](r) = δEXC[n]/δn(r) the exchange-correlation potential. Note that within the Kohn-Sham framework at finite temperatures, several quantities including vS(r), ϵj, ϕj, n, TS, SS, and EXC are technically temperature dependent; we omit to label this temperature dependency explicitly in the following for the sake of brevity. The Kohn–Sham formalism of DFT is formally exact if the correct form of the exchange-correlation functional EXC[n] was known. In practice, approximations of the exchange-correlation functional are employed. There exists a plethora of useful functionals both for the ground state (such as the LDA11,53, PBE54,55,56, and SCAN57 functionals) and at finite temperature58,59,60,61. Such functionals draw on different ingredients for approximating the exchange-correlation energy. Some rely only on the electronic density (e.g., LDA), while others incorporate density gradients (e.g., PBE) or even the kinetic energy density (e.g., SCAN). Consequently, functionals differ in their provided accuracy and application domain like molecules or solids.
The calculation of dynamical properties is enabled in this framework via the estimation of the atomic forces, which are then used to time-propagate the ions in a process called DFT-MD. The forces are evaluated via the total free energy as \(-\partial A[n](\underline{{{{\bf{R}}}}})/\partial \underline{{{{\bf{R}}}}}\), where the total free energy A is obtained from Eq. (7) as \(A[n](\underline{{{{\bf{R}}}}})=\Omega [n]+\mu L\). While this framework can be employed to calculate a number of (thermodynamic) materials properties62, the treatment of systems of more than roughly a thousand atoms becomes computationally intractable due to the L2 to L3 scaling typically observed when running DFT calculations for systems in this size range. Therefore, current research efforts are increasingly focused on the combination of ML and DFT methods23.
DFT surrogate models
ML comprises a number of powerful algorithms, that are capable of learning, i.e., improving through data provided to them. Within DFT and computational materials science in general, ML is often applied in one of two settings, as shown in ref. 23. Firstly, ML algorithms learn to predict specific properties of interest (e.g., structural or electronic properties) and thus bypass the need to perform first-principles simulations for investigations across vast chemical parameter spaces. Secondly, ML algorithms may provide direct access to atomic forces and energies, and thus accelerate dynamical first-principles simulations drastically, resulting in ML interatomic potentials for MD simulations.
We have recently introduced an ML framework that does not fall in either category, as it comprises a DFT surrogate model that replaces DFT for predicting a range of useful properties37. Our framework directly predicts the electronic structure of materials and is therefore not restricted to singular observables. Within this framework, the central variable is the local density of states (LDOS) defined by
The merit of using the LDOS as a central variable is that it determines both the electronic density as
and the density of states (DOS) as
As opposed to related work63, we use these two quantities to calculate the total free energy drawing on a reformulation of Eq. (7), which expresses all energy terms dependent on the KS wave functions and eigenvalues in terms of the DOS. More precisely, by employing the band energy
and reformulating the electronic entropy in terms of the DOS, i.e.,
the total free energy A can be expressed as
where D and n are functionals of the LDOS and vXC(r) = δEXC[n[d](r)]/δn[d](r).
In our framework, the LDOS is learned locally. For each point in real space, the respective LDOS (a vector in the energy domain) is predicted separately from adjacent points. Non-locality enters this prediction through the descriptors that serve as input to the ML algorithm. Here, we chose bispectrum descriptors30 denoted as B. In contrast to their usual application as a basis for interatomic potentials, these descriptors are employed to encode local information on atomic neighborhoods at each point in space. This is done by evaluating the total density of neighbor atoms via a sum of delta functions
In Eq. (16), the sum is performed over all k atoms within a cutoff distance \({R}_{{{{\rm{cut}}}}}^{{\nu }_{k}}\) using a switching function fc that ensures smoothness of atomic contributions at the edges of the sphere with radius \({R}_{{{{\rm{cut}}}}}^{{\nu }_{k}}\). These atoms are located at position rk relative to the grid point r, while the chemical species νk enters the equation via the dimensionless weights \({w}_{{\nu }_{k}}\). The thusly defined density is then expanded into a basis of 4D hyperspherical harmonic functions, eventually yielding the descriptors B(J, r) with a feature dimension J (see refs. 37,30). Constructing descriptors in such a way introduces two hyperparameters, \({R}_{{{{\rm{cut}}}}}^{{\nu }_{k}}\), which determines the radius from which information is incorporated into the descriptors and \({J}_{\max }\), which determines the number of hyperspherical harmonics used for the expansion, i.e., the dimensionality of the descriptor vectors. Each of the hyperparameters must be precisely calibrated to adequately represent the underlying physical properties of the system, resulting in useful models. Inappropriate selections for these hyperparameters can lead to poorly performing models. If \({R}_{{{{\rm{cut}}}}}^{{\nu }_{k}}\) is set to a small value, the descriptors generated may not accurately capture non-local effects. On the other hand, if it is set to a large value, the descriptors may encode information from the entire simulation cell, resulting in an overly complex learning task. Similarly, a small value of \({J}_{\max }\) produces low-dimensional descriptor vectors that may not capture all features of the atomic density, while a large value introduces additional noise, thereby complicating the training process.
One can identify suitable values for these hyperparameters using traditional grid search methods. However, as the optimal values are linked to the system’s physics, similarities and differences in the local electronic structure should be adequately resolved by the descriptors. Recently, we demonstrated an algorithm for identifying such optimal values in ref. 64.
A mapping from B(J, r) to d(ϵ, r) is now performed via a neural network (NN), M, i.e.,
where \(\tilde{d}\) is the approximate LDOS. After performing such a network pass for each point in space, the resulting approximate LDOS can be post-processed into the observables mentioned above.
We employ NNs, because they are, in principle, capable of approximating any given function65. In the present case, we employ feed-forward NNs66 which consist of a sequence of layers containing individual artificial neurons67 that are fully connected to each neuron in subsequent layers. Each layer is a transformation of the form
that maps x from layer ℓ to ℓ + 1 by addition of a bias vector b, matrix multiplication with a weight matrix W, and an activation function Φ. For the DFT surrogate models discussed here, the input to the first transformation x0 is B(J, r) for a specific point in space r; the output of the last layer \({{{{\bf{x}}}}}^{{{{{L}}}}}\) is d(ϵ, r) for the same r. The number of layers \({{{{L}}}}\) and activation function Φ have to be determined through prior hyperparameter optimization, among other hyperparameters such as the width of the individual layers. In ref. 64 we show how such a hyperparameter optimization can be drastically improved upon in terms of computational effort, while the hyperparameters employed for this study are detailed in “Machine learning computational details”. For each architecture of the NN, the weights and biases have to be optimized using gradient-based updates in a process called training based on a technique called backpropagation68 which is carried out using gradients averaged over portions of the data (so-called mini-batches); other technical parameters include stopping criteria for the early stopping of the model optimization and the learning rate for the gradient-based updates69.
Unlike DFT, the great majority of operations in our DFT surrogate model have a computational cost that naturally scales linearly with system size: (1) the descriptors are evaluated independently at each point on the computational grid using algorithms in LAMMPS that take advantage of the local dependence of the descriptors on the atomic positions in order to maintain linear scaling; (2) the NN is evaluated independently at each grid point in order to obtain the LDOS at each point; (3) the DOS is evaluated by a reduction over grid points, the Fermi level is found, and Eb and SS are evaluated; (4) the density is calculated independently at each grid point; and (5) three-dimensional Fast Fourier transforms, which are implemented efficiently in Quantum ESPRESSO, are used to evaluate EH from the density. The remaining terms are EXC, vXC, and the ion-ion interaction energy. The exchange-correlation terms can almost be evaluated independently at each point (using Fast Fourier transforms to evaluate gradients if necessary), but the pseudo-potentials that we use include non-linear core corrections, which require the addition of a “core density” centered on each atom to the density used to calculate EXC and vXC. Likewise, the ion-ion interaction energy can be evaluated efficiently using Fast Fourier transforms if we can compute the sum of non-overlapping charge distributions containing the appropriate ionic charge centered on each atom. The key to calculating these terms with a computational cost that scales linearly with system size is an efficient algorithm to evaluate the structure factor.
If F(r) is some periodic function represented by its values on the computational grid, its Fast Fourier transform \(\tilde{F}({{{\bf{G}}}})\) gives its representation in the basis of plane-waves \(\exp (i{{{\bf{G}}}}\cdot {{{\bf{r}}}})\), where the reciprocal lattice vectors G form a reciprocal-space grid with the same dimensions as the computational grid. The structure factor is defined as
where the summation over atom positions Rα runs over all atoms within one copy of the periodically repeated computational cell. The structure factor is very useful because
can be efficiently evaluated as the inverse Fourier transform of \({\tilde{F}}^{{{{\rm{S}}}}}=\tilde{S}({{{\bf{G}}}})\tilde{F}({{{\bf{G}}}})\). Thus, the structure factor can be used to evaluate the non-linear core correction density and the ion-ion interaction energy when evaluating the DFT total energy. However, the straightforward evaluation of \(\tilde{S}({{{\bf{G}}}})\) on the grid of G vectors scales as the square of the system size. We circumvent this bottleneck by taking advantage of the real-space localization properties of the Gaussian function G(r) in order to efficiently evaluate GS(r) within the LAMMPS code40. Then, within Quantum ESPRESSO, we use a fast Fourier transformation to calculate \({\tilde{G}}^{{{{\rm{S}}}}}({{{\bf{G}}}})\), and the structure factor is obtained as
A suitable choice of the Gaussian width for G(r) allows us to minimize aliasing errors due to Fourier components beyond the Nyquist limit of the computational grid, while also maintaining good precision in the above division.
Data analysis
We assess whether our models are employed in an interpolative setting when applied to larger cells. To that end, we analyze the radial distribution function, which is defined as
It is the average ion density in a shell [r, r + dr] of volume V(r) around a reference ion at r = 0, relative to an isotropic system of density ρ = N/V70. The radial distribution function is often used to identify different phases of a material and, in our case, it can be used to verify that simulation cells with differing numbers of atoms are equivalent in their ion distribution up to a certain cutoff radius. For technical reasons, there exists an upper radius up to which g is well-defined, which is a result of the minimum image convention71 and which amounts to half the cell edge length in case of cubic cells. For small cells, the employed cutoff radius lies slightly beyond this radius, but this does not affect model inference, since periodic boundary conditions are applied for the calculation of the bispectrum descriptors.
Training data
Increasingly larger DFT-MD simulations at 298 K have been performed to acquire atomic configurations for simulation cells containing 256 to 2048 Beryllium atoms. DFT-MD calculations up to 512 atoms have been carried out using Quantum ESPRESSO, while simulations for 1024 and 2048 atoms have been performed using VASP45,72,73. In either case, DFT-MD simulations have been performed at the Γ-point, using a plane-wave basis set with an energy cutoff of 40 Ry (Quantum ESPRESSO) or 248 eV (VASP), and an ultrasoft pseudopotential74 (Quantum ESPRESSO) or a PAW pseudopotential75,76 (VASP). The resulting trajectories have been analyzed with a method akin to the equilibration algorithm outlined in prior work77, although here equilibration thresholds have been defined manually. Thereafter, snapshots have been sampled from these trajectories such that the minimal euclidean distance between any two atoms within the last sampled snapshot and potentially next sampled snapshots lies above the empirically determined threshold of 0.125 Å. The resulting data set of Beryllium at room temperature includes ten configurations per system size, except for 256 atoms, where a larger number of configurations is needed to enable the training and verification of models. For all of these configurations, DFT calculations have been carried out with Quantum ESPRESSO, using the aforementioned cutoff and pseudopotential. The Brillouin zone has been sampled by Monkhorst–Pack78 sampling, with the number of k-points given in Table 1.
The employed calculation parameters have been determined via a convergence analysis with a threshold of 1 meV atom−1, except for Beryllium systems with 2048 atoms, where only Γ point calculations have been performed due to computational constraints.
The values in Table 1 refer to those DFT calculations that were performed to gather reference energies and densities. To calculate the LDOS, one has to employ larger k-grids, as the discretiatzion of k-space with a finite number of points in k-space can introduce errors and features into the (L)DOS that are unphysical. As discussed in prior work37,64, such features can be removed by employing a larger number of k-points than for typical DFT simulations. The correct k-grid has to be determined through a convergence test such that no unphysical oscillations occur in the (L)DOS. By applying an established analysis37,64, we have determined 12 × 6 × 6 as a suitable k-grid for 256 Beryllium atoms. Again, Monkhorst–Pack sampling has been used.
In order to assess the scaling of DFT for Fig. 3a of the main manuscript, we kept a constant k-grid were possible, in comparison to the adapted k-grids used for the reference data calculation used for Fig. 4. More specifically, in order to reflect realistic simulation settings, we employed a 3 × 3 × 3 grid, i.e., a k-grid consistent with 1024 atoms, the largest number of atoms for which k-point converged simulations could be performed. The same number of k-points was used for 128, 256, and 512 atoms. Performing a DFT calculation for 2048 atoms was impossible due to the large memory demand and the computational resources available. We, therefore, performed a singular 2048 atoms calculation with a 4 × 2 × 2k-grid, utilizing more k-points in the x-direction, since the 2,048 atom cells are extended in that direction compared to the 1024 atom cells. Overall, this change in k-grid leads to only a small deviation of the observed ~N3 behavior.
Machine learning computational details
For all ML experiments, the architecture and hyperparameters discussed in the original MALA publication37 have been employed. One training and one validation snapshot have been used in the 256-atom case. Training was performed on a single Nvidia Tesla V100, and five models were trained using different random initializations to assess the robustness of the approach. The average duration of model training was 51.40 min. It is worth noting that data generation time, namely the time required to perform DFT calculations, must also be taken into account. For the models trained in this study, two training snapshots were needed, each calculated on 144 Intel Xeon CPUs, with a respective calculation time of 20.17 and 19.20 h. This relatively high computational cost can be attributed to the large k-grids required for accurate LDOS sampling as previously specified. These DFT calculations can be executed in parallel, resulting in a total training time of 21.02 h for a size-transferable model of this magnitude.
The timings for inference, i.e., utilizing the ML model to perform predictions, are detailed in Table 2. These timings complement the data presented in Fig. 3.
Data availability
Training data of the Beryllium system is publicly available79. Please note that this published data set79 includes a larger data set of the Beryllium system as it has been used in multiple publications. In this manuscript, only a subset of Beryllium configurations has been used (256 atoms: 0–30; 512 atoms: 5–14; 1024 atoms: 0–9; 2048 atoms: 0-9).
References
Kang, K., Meng, Y. S., Bréger, J., Grey, C. P. & Ceder, G. Electrodes with high power and high capacity for rechargeable lithium batteries. Science 311, 977–980 (2006).
Lu, J. et al. A lithium–oxygen battery based on lithium superoxide. Nature 529, 377–382 (2016).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Hannagan, R. T. et al. First-principles design of a single-atom–alloy propane dehydrogenation catalyst. Science 372, 1444–1447 (2021).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Nakashima, P. N. H., Smith, A. E., Etheridge, J. & Muddle, B. C. The bonding electron density in aluminum. Science 331, 1583–1586 (2011).
Thomas, L. H. The calculation of atomic fields. Math. Proc. Camb. Philos. Soc. 23, 542–548 (1927).
Fermi, E. Zur Quantelung des idealen einatomigen gases. Z. Physik 36, 902–912 (1926).
Dirac, P. A. M. Note on exchange phenomena in the Thomas atom. Math. Proc. Camb. Philos. Soc. 26, 376–385 (1930).
Teller, E. On the stability of molecules in the Thomas-Fermi theory. Rev. Mod. Phys. 34, 627–631 (1962).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Kohn, W. Nobel Lecture: electronic structure of matter—wave functions and density functionals. Rev. Mod. Phys. 71, 1253–1266 (1999).
Pople, J. A. Nobel Lecture: quantum chemical models. Rev. Mod. Phys. 71, 1267–1274 (1999).
Jones, R. O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 87, 897–923 (2015).
de Pablo, J. J. et al. New frontiers for the materials genome initiative. npj Comput. Mater. 5, 41 (2019).
Lejaeghere, K. et al. Reproducibility in density functional theory calculations of solids. Science 351, aad3000 (2016).
Medvedev, M. G., Bushmarinov, I. S., Sun, J., Perdew, J. P. & Lyssenko, K. A. Density functional theory is straying from the path toward the exact functional. Science 355, 49–52 (2017).
Kirkpatrick, J. et al. Pushing the frontiers of density functionals by solving the fractional electron problem. Science 374, 1385–1389 (2021).
Pederson, R., Kalita, B. & Burke, K. Machine learning and density functional theory. Nat. Rev. Phys 4, 357–358 (2022).
Lignères, V. L. & Carter, E. A. An introduction to orbital-free density functional theory. In Handbook of Materials Modeling: Methods, 137–148 (Springer Netherlands, 2005).
Yang, W. Direct calculation of electron density in density-functional theory. Phys. Rev. Lett. 66, 1438–1441 (1991).
Goedecker, S. & Colombo, L. Efficient linear scaling algorithm for tight-binding molecular dynamics. Phys. Rev. Lett. 73, 122–125 (1994).
Fiedler, L., Shah, K., Bussmann, M. & Cangi, A. Deep dive into machine learning density functional theory for materials science and chemistry. Phys. Rev. Mater. 6, 040301 (2022).
Rupp, M., Tkatchenko, A., Müller, K.-R. & von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012).
Schmidt, J. et al. Predicting the thermodynamic stability of solids combining density functional theory and machine learning. Chem. Mater. 29, 5090–5103 (2017).
Friederich, P., dos Passos Gomes, G., De Bin, R., Aspuru-Guzik, A. & Balcells, D. Machine learning dihydrogen activation in the chemical space surrounding Vaska’s complex. Chem. Sci. 11, 4584–4601 (2020).
Musil, F. et al. Machine learning for the structure–energy–property landscapes of molecular crystals. Chem. Sci. 9, 1289–1300 (2018).
Ye, W., Chen, C., Wang, Z., Chu, I.-H. & Ong, S. P. Deep neural networks for accurate predictions of crystal stability. Nat. Commun. 9, 3800 (2018).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Shapeev, A. V. Moment tensor potentials: a class of systematically improvable interatomic potentials. Multiscale Model. Simul. 14, 1153–1173 (2016).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Zhang, L., Han, J., Wang, H., Car, R. & E, W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Brockherde, F. et al. Bypassing the Kohn-Sham equations with machine learning. Nat. Commun. 8, 872 (2017).
Tsubaki, M. & Mizoguchi, T. Quantum deep field: data-driven wave function, electron density generation, and atomization energy prediction and extrapolation with machine learning. Phys. Rev. Lett. 125, 206401 (2020).
Mills, K. et al. Extensive deep neural networks for transferring small scale learning to large scale systems. Chem. Sci. 10, 4129–4140 (2019).
Ellis, J. A. et al. Accelerating finite-temperature Kohn-Sham density functional theory with deep neural networks. Phys. Rev. B 104, 035120 (2021).
Kohn, W. Density functional and density matrix method scaling linearly with the number of atoms. Phys. Rev. Lett. 76, 3168–3171 (1996).
Cangi, A. et al. MALA. Zenodo, https://doi.org/10.5281/zenodo.5557254 (2021).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 271, 108171 (2022).
Paszke, A. et al. PyTorch: An imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems, vol. 32 (Curran Associates, Inc., 2019).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Condens. Matter Phys. 21, 395502 (2009).
Wood, M. A., Cusentino, M. A., Wirth, B. D. & Thompson, A. P. Data-driven material models for atomistic simulation. Phys. Rev. B 99, 184305 (2019).
Wilkins, D. M. et al. Accurate molecular polarizabilities with coupled cluster theory and machine learning. Proc. Natl. Acad. Sci. U.S.A. 116, 3401–3406 (2019).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Nakata, A. et al. Large scale and linear scaling DFT with the CONQUEST code. J. Chem. Phys. 152, 164112 (2020).
Bowler, D. R. & Miyazaki, T. Calculations for millions of atoms with density functional theory: linear scaling shows its potential. J. Phys. Condens. Matter 22, 074207 (2010).
Daw, M. S. & Baskes, M. I. Embedded-atom method: derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 29, 6443–6453 (1984).
Agrawal, A., Mishra, R., Ward, L., Flores, K. M. & Windl, W. An embedded atom method potential of beryllium. Model. Simul. Mat. Sci. Eng. 21, 085001 (2013).
Mermin, N. D. Thermal properties of the inhomogeneous electron gas. Phys. Rev. 137, A1441–A1443 (1965).
Born, M. & Oppenheimer, R. Zur Quantentheorie der Molekeln. Ann. Phys. 389, 457–484 (1927).
Toda, M., Kubo, R., Kubo, R., Saitō, N. & Hashitsume, N. Statistical Physics: Equilibrium Statistical Mechanics (Springer Berlin, 1983).
Ceperley, D. M. & Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566–569 (1980).
Perdew, J. P. & Yue, W. Accurate and simple density functional for the electronic exchange energy: generalized gradient approximation. Phys. Rev. B 33, 8800–8802 (1986).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249 (1992).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Brown, E. W., DuBois, J. L., Holzmann, M. & Ceperley, D. M. Exchange-correlation energy for the three-dimensional homogeneous electron gas at arbitrary temperature. Phys. Rev. B 88, 081102 (2013).
Karasiev, V. V., Chakraborty, D., Shukruto, O. A. & Trickey, S. B. Nonempirical generalized gradient approximation free-energy functional for orbital-free simulations. Phys. Rev. B 88, 161108 (2013).
Karasiev, V. V., Sjostrom, T., Dufty, J. & Trickey, S. B. Accurate homogeneous electron gas exchange-correlation free energy for local spin-density calculations. Phys. Rev. Lett. 112, 076403 (2014).
Groth, S. et al. Ab initio exchange-correlation free energy of the uniform electron gas at warm dense matter conditions. Phys. Rev. Lett. 119, 135001 (2017).
Iftimie, R., Minary, P. & Tuckerman, M. E. Ab initio molecular dynamics: concepts, recent developments, and future trends. Proc. Natl. Acad. Sci. U.S.A. 102, 6654–6659 (2005).
Chandrasekaran, A. et al. Solving the electronic structure problem with machine learning. npj Comput. Mater. 5, 22 (2019).
Fiedler, L. et al. Training-free hyperparameter optimization of neural networks for electronic structures in matter. Mach. Learn. Sci. Technol. 3, 045008 (2022).
Hornik, K. Approximation capabilities of multilayer feedforward networks. Neural Netw. 4, 251–257 (1991).
Minsky, M. & Papert, S. A. Perceptrons. An Introduction to Computational Geometry (MIT Press, 2017).
Rosenblatt, F. The Perceptron: A Perceiving and Recognizing Automaton (Project PARA). (Cornell Aeronautical Laboratory, 1957).
Rumelhart, D. E., Hinton, G. E. & Williams, R. J. Learning representations by back-propagating errors. Nature 323, 533–536 (1986).
Goodfellow, I., Bengio, Y. & Courville, A. Deep Learning (MIT Press, 2016).
Allen, M. P. & Tildesley, D. J. Computer Simulation of Liquids (Oxford University Press, 1989).
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H. & Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 21, 1087–1092 (1953).
Kresse, G. & Furthmüller, J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Dal Corso, A. Pseudopotentials periodic table: from H to Pu. Comput. Mater. Sci. 95, 337–350 (2014).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Fiedler, L. et al. Accelerating equilibration in first-principles molecular dynamics with orbital-free density functional theory. Phys. Rev. Res. 4, 043033 (2022).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Fiedler, L. & Cangi, A. LDOS/SNAP data for MALA: Beryllium at 298K. RODARE, https://doi.org/10.14278/rodare.1834 (2022).
Fiedler, L. et al. Scripts and Models for “Predicting electronic structures at any length scale with machine learning”. RODARE, https://doi.org/10.14278/rodare.1851 (2022).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mat. Sci. Eng. 18, 015012 (2009).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
The authors are grateful to the Center for Information Services and High Performance Computing [Zentrum für Informationsdienste und Hochleistungsrechnen (ZIH)] at TU Dresden for providing its facilities for high throughput calculations. We also gratefully acknowledge Alexander Debus for providing a CPU allocation on the taurus HPC system of ZIH at TU Dresden. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly-owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. This work was in part supported by the Center for Advanced Systems Understanding (CASUS) which is financed by Germany’s Federal Ministry of Education and Research (BMBF) and by the Saxon state government out of the State budget approved by the Saxon State Parliament.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.F. performed all Beryllium-related calculations (DFT-MD, DFT, and MALA), code integration into the MALA code, and data visualization. N.M. and D.V. implemented the parallelization of the total energy evaluation, and N.M. eliminated scaling bottlenecks in the total energy evaluation. S.S. carried out the extrapolation transferability analysis. A.T. developed the parallelization of the descriptor calculation. S.R. and A.C. contributed to the theory and development of the MALA framework, supported data analysis, and supervised the overall project. All authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fiedler, L., Modine, N.A., Schmerler, S. et al. Predicting electronic structures at any length scale with machine learning. npj Comput Mater 9, 115 (2023). https://doi.org/10.1038/s41524-023-01070-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01070-z
This article is cited by
-
Machine-learning potentials for nanoscale simulations of tensile deformation and fracture in ceramics
npj Computational Materials (2024)
