
Abstract
Mitochondria are the main oxygen consumers in cells and as such are the primary organelle affected by hypoxia. All hypoxia pathology presumably derives from the initial mitochondrial dysfunction. An early event in hypoxic pathology in C. elegans is disruption of mitochondrial proteostasis with induction of the mitochondrial unfolded protein response (UPRmt) and mitochondrial protein aggregation. Here in C. elegans, we screen through RNAis and mutants that confer either strong resistance to hypoxic cell death or strong induction of the UPRmt to determine the relationship between hypoxic cell death, UPRmt activation, and hypoxia-induced mitochondrial protein aggregation (HIMPA). We find that resistance to hypoxic cell death invariantly mitigated HIMPA. We also find that UPRmt activation invariantly mitigated HIMPA. However, UPRmt activation was neither necessary nor sufficient for resistance to hypoxic death and vice versa. We conclude that UPRmt is not necessarily hypoxia protective against cell death but does protect from mitochondrial protein aggregation, one of the early hypoxic pathologies in C. elegans.
Similar content being viewed by others

Introduction
Hypoxia, when extreme, will kill aerobic cells. The pathways whereby cells go from healthy to dead when they encounter hypoxia are many and complex. By the time cells are dying from hypoxia, virtually every energy dependent process in the cell is dysfunctional. Pathological processes that occur early in hypoxic exposure when the pathology may still be reversible are of particular interest. This pathologic time course certainly varies by cell type and organism. In mammals, central nervous system neurons and cardiac myocytes are exquisitely sensitive to hypoxia, and during complete ischemia begin dying in only a few minutes after dissolved oxygen is depleted [1, 2]. On the other hand, peripheral neurons and skeletal myocytes can endure hours of a similar reduction of oxygen and fully recover. Some organisms can withstand prolonged severe hypoxia while others cannot. Certain hibernating fish and reptiles modulate anabolic and catabolic pathways and can tolerate near anoxic conditions over the winter months, while living in normal oxygen levels in the summer [3]. The primary cellular determinants of this enormous range in hypoxia tolerance are not well defined.
Mitochondria lie at the center of hypoxic biology and are logically the first organelle affected when oxygen levels fall below a crucial threshold. The vast majority of oxygen in metazoan cells is consumed by mitochondrial cytochrome C oxidase enabling the formation of ATP by ATP synthase. When oxygen levels fall, cytochrome C oxidase function fails, ATP levels decrease, and reactive oxygen species (ROS) are produced [4]. Besides ROS, the ATP deficit generated by hypoxia creates havoc throughout the cell including in the mitochondria where ATP-dependent processes such as the proton gradient, protein transport, synthesis, and folding are disrupted. The degree to which the failure of each of these processes contributes to hypoxic cell death is unclear.
In the nematode C. elegans, we and others have identified mutants and RNAis that protect worms from hypoxic cellular and organismal injury [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. Not surprisingly, what is clear from these studies is that multiple pathways regulate hypoxic sensitivity. However, to what degree the pathological state of the mitochondria influences these pathways and vice versa has not been systematically examined. We have previously shown that hypoxia disrupts mitochondrial proteostasis [6, 26]. This disruption is evident by hypoxic induction of the mitochondrial unfolded protein response UPRmt [26], a conserved adaptive pathway that responds to an increase in mitochondrial unfolded protein levels by increasing the synthesis of mitochondrial chaperones, proteases, and other proteins to restore proteostasis [27]. Hypoxia also promotes aggregation of mitochondrial proteins [6, 26]. Both UPRmt activation and hypoxia-induced mitochondrial protein aggregation (HIMPA) occur well before there is evidence of cell death in C. elegans or permanent behavioral deficits [6, 12]. Thus, disruption of mitochondrial proteostasis occurs relatively early in the hypoxic pathological cascade, but the consequence of disruption of mitochondrial proteostasis and HIMPA on downstream cellular pathology, most importantly cell death is unclear. Even the relationship of the UPRmt to HIMPA is not entirely clear. Activation of the UPRmt by three RNAis targeting mitochondrial proteins reduced HIMPA and protected from hypoxic death [6]. However, loss of function of atfs-1, a gene which is thought to be essential for UPRmt activation, also decreased HIMPA while atfs-1 gain-of-function mutants increased HIMPA [6]. Thus, whether the UPRmt promotes HIMPA or inhibits it is unclear with the relatively small number of UPRmt activators examined. Here, we make use of the wide variety of C. elegans RNAis and mutants that have been shown to be hypoxia protective, or to induce the UPRmt to address three fundamental questions about the relationship between mitochondrial proteostasis and hypoxic injury. Is activation of the UPRmt a necessary component of hypoxia protective mechanisms? Is UPRmt activation itself hypoxia protective? Does the UPRmt promote or reduce HIMPA?
Results
UPRmt activation not necessary for hypoxia resistance
We have previously performed a forward genome-wide screen in Caenorhabditis elegans for genes, whose inactivation by RNAi conferred a hypoxia resistance phenotype (HypR) and identified about 200 such genes [9]. To explore the hypothesis that activation of the UPRmt might be a common or even invariant component of the mechanism underlying hypoxia resistance of these RNAis, we screened through all the HypR RNAis that we could recover from the library and confirm their identity by sequence. Hundred and sixty-one of the HypR RNAi were confirmed and tested for activation of the mitoUPR under normoxic conditions as assessed by the fluorescence levels of the mitoUPR transcriptional reporter hsp-6p::GFP (hsp-6 is the ortholog of human mitochondrial chaperone HSP70 and is a canonical component of the UPRmt) [28,29,30,31]. RNAi knockdown of only five genes reproducibly induced the mitoUPR reporter (Fig. 1A). Thus, mitoUPR activation, at least as assessed by the hsp-6 reporter, is by no means necessary for hypoxia resistance.

A Fluorescence screen of RNAis for UPRmt activation. hsp-6p::GFP induction was quantified for 161 Hyp RNAi clones with the functional categories as originally assigned [9]. Fold induction (mean) of GFP fluorescence is relative to empty vector (EV) RNAi (L4440) (n = 10). Expression of GFP over 2-fold (blue dash line) of empty vector is considered to be positive. Five positive hits confirmed in 2nd screen are shown in red. B Examples of hsp-6p::GFP expression induced by knockdown of the five positive RNAi clones. zcIs13[hsp-6p::GFP] worms were placed onto RNAi bacteria from egg and imaged as young adults 3 days later. Bar, 0.1 mm. C Quantification of GFP expression by corresponding RNAis as in B. Data (mean ± SD) were obtained from four independent experiments, n = 30 animals; ***p < 0.001 with pooled data shown and p-values were calculated using unpaired t-test. D Examples of zcIs13[hsp-6p::GFP] GFP expression induced by knockdown of positive five RNAi clones in atfs-1(tm4919) background. Worms were placed onto RNAi bacteria from egg and imaged 3 days later. E Quantification of GFP expression by corresponding RNAis in atfs-1(tm4919) background. Data (mean ± SD) were obtained from five independent experiments, n = 30 animals. F Examples of HIMPA in body wall muscles after hypoxia of indicated RNAi treated gcIs46 worms. Green, UCR-11::GFP. Arrow, UCR-11::GFP aggregate. Bar, 2 μm. G Quantification of aggregate numbers as in F. n = 15 animals from three independent experiments; ***p < 0.001. with pooled data shown. P-values were calculated using unpaired t-test. H Quantification of HIMPA by UCR-11::GFP after hypoxia in body wall muscles of indicated RNAi treated atfs-1(tm4919); gcIs46 worms. n = 15 animals from three independent experiments; ***p < 0.001. with pooled data shown. P-values were calculated using unpaired t-test. I Hypoxia resistance is maintained in atfs-1(lf) background. Wild-type (N2) or atfs-1(tm4919) treated with indicated RNAis and hypoxic survival was scored. n = 9 plates of worms (>100 worms/plate) from three independent trials. *p < 0.05, unpaired t-test.
The five RNAis that induced the mitoUPR were against pept-1, sucl-2, C33F10.12, pdha-1, and R04F11.2 (Fig. 1A and Supplementary Table S1). pept-1 encodes an oligopeptide transmembrane transporter, localized to the apical plasma membrane in the intestine [32]. Reduction of function of pept-1 has been found to reduce protein synthesis rates and levels of essential amino acids [5, 33]. A pept-1 loss of function mutant was recently isolated in a screen for hypoxia resistant mutant worms, and its essential role in normal rates of protein synthesis was confirmed [5]. How loss of function of pept-1 activates the UPRmt is unclear although recently the UPRmt has been shown to regulate mitochondrial protein synthesis that would rely on amino acids, whose transport depends on pept-1. On the other hand, sucl-2, C33F10.12, pdha-1, R04F11.2 all encode mitochondrial proteins that form or interact with multi-subunit proteins, where perturbations in stoichiometry have been shown to induce the UPRmt [28, 30, 34]. Inactivation of these five genes by RNAi led to a range of UPRmt reporter induction (Fig. 1B, C), from 2.3 to 13.7-fold that had no correlation with the previously reported levels of hypoxia resistance where pept-1(RNAi) was most resistant and sucl-2(RNAi) least resistant to hypoxia [9]. atfs-1 is a transcription factor required for UPRmt activation [35, 36]. The induction of hsp-6p::GFP by the five UPRmt-activating HypR RNAis was fully suppressed by an atfs-1 loss-of-function mutation (Fig. 1D, E), indicating that the induction of hsp-6p::GFP by the five Hyp RNAis is acting through the canonical UPRmt pathway.
Hypoxia induces mitochondrial protein aggregation (HIMPA) [6, 26]. We found that inactivation of all five UPRmt-activating HypR genes strongly lowered HIMPA as assessed by the ucr-11::GFP mitochondrial protein aggregation reporter [6] (Fig. 1F, G). As shown previously, atfs-1(lf) also reduced the aggregate levels; only pept-1(RNAi) significantly reduced aggregates further in the atfs-1(lf) background indicating that at least part of the effect of pept-1(RNAi) is independent of atfs-1 (Fig. 1H). atfs-1(lf) did not suppress the hypoxia resistance of any of the five RNAis; thus, neither atfs-1 function nor UPRmt activation is essential to their mechanisms of hypoxia resistance (Fig. 1I). These results suggest that while resistance to hypoxic cell death and HIMPA may be mechanistically associated and that a functional UPRmt may be critical for aggregate formation, the UPRmt is not required for protection from hypoxia and in general activation of the UPRmt does not appear to commonly accompany a hypoxia-resistant phenotype in C. elegans.
UPRmt activation reduces HIMPA
To examine more broadly the role of the UPRmt in HIMPA, we tested 12 RNAis previously shown to activate the UPRmt (Supplementary Table S2) [37]. We first confirmed that all 12 RNAis did indeed activate the UPRmt as measured by atfs-1-dependent induction of the hsp-6::GFP transcriptional reporter (Fig. 2A, C). All 12 UPRmt-activating RNAis strongly reduced HIMPA (Fig. 2B). Unlike for the pept-1(RNAi), none of the 12 RNAis significantly reduced HIMPA further in the atfs-1(lf) background (Fig. 2D). Again, atfs-1(lf) paradoxically reduced HIMPA in the no RNAi empty vector condition but not to the same degree of reduction as the UPRmt RNAis. The mitochondrial translation inhibitors doxycycline and meclocycline induce the UPRmt in an atfs-1-dependent manner (Supplementary Fig. S1) [26, 38]. Likewise, doxycycline and meclocycline reduced HIMPA and did not further reduce the aggregate levels in atfs-1(lf) animals (Fig. 2E). Thus both genetic and pharmacological UPRmt activators reduce HIMPA, while at the same time the UPRmt master regulator gene atfs-1 paradoxically promotes HIMPA, as previously reported [6].

A UPRmt-activating RNAis as assessed by hsp-6p::GFP expression. hsp-6p::GFP induction was quantified for 12 RNAis previously shown to induce the UPRmt. Fold induction of GFP fluorescence is the mean fluorescence relative to empty vector RNAi (L4440) (n = 10). Data were obtained from 10–36 individual animals for all panels; mean ± SD; **p < 0.01, ****p < 0.0001, unpaired t-test. B UCR-11:GFP HIMPA reduced by the UPRmt activating RNAis. C UPRmt activation is partially or completely blocked in the atfs-1(tm4919 lf) animals. D atfs-1(lf) reduces aggregates and suppresses a further reduction in aggregates by UPRmt-activating RNAis. E Mitochondrial translation inhibitors (doxycycline and meclocycline) reduce HIMPA in a non-additive manner along with atfs-1(lf). **p < 0.01, ***p < 0.001, ****p < 0.0001, n or ns not significant versus EV @ Bonferroni-corrected p < 0.01, unpaired t-test.
Activation of UPRmt does not necessarily confer hypoxia resistance
We tested whether the 12 UPRmt-activating RNAis were protective from hypoxic organismal death like the five UPRmt-activating HypR RNAis and like doxycycline and meclocycline [9, 26, 38]. Five RNAis (letm-1, pdhb-1, timm-17B1, tin-44, and tomm-22) produced significant hypoxia resistance (Fig. 3A–E); five RNAis (cco-1, dnj-21, mrpl-44, lpd-9, and wah-1) showed no significant effect on hypoxic sensitivity (Fig. 3F–J); 2 RNAis (dlst-1, F15D3.6) produced hypersensitivity to hypoxia (Fig. 3K, L). Interestingly, unlike for the UPRmt-activating RNAis identified in our screen for strongly hypoxia resistant RNAis (Fig. 1), the relatively weak hypoxia resistance of these UPRmt-activating RNAis was atfs-1-dependent (Fig. 3M–Q). Thus, our data shows that activation of the UPRmt may in some genetic contexts confer atfs-1-dependent mild hypoxia protection, but this is by no means a necessary consequence of UPRmt activation.

The twelve UPRmt-activating RNAis are tested for hypoxic sensitivity in the wild type background A–L and in the atfs-1(lf) mutant background M–Q; significance @ p < 0.0001, F-test, simultaneous nonlinear regression; data for each point is from 3 to 12 independent trials with at least 100 animals/trial, mean +/− sem. A–E Five RNAis produced significant hypoxia resistance; F–J Five RNAis have no significant effect on hypoxic sensitivity; and K, L Two RNAis produced significant hypersensitivity to hypoxia. M–Q The hypoxia resistance of the five RNAis is suppressed by atfs-1(lf).
TMRE stains mitochondrial aggregates
While examining the effect of hypoxia on mitochondrial membrane potential, we discovered that the voltage sensitive mitochondrial dye tetramethylrhodamine ethyl ester (TMRE) not only stained functional mitochondria, but also stained hypoxia-induced mitochondrial protein aggregates (Fig. 4A–C). This finding in the wild type N2 strain importantly demonstrates that aggregates occur in an animal with native mitochondrial proteins. We then compared the aggregates visualized with TMRE to those visible with the ucr-11::GFP reporter. The TMRE-stained aggregates strongly colocalized with UCR-11::GFP aggregates (Fig. 4D–F). Besides demonstrating importantly that the UCR-11::GFP aggregates are not an artifact of an overexpressed and GFP-tagged protein, TMRE staining can be used in all C. elegans strains to detect mitochondrial aggregates and potentially in other models of hypoxic injury.

A Wild type worms exposed to 12 h of hypoxia, stained with TMRE and mounted immediately. TMRE stains the mitochondria and HIMPA. B Normoxic wild type worms (not exposed to hypoxia) stained with TMRE and mounted immediately. Few TMRE-stained aggregates in normoxic wild type animals. C Quantitation of aggregates in wild type worms. Mean ± SD of ten animals ****p < 0.0001, unpaired t-test. D HIMPA visible by GFP in gcIs46[UCR-11::GFP]. E HIMPA visible with TMRE staining in gcIs46. F Merge of D and E. 79.7% of UCR-11::GFP aggregates colocalized with TMRE aggregates in muscle (n = 1472). 93.5% of TMRE aggregates in muscle colocalized with UCR-11::GFP aggregates (n = 1255). Number of images = 50. Scale bars = 2 µm.
HIMPA blocked by hypoxia resistance without UPRmt activation
We have previously identified two distinct mechanisms, the insulin/IGF-receptor pathway and the translation machinery, that regulate cytoplasmic proteostasis and profoundly modulate hypoxic survival. The insulin/IGF-receptor pathway has been shown to strongly regulate hypoxic sensitivity, aging, and proteostasis in C. elegans [12, 23, 39, 40]. daf-2 encodes the C. elegans insulin/IGF receptor, and daf-2 reduction-of-function mutants are highly resistant to hypoxic injury via a mechanism that requires the FOXO transcription factor DAF-16 [12, 23]. To test whether the daf-2 pathway affected HIMPA, we built the daf-2(e1370lf);gcIs46[ucr-11p::GFP] strain, confirmed that it was strongly hypoxia resistant (Fig. 5A), and then scored for HIMPA. daf-2(e1370) strongly reduced HIMPA as measured by UCR-11::GFP and TMRE (Fig. 5B–E, G, H). HIMPA and hypoxic sensitivity were restored in the daf-2(e1370);daf-16(mu86) double mutant strain (Fig. 5A, F, H). daf-2 RNAi, which is strongly protective from hypoxic injury [10], did not activate the UPRmt and therefore the effect of daf-2(rf) on reducing aggregates is not explained by UPRmt activation (Supplementary Fig. S2).

A As previously reported [12], daf-2(e1370) animals are hypoxia resistant in a daf-16 dependent manner and in the gcIs46 background (mean ± SD, n = 4–12 independent trials). B, C Comparison of gcIs46::GFP HIMPA in the wild type B and daf-2(e1370) background C. D–F Comparison of HIMPA by TMRE in wild type (N2), daf-2(e1370), and in daf-2(e1370);daf-16(mu86). G Quantitation of GFP HIMPA in gcIs46 and daf-2(e1370);gcIs46 (mean ± SD of 18 animals). H Quantitation of TMRE HIMPA in the strains with the noted genotypes (mean ± SD of 11–24 animals). ****p < 0.0001, unpaired t-test. Scale bars = 2 µm.
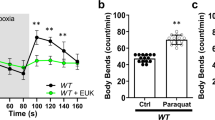
The translation machinery has been repeatedly implicated in C. elegans hypoxic sensitivity [5, 8, 9, 11]. Translation machinery mutations and RNAis that reduce protein synthesis are strongly hypoxia protective [5, 11]. tars-1(gc52) and rars-1(gc47) carry reduction of function mutations in threonyl-aminoacyl and arginyl-aminoacyl tRNA synthetases, respectively, and are strongly resistant to hypoxic death (Fig. 6A), as previously shown [5, 11]. Both translation machinery mutants fully blocked HIMPA as measured by both UCR-11::GFP and TMRE (Fig. 6B–E, G, H). A combination of loss-of-function mutations in two negative regulators of the translation machinery, larp-1 and ncl-1 [5], suppresses the hypoxia resistance of tars-1(gc52) (Fig. 6A) as previously reported [5] and likewise restores HIMPA (Fig. 6F, H). As was seen for daf-2, RNAi knockdown of rars-1 did not activate UPRmt (Supplementary Fig. S2), demonstrating the effect on rars-1 reduction-of-function on hypoxic death and HIMPA was not due to an activation of the UPRmt. These results indicate that hypoxia protective mechanisms of daf-2/daf-16 signaling and reduction of translation do not require UPRmt activation yet protect from HIMPA.

A As previously reported [5, 11], rars-1 (gc47) and tars-1(gc52) animals are resistant to hypoxia and the resistance of tars-1(gc52) is maintained in a gcIs46 background and is suppressed by larp-1(q783)ncl-1(gc53) (mean ± SD of 6–12 independent trial with >100 animals/trial). B, C Comparison of gcIs46::GFP HIMPA in the wild type (B) and tars-1(gc52) backgrounds (C). D–F Comparison of HIMPA by TMRE in wild type (N2), tars-1(gc52), and in tars-1(gc52); larp-1(q783)ncl-1(gc53) animals G Quantification of GFP aggregates in gcIs46 and tars-1(gc52);gcIs46 (mean ± SD of 12–19 animals). H Quantification of TMRE aggregates in the strains with the noted genotypes (mean ± SD of 10–28 animals).****p < 0.0001, unpaired t-test. Scale bars = 2 µm.
Discussion
Our study shows that a hypoxia resistant phenotype in C. elegans is not commonly accompanied by UPRmt activation and that UPRmt activation is not always hypoxia protective. In other words, UPRmt activation is neither necessary nor sufficient for protection from hypoxic cell death in C. elegans. However, among RNAis selected solely for their ability to induce the UPRmt we did find a strong enrichment for hypoxia resistance, where five of the twelve UPRmt-activating RNAis conferred hypoxia resistance. By comparison, in a screen of over 16,000 RNAis, only 199 were found to reproducibly produce hypoxia resistance [9]. While these twelve are a small sample and the whole genome RNAi screen deliberately ignored weak hypoxia resistance phenotypes, this approximately 30-fold enrichment for hypoxia resistance suggests that UPRmt activation may provide hypoxia protection in certain genetic backgrounds.
We also examined the effect of hypoxia resistance and UPRmt activation on HIMPA. We found that resistance to hypoxic death and resistance to HIMPA were invariantly associated. All RNAis or mutants that were resistant to hypoxic death reduced HIMPA. Importantly, the strongly hypoxia resistant daf-2, tars-1, and rars-1 mutants or RNAis block HIMPA without activating the UPRmt. Thus, UPRmt activation is neither necessary for resistance to hypoxia nor for abrogation of HIMPA. We also found that all UPRmt-activating RNAis as well as doxycycline and meclocycline, which strongly activate the UPRmt, brought hypoxia-induced mitochondrial aggregates to near background levels. To be clear, both hypoxia-resistant and non-hypoxia resistant UPRmt-activating RNAis strongly reduced HIMPA. The effect of the RNAis on activation of the UPRmt was mostly if not completely dependent on a functional atfs-1 gene. This was to be expected as hsp-6 activation is dependent on ATFS-1 [34, 36].
The reduction in the aggregates by both UPRmt activation and atfs-1 loss-of-function is a puzzling paradox, albeit consistent with our previously published results [6]. We previously showed that atfs-1 RNAi and the atfs-1(tm4919) loss-of-function mutant reduced HIMPA and an atfs-1 gain-of-function mutant increased aggregates [6]. We had also previously shown that cco-1, letm-1, and F02A9.4 RNAis reduced HIMPA, but we attributed this effect to their hypoxia resistance or reduction in the levels of their corresponding proteins that might form the aggregates themselves [6]. We found here that of these three only letm-1 RNAi reproducibly conferred hypoxia resistance yet all three RNAis, indeed all of the UPRmt activating RNAis, strongly reduced HIMPA. Thus, clearly hypoxia resistance is not necessary for this effect. To reconcile that both atfs-1(lf) and atfs-1-dependent UPRmt activation reduce aggregates, we hypothesize that ATFS-1 functions both to inhibit HIMPA by activating the UPRmt and to promote HIMPA through some other unknown mechanism (Fig. 7). ATFS-1 could both function to reduce aggregates by activation of its downstream UPRmt targets, and in the absence of UPRmt activation function to promote HIMPA through an unknown mechanism. The fact that the level of activation of the hsp-6::GFP transcriptional reporter does not correlate with the ability to suppress hypoxia-induced aggregates (Fig. 2A, B) suggests that the HSP-6 chaperone levels are not directly responsible for the effect of the UPRmt on aggregation, or that a very low threshold of hsp-6 induction suppresses aggregation fully. Which atfs-1-regulated protein(s) might be responsible for promoting HIMPA is unknown; ATFS-1 has been shown to control the synthesis of a large number of proteins involved in mitochondrial proteostasis and mitochondrial function as well as some non-mitochondrial proteins [34, 41, 42]. Likewise, cytoplasmic and ER proteostasis pathways regulate the UPRmt and mitochondrial proteostasis [42]. Thus, ATFS-1 may function in concert with one of those non-UPRmt pathways to regulate HIMPA directly.

ATFS-1, the UPRmt master regulator, both reduces HIMPA by activating the UPRmt and promotes HIMPA by an unknown mechanism. HIMPA may contribute to but is not required for cellular and organellar death.
An important technical advance of this work is the discovery that TMRE stains HIMPA. TMRE is a lipophilic cationic dye that accumulates in the matrix of viable mitochondrial, where the membrane potential is highly negative relative to the intermembrane space [43, 44]. Besides membrane potential driven accumulation, TMRE has been shown to bind to both surfaces of the inner membrane; however, whether it is binding to lipids, proteins, or both at the inner mitochondrial membrane is unclear [43]. The aggregates appear to be in the matrix of the mitochondria although the resolution is inadequate to be certain. Other than UCR-11::GFP which strongly colocalizes with the TMRE-stained aggregates, we have no empirical evidence for what composes the TMRE-aggregates. In principle, TMRE can bind non-specifically to hydrophobic and negatively charged surfaces. Thus, denatured mitochondrial proteins, lipids, and even nucleic acids are reasonable candidates. Regardless of the nature of the binding target, the discovery that TMRE binds to hypoxia-induced mitochondrial aggregates rules out that HIMPA is an artifact of the GFP reporter overexpression. TMRE can be used to examine for HIMPA in any hypoxic/ischemic model without the need for expression of a tagged protein, electron microscopy, or immunofluorescence.
In summary, resistance to hypoxic organismal death and resistance to hypoxia-induced mitochondrial protein aggregation are strongly correlated. This suggests that HIMPA may be an early hypoxic pathology along the pathway to eventual hypoxic cell death. However, it is important to emphasize that we have not directly measured any aspect of mitochondrial function to determine whether HIMPA correlates with abnormalities in mitochondrial function. On the other hand, activation of the UPRmt is neither necessary nor sufficient for hypoxia resistance but may weakly promote hypoxic survival in certain contexts. Activation of the UPRmt did invariably reduce HIMPA whether or not it conferred resistance to hypoxic death. This effect of the UPRmt favors a model, where the UPRmt directly abrogates mitochondrial protein aggregation during hypoxia. Future investigation will continue to define the breadth of mechanisms that protect cells from hypoxic death, to define the components of the UPRmt that inhibit HIMPA, and the ATFS-1-regulated mechanism that promotes it.
Materials and methods
C. elegans strains and culture methods
C. elegans strains were cultured and maintained at 20 °C on NGM agar with OP50 E. coli food unless otherwise noted [45]. The N2 (Bristol) strain was the standard wild-type strain from the Caenorhabditis Genetics Center (CGC, University of Minnesota). The mitochondrial UPR transcriptional reporter strain SJ4100 (zcIs13[hsp-6p::GFP]) was obtained from the CGC. gcIs46 [ucr-11::GFP] and atfs-1(tm4919);gcIs46 were generated as previously described [6]. atfs-1(tm4919);zcIs13, daf-2(e1370);gcIs46, rars-1(gc47);gcIs46, and tars-1(gc53);gcIs46 were generated in this study by routine genetic crosses and confirmation by phenotypes and/or PCR genotyping.
Hypoxic incubations and hypoxic death
Synchronized young adult worms were subjected to hypoxia as described previously except that hypoxic incubation temperature was 26–26.5 °C [12, 46]. Briefly, each plate of worms was washed into a 1.5 ml tube with 1 ml of M9 buffer (22 mM KH2PO4, 22 mM Na2HPO4, 85 mM NaCl, and 1 mM MgSO4), and after the worms settled by gravity, all but 100 μl of M9 was removed. The tubes were then placed in the anaerobic chamber (Forma Scientific, Waltham, MA, USA) for incubation times ranging from 10 to 34 h as indicated. Oxygen tension was always ≤ 0.3%. Following the hypoxic incubation unless otherwise noted, worms were transferred using glass Pasteur pipettes onto seeded NGM plates and recovered at 20 °C for 24 h. Normoxic incubations were otherwise identical except performed in a 26.5 °C room air incubator. Organismal death was scored as previously described [46]. Briefly, animals were scored as dead if pharyngeal pumping, spontaneous and evoked movement (touching with a platinum wire) were not observed. For the mitochondrial aggregates assay, synchronized worms were kept on NGM plates and placed into anaerobic chamber for 12 h and then mounted for confocal imaging on glass slides [6].
RNAi experiments
The bacterial RNAi feeding strain L4440 carrying the empty RNAi vector strain was from the Ahringer C. elegans RNAi library (MRC Gene service, Swindon, UK) and was used as the negative control (Empty Vector-EV) for all RNAi experiments [47]. Bacterial strains containing RNAi plasmids were cultured and induced with 0.1% β-lactose in 100 mg/ml ampicillin for 24 h at 23 °C as described previously [9]. Worms were synchronized on RNAi plates for 3.5 days (N2) or 4.5 days (rars-1(gc47) background) until reaching adulthood; worms not reaching adulthood were excluded.
Mitochondrial UPR reporter and screening
SJ4100 (zcIs13[hsp-6p::GFP]) was used as a reporter for mitochondrial UPR activation [28]. Animals were synchronized from eggs by bleaching gravid adults, allowed to develop to adulthood, and then mounted on agar pads for imaging. All UPRmt fluorescent reporter determinations were performed with worms exposed only to normoxic conditions. Briefly, a minimum of ten worms were mounted onto an agar pad on a standard microscope slide and imaged on a Zeiss axioskope 2 at ×10 magnification. Using ImageJ software (https://imagej.nih.gov/ij/), each worm was outlined and the fluorescence intensity within the area was measured and background intensity was then subtracted to obtain the total intensity/worm as previously described [26].
Chemical treatment and TMRE staining
Concentrated stocks of doxycycline (225 mM) and meclocycline (294 mM) in DMSO (Dimethyl sulfoxide) (Sigma-Aldrich Corp, St. Louis, MO, USA), were diluted to a final concentration of 100 μM with nuclease-free, deionized water and 300 µl was spread evenly onto a NGM plate seeded with OP50 onto which were placed synchronized eggs allowed to develop into 1-day-old adults. Plates containing TMRE (ThermoFisher, Waltham, MA, USA) were made the same way as described above with 300 µl at a final concentration of 5 μM spread onto the plate. For TMRE staining, 1-day-old synchronized worms were transferred onto the TMRE-containing plates for 2 h before the plates were placed into the anaerobic chamber for 12 h for hypoxic exposure.
Aggregate imaging and counting
Images of UCR-11::GFP and TMRE staining were acquired with confocal microscopy as previously described [6, 26]. Briefly, paralysis was produced by mounting worms in a solution of 50 mM levamisole (Sigma-Aldrich Corp., St. Louis, MO, USA) in M9 prior to imaging. Images were acquired from at least 5–10 randomly selected worms at 1024 × 1024 resolution using a 63× objective with 8 × zoom producing a 23.07 × 23.07 μm image (defined as one high power field, HPF). All images were acquired as a ten slice Z-stack with scan speed of 800–1800 Hz and flattened as a maximum intensity projection prior to analysis. The wavelength of the laser to capture the signal for GFP and TMRE signals was 500–530 nm and 620–700 nm, respectively. Aggregates were counted per HPF by an observer blinded to condition.
Statistics
Two-sided unpaired or paired, as appropriate, t-tests were used for statistical comparisons and all variances were of a similar magnitude except for death versus hypoxic exposure time curves (Fig. 3), where simultaneous curve-fitting was performed [48, 49]. Simultaneous curve-fitting forced the test RNAi death curve fit and the L4440 empty vector control curve fit to the same hypoxic exposure time producing 50% death (LT50) and F-tests were performed for statistical significance of the change in variance of the LT50 versus fitting each curve independently. Statistics were calculated using GraphPad Prism 6.01 (San Diego, CA, USA) for simultaneous curve-fitting, t-tests, or Excel 2007 for t-tests (Microsoft, Redmond, WA). Values are expressed as mean ± SD of at least three independent experiments. A minimum p value of ≤0.05 was considered significant and was Bonferroni-corrected for multiple comparisons where appropriate. Sample sizes for technical replicates for number of animals was based on the variability from animal to animal for fluorescence-based assays and was always ≥10 or a minimum of 30 animals, where a percent of the population was scored. Based on our previous experience with hypoxic death population assays biological replicates were at least three per condition, and if significant variability was observed among replicates more were performed until clear reproducibility was achieved. In terms of randomization, strains were not randomized as they are genetically identical and raised under environmentally identical conditions.
Data availability
The raw data from the screen of the RNAi strain set are available upon request. All other data generated and analyzed during this study are included in the published article. The unique C. elegans strains generated for this study are available upon request.
References
Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15:1239–53.
Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:343–9.
Storey KB, Storey JM. Metabolic rate depression: the biochemistry of mammalian hibernation. Adv Clin Chem. 2010;52:77–108.
Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009;20:332–40.
Itani OA, Zhong X, Tang X, Scott BA, Yan JY, Flibotte S, et al. Coordinate regulation of ribosome and tRNA biogenesis controls hypoxic injury and translation. Curr Biol. 2021;31:128–37. e5
Kaufman DM, Wu X, Scott BA, Itani OA, Van Gilst MR, Bruce JE, et al. Ageing and hypoxia cause protein aggregation in mitochondria. Cell Death Differ. 2017;24:1730–8.
Mao XR, Kaufman DM, Crowder CM. Nicotinamide mononucleotide adenylyltransferase promotes hypoxic survival by activating the mitochondrial unfolded protein response. Cell Death Dis. 2016;7:e2113.
Scott B, Sun CL, Mao X, Yu C, Vohra BP, Milbrandt J, et al. Role of oxygen consumption in hypoxia protection by translation factor depletion. J Exp Biol. 2013;216:2283–92.
Mabon ME, Mao X, Jiao Y, Scott BA, Crowder CM. Systematic identification of gene activities promoting hypoxic death. Genetics 2009;181:483–96.
Mabon ME, Scott BA, Crowder CM. Divergent mechanisms controlling hypoxic sensitivity and lifespan by the DAF-2/insulin/IGF-receptor pathway. PLoS ONE. 2009;4:e7937.
Anderson LL, Mao X, Scott BA, Crowder CM. Survival from hypoxia in C. elegans by inactivation of aminoacyl-tRNA synthetases. Science 2009;323:630–3.
Scott BA, Avidan MS, Crowder CM. Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science 2002;296:2388–91.
Heimbucher T, Hog J, Gupta P, Murphy CT. PQM-1 controls hypoxic survival via regulation of lipid metabolism. Nat Commun. 2020;11:4627.
Iranon NN, Jochim BE, Miller DL. Fasting prevents hypoxia-induced defects of proteostasis in C. elegans. PLoS Genet. 2019;15:e1008242.
Doshi S, Price E, Landis J, Barot U, Sabatella M, Lans H, et al. Neuropeptide signaling regulates the susceptibility of developing C. elegans to anoxia. Free Radic Biol Med. 2018;131:197–208.
Pena S, Sherman T, Brookes PS, Nehrke K. The mitochondrial unfolded protein response protects against anoxia in caenorhabditis elegans. PLoS ONE. 2016;11:e0159989.
Ladage ML, King SD, Burks DJ, Quan DL, Garcia AM, Azad RK, et al. Glucose or altered ceramide biosynthesis mediate oxygen deprivation sensitivity through novel pathways revealed by transcriptome analysis in Caenorhabditis elegans. G3. 2016;6:3149–60.
LaMacchia JC, Frazier HN, 3rd, Roth MB. Glycogen fuels survival during hyposmotic-anoxic stress in Caenorhabditis elegans. Genetics. 2015;201:65–74.
Garcia AM, Ladage ML, Dumesnil DR, Zaman K, Shulaev V, Azad RK, et al. Glucose induces sensitivity to oxygen deprivation and modulates insulin/IGF-1 signaling and lipid biosynthesis in Caenorhabditis elegans. Genetics. 2015;200:167–84.
Fawcett EM, Hoyt JM, Johnson JK, Miller DL. Hypoxia disrupts proteostasis in Caenorhabditis elegans. Aging Cell. 2015;14:92–101.
Flibotte JJ, Jablonski AM, Kalb RG. Oxygen sensing neurons and neuropeptides regulate survival after anoxia in developing C. elegans. PLoS ONE. 2014;9:e101102.
Menuz V, Howell KS, Gentina S, Epstein S, Riezman I, Fornallaz-Mulhauser M, et al. Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science 2009;324:381–4.
Mendenhall AR, LaRue B, Padilla PA. Glyceraldehyde-3-phosphate dehydrogenase mediates anoxia response and survival in Caenorhabditis elegans. Genetics 2006;174:1173–87.
Nystul TG, Goldmark JP, Padilla PA, Roth MB. Suspended animation in C. elegans requires the spindle checkpoint. Science 2003;302:1038–41.
Jiang H, Guo R, Powell-Coffman JA. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci USA. 2001;98:7916–21.
Kaufman DM, Crowder CM. Mitochondrial proteostatic collapse leads to hypoxic injury. Curr Biol. 2015;25:2171–6.
Naresh NU, Haynes CM. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb Perspect Biol. 2019;11:a033944.
Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–66.
Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006;174:229–39.
Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–80.
Durieux J, Wolff S, Dillin A. The cell non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91.
Meissner B, Boll M, Daniel H, Baumeister R. Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. J Biol Chem. 2004;279:36739–45.
Geillinger KE, Kuhlmann K, Eisenacher M, Giesbertz P, Meyer HE, Daniel H, et al. Intestinal amino acid availability via PEPT-1 affects TORC1/2 signaling and the unfolded protein response. J. Proteome Res. 2014;13:3685–92.
Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. 2015;58:123–33.
Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–40.
Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012;337:587–90.
Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, et al. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun. 2014;5:3483.
Sun CL, Zhang H, Liu M, Wang W, Crowder CM. A screen for protective drugs against delayed hypoxic injury. PLoS ONE. 2017;12:e0176061.
Kenyon CJ. The genetics of ageing. Nature 2010;464:504–12.
Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015;161:919–32.
Shpilka T, Du Y, Yang Q, Melber A, Uma Naresh N, Lavelle J, et al. UPR(mt) scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nat Commun. 2021;12:479.
Anderson NS, Haynes CM. Folding the mitochondrial UPR into the integrated stress response. Trends Cell Biol. 2020;30:428–39.
Scaduto RC Jr., Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999;76:469–77.
Teodoro JS, Machado IF, Castela AC, Rolo AP, Palmeira CM. The evaluation of mitochondrial membrane potential using fluorescent dyes or a membrane-permeable cation (TPP(+)) electrode in isolated mitochondria and intact cells. Methods Mol. Biol. 2020;2184:197–213.
Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974;77:71–94.
Sun CL, Kim E, Crowder CM. Delayed innocent bystander cell death following hypoxia in Caenorhabditis elegans. Cell Death Differ. 2014;21:557–67.
Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003;421:231–7.
Waud DR. On biological assays involving quantal responses. J Pharmacol Exp Ther. 1972;183:577–607.
DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–102.
Acknowledgements
We thank Barbara Scott for her technical contributions to the work.
Funding
The work was supported by National Institute of Neurological Disorders and Stroke R01NS109088 (C.M.C., and M.V.G.), R01NS100350 (C.M.C.) and R01GM129034 (M.V.G.).
Author information
Authors and Affiliations
Contributions
C.S., M.V.G., and C.M.C. conceived the study. J.Y., C.S., and S.S. performed the experiments. C.S., M.V.G., J.Y., and C.M.C. wrote the manuscript with input from all co-authors.
Corresponding author
Ethics declarations
Ethics statement
There is no requirement for ethical approval of this work as it did not involve the study of humans, human material, or vertebrate animals.
COMPETING INTERESTS
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by D. Bano
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, J., Sun, CL., Shin, S. et al. Effect of the mitochondrial unfolded protein response on hypoxic death and mitochondrial protein aggregation. Cell Death Dis 12, 711 (2021). https://doi.org/10.1038/s41419-021-03979-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03979-z
This article is cited by
-
Hypoxia-induced mitochondrial stress granules
Cell Death & Disease (2023)
