
Abstract
Ferroptosis is a non-apoptotic form of cell death characterized by overwhelming iron-dependent lipid peroxidation, which contributes to a number of pathologies, most notably tissue ischemia/reperfusion injury, neurodegeneration and cancer. Cysteine availability, glutathione biosynthesis, polyunsaturated fatty acid metabolism and modulation of the phospholipidome are the key events of this necrotic cell death pathway. Non-enzymatic and enzymatic lipoxygenase (LOX)-mediated lipid peroxidation of lipid bilayers is efficiently counteracted by the glutathione (GSH)/glutathione peroxidase 4 (GPX4) axis. Preliminary studies suggest that bursting ferroptotic cells release pro-inflammatory damage-associated molecular patterns (DAMPs) that trigger the innate immune system as exemplified by diseased kidney and brain tissues where ferroptosis contributes to organ demise in a predominant manner. The GSH/GPX4 node is known to control the activities of LOX and prostaglandin-endoperoxide synthase (PTGS) via the so-called peroxide tone. Since LOX and PTGS products do have pro- and anti-inflammatory effects, one may speculate that these enzymes contribute to the ferroptotic process on several levels in cell-autonomous and non-autonomous ways. Hence, this review provides the reader with an outline on what is currently known about the link between ferroptosis and necroinflammation and discusses critical events that may alert the innate immune system in early phases when cells become sensitized towards ferroptosis.
Similar content being viewed by others
Facts
-
Ferroptosis is a regulated form of necrotic cell death characterized by iron-dependent lipid peroxidation.
-
GPX4 is the key regulator of ferroptosis by quenching lipid peroxidation.
-
Initial data suggest that ferroptotic cells trigger the innate immune system in mouse models of brain and kidney disease.
-
The glutathione (GSH)/GPX4 node controls LOX and PTGS activities via the so-called peroxide tone.
Open questions
-
What is the nature of DAMPs released by ferroptotic cells?
-
What is the sequence of events causing a full-blown immune cell activation?
-
Does an impaired glutathione (GSH)/GPX4 axis prior to ferroptosis contribute to an activation of the innate immune system via increased LOX/PTGS signaling?
-
Are cancer cells undergoing ferroptosis immunogenic?
-
Is there a role of ferroptosis in physiological contexts?
Ferroptosis is a regulated necrotic cell death routine characterized by the iron-dependent generation of lethal amounts of lipid hydroperoxides. It clearly differs from other forms of cell death such as apoptosis, necroptosis and pyroptosis, in morphological, biochemical and genetic terms [1]. First discovered in the context of cancer through ferroptosis inducing agents (FIN) able to elicit ferroptotic cell death in certain tumor cells, ferroptosis is now widely accepted to contribute to various disease processes related to ischemia/reperfusion injury, neurodegeneration and tissue demise [2]. Yet, to the best of our knowledge very little (if at all) is known about the role of ferroptosis in physiological processes or development [3].
Pathological conditions arising from tissue injury, degeneration and cancer are often linked to inflammatory responses. Therefore, mechanisms must be in place for immune cells to discriminate between different forms of cell death and conversely detect signals sent out by dying cells for eliciting immune responses [4]. During the normal physiological context of tissue homeostasis and development, cells dying by apoptosis generally retain plasma membrane integrity, which allows for their rapid clearance by macrophages and/or other phagocytotic cells in vivo thereby avoiding autoimmune reactions. In contrast, cells dying via necrotic mechanisms, regulated or accidental, are characterized by plasma membrane rupture and release of intracellular components such as damage-associated molecular patterns (DAMPs), an immunogenic process, also known as necroinflammation. In the context of tissue injury and degeneration it was hypothesized that DAMP accumulation can trigger both tissue inflammation and the activation of further pathways of regulated cell death in an auto-amplification manner [5]. However, the exact sequence causing the events of cell death and immune cell activation still remains elusive.
In tumor cells, ferroptosis is generally triggered by pharmacological means through FINs. Due to restricted availability of metabolically stable FINs for the use of in vivo studies in tumor-bearing mice, it remains to be investigated whether tumor cells succumbing to ferroptosis are (i) immunogenic per se, (ii) enough immunogenic to achieve proper anti-tumor immune responses or (iii) immunosuppressive by encouraging accumulation of immunosuppressive cells in the long run. As to date mere circumstantial evidence for modification of the immune system by cells that die via ferroptosis is available, further studies are warranted.
The ferroptotic death pathway
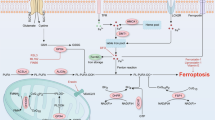
The most upstream player in the ferroptosis signaling cascade is the transmembrane cystine–glutamate antiporter system xc−, consisting of a light chain, xCT (SLC7A11), and a heavy chain, 4F2 (SLC3A2) (Fig. 1). System xc− belongs to the family of heterodimeric amino acid transporters and functions by exchanging extracellular cysteine/cystathionine for intracellular glutamate in a 1:1 ratio [6, 7]. Pharmacologically, system xc− can be inhibited by high concentrations of extracellular glutamate, erastin, sulfasalazine or sorafenib [1, 8,9,10]. Once inside the cell, cystine is reduced by glutathione (GSH) or thioredoxin reductase 1 (TXNRD1) [11] to cysteine, for further biosynthesis of the tripeptide GSH in a two-step reaction involving γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthase (GSS) [12]. The enzyme γ-GCS can be inhibited by L-buthionine-(S,R)-sulfoximine (BSO) thereby causing depletion of GSH and ferroptotic cell death. Alternatively, intracellular cysteine may be obtained via the transsulfuration pathway [13], while intracellular glutamate for GSH synthesis can be provided by conversion of glutamine through glutaminase 2 (GLS2) [14].

The core mechanisms controlling ferroptotic cell death. System xc- (a heterodimeric amino acid transporter consisting of 4F2 heavy chain and xCT (SLC7A11) light chain—the latter is shown) is responsible for the uptake of extracellular cystine. Cysteine can also be taken up by the neutral amino acid transporter ASC (not shown), which is highly relevant for in vivo conditions. Cysteine is the substrate-limiting step for glutathione biosynthesis. GSH provides electrons to the key regulator of ferroptosis, GPX4, which is one of the most efficient enzymes in reducing lipid peroxides (–OOH) in membranes to the corresponding alcohols (–OH). Recently, additional enzymes, such as ACSL4 and LPCAT that are directly involved in shaping the cellular phospholipidome, have been added to the list of enzymes contributing to ferroptosis. Oxidation of lipid bilayers during ferroptosis may occur both in an enzymatic (i.e. LOX) and non-enzymatic, autoxidative manner (i.e. radical-mediated; symbolized by the red arrow). Ferroptosis inhibitors are depicted by green boxes, whereas ferroptosis-inducing agents are marked by red boxes. ACSL4 acyl-CoA synthetase long-chain family member 4, GPX4 glutathione peroxidase 4, GSH reduced glutathione, GSSG oxidized glutathione, LOX lipoxygenase, LPCAT lysophosphatidylcholine acyltransferase, PUFA polyunsaturated fatty acid, BSO L-buthionine-sulfoximine
GSH is the main substrate of glutathione peroxidase 4 (GPX4), currently regarded as the key ferroptosis regulator [15, 16]. GPX4 is the only enzyme capable of effectively reducing phospholipid and cholesterol hydroperoxides to their corresponding alcohols using two equivalents of GSH [17]. Subsequently, oxidized GSH (GSSG) is recycled to its reduced form by glutathione reductase (GSR) at the expense of NADPH/H+. Several GPX4 inhibitors have been reported to date, among them (1 S, 3 R)-RSL3 (RSL3) [16], ML162 [18] and altretamine [19] thereby causing ferroptotic cell death. Genetic inactivation [15, 20] or inhibition of GPX4 by pharmacological means [16] results in the accumulation of high amounts of lipid peroxidation products, a hallmark of ferroptosis, also referred to as the lethal lipid signal [21]. Using lipidomics studies, attempts have been made to characterize the lethal lipid signal, which led to the identification of arachidonic (AA; C20:4) and adrenic acid (AdA; C22:4) containing phosphatidylethanolamine (PE) species that are specifically oxidized during ferroptosis execution in vivo and exactly these lipid peroxidation signals could be inhibited using the ferroptosis inhibitor liproxstatin-1 (Lip1) [21]. The enzymes, acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are involved in the incorporation of polyunsaturated fatty acids (PUFA) into membranes (Fig. 1). Conversely, genetic deletion of Acsl4 and to some degree Lpcat3 causes ferroptosis resistance [21,22,23]. The thiazolidinedione class of compounds represented by the insulin sensitizers rosiglitazone and pioglitazone efficiently inhibit ACSL4 and therefore prevent ferroptosis in vitro and in vivo [23].
In general, accumulation of phospholipid hydroperoxides in cells is known to occur via enzymatic pathways involving either GPX4 dysfunction (see above), lipoxygenase (LOX) -mediated oxidation and/or via non-enzymatic, radical-mediated autoxidation [24, 25]. Owing to the discovery that single deletion of Alox15 fails to rescue from acute renal failure caused by Gpx4 knockout induced ferroptosis in vivo, it was suggested that not a single but multiple LOX isoforms may be involved in PUFA peroxidation and subsequent cell death by ferroptosis in mouse kidneys [15]. Recently, experimental evidence has been provided that the ferroptosis inhibitors Lip1 and Ferrostatin-1 (Fer1) and to a smaller extend α-tocopherol (αTOC) are highly efficient inhibitors of lipid autoxidation by acting as radical-trapping antioxidants (RTAs) [26, 27].
The mevalonate pathway has also been implicated in ferroptosis modulation. The ferroptosis inducing compound FIN56 presumably activates squalene synthase (SQS) resulting in coenzyme Q10 (CoQ10) depletion [28]. In addition, statins, known as HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase) inhibitors, sensitize cells to FIN56 induced ferroptosis either by depletion of CoQ10 or through their effects on isopentenylation of the selenocysteine-charged tRNA (Trsp, nuclear encoded tRNA selenocysteine 2 (anticodon TCA)) as part of the GPX4 selenoprotein biosynthesis pathway [28, 29].
Association of the ferroptosis pathway with iron dependence originated from the observation that erastin- and RSL3-induced cell death can be rescued by iron chelation [30, 31]. Additionally, the iron metabolism master regulator iron response element binding protein 2 (IREB2) [1] and transferrin [14] have been associated with ferroptosis modulation. Upon ferroptosis induction, autophagy is activated to degrade the iron storage protein ferritin, mediated by the cargo receptor nuclear receptor coactivator 4 (NCOA4). The NCOA4-mediated autophagic degradation of ferritin, a process termed ferritinophagy, maintains the cellular labile iron pool and subsequently sensitizes cells to ferroptosis [32].
Recently, FINO2 and its analogues were reported as new class of ferroptosis initiators. Compared to erastin, RSL3 and FIN56, FINO2 does not inhibit system xc− or GPX4 and does not result in depletion of GPX4 protein. In contrast, FINO2 was shown to induce ferroptosis through a combination of direct iron oxidation, widespread peroxidation of phospholipids and indirect GPX4 inactivation in a yet unknown manner [33].
Links between ferroptosis and degenerative diseases
A direct link between ferroptosis and human disease is still lacking. However, knockout studies performed in mice have greatly aided in our current understanding of the in vivo relevance of ferroptosis. While the whole body knockout of Gpx4 is early embryonic lethal [34], conditional mouse models have been used to delineate the role of GPX4 in a tissue-specific manner. GPX4 plays a crucial role in protecting neuronal populations in hippocampus, cortex, cerebellum and motor neurons [20, 35,36,37,38]. This protection appears to be neuronal subtype specific, as pro-opiomelanocortinergic and dopaminergic neurons are resistant to GPX4 depletion and ferroptosis induction [39]. Moreover, GPX4 has been shown to be essential for photoreceptor cells, endothelial cells, CD8 positive T-cells, kidney tubular cells and hepatocytes [15, 40,41,42,43].
The ferroptosis inhibitors Lip1 and Fer1 protect from ischemia/reperfusion injury in kidney, liver and brain [15, 36, 44, 45], acute renal failure [15], neurodegeneration [36], intracerebral hemorrhage [46], and liver hemochromatosis [47] in vivo. Furthermore, ferroptosis inhibitors have been successfully used in cellular or ex vivo models of Huntington’s disease, periventricular leukomalacia, myocardial infarction and neuronal transdifferentiation [14, 48, 49].
Ferroptosis and cancer
Ferroptosis was first discovered as a cell death pathway that can be induced in a subset of tumor types and engineered cell lines overexpressing oncogenic RAS by the FINs erastin and RSL3 [1, 16]. Interestingly, ultrasmall poly(ethylene glycol) (PEG)-coated silica nanoparticles, were also shown to induce ferroptosis in cancer cells and tumor-bearing mice, in a manner that could be reversed by the administration of Lip1 [50].
As many cancer cells display high levels of reactive oxygen species (ROS), they generally depend on maintenance of GSH levels for survival and proliferation [51]. Under elevated ROS conditions, endogenous cysteine production was deemed insufficient for GSH synthesis, therefore requiring uptake of extracellular cysteine/cystine via system xc−. Thus, administration of a recombinant cyst(e)inase enzyme resulted in depletion of the extracellular cysteine/cystine pool in mice and non-human primates, selectively causing cell death in cancer cells due to depletion of intracellular GSH [51]. Of note, it was demonstrated that the multikinase inhibitor sorafenib induces ferroptosis in cancer cells by acting as system xc− inhibitor [10, 52]. Recent evidence was also provided that ferroptosis can be modulated by the bona fide tumor suppressor protein p53 [53,54,55,56,57], and possible links between p53 and system xc− expression/activity have been reported [53, 57].
Moreover, it was shown that cancer cell lines displaying a therapy-resistant high-mesenchymal cell state are highly dependent on lipid metabolizing enzymes associated with the ferroptosis pathway. Thus, inactivation of GPX4 in these cell lines rendered cells vulnerable to ferroptotic cell death, whereas epithelial state cells remained unaffected [29]. Similarly, cells persisting treatment with certain kinase inhibitors acquired dependency on GPX4. Therefore, loss of GPX4 by genetic inactivation or pharmacological inhibition resulted in ferroptosis in vitro and prevented relapse in vivo [58]. Another study correlated the dedifferentiation status of melanoma cell lines associated with BRAF kinase inhibitor as well as cytokine resistance to ferroptosis sensitivity [59]. In terms of immunotherapy-associated cytokine-induced dedifferentiation, the authors stimulated melanoma cell lines with tumor necrosis factor α (TNFα) and interferon γ (IFNγ) (T cell secreted cytokines) in vitro and discovered that co-treatment with erastin or RSL3 resulted in increased cell death, compared to vehicle or cytokine controls [59]. These studies suggest that targeting ferroptosis could be a novel way to develop drugs in order to overcome several mechanisms of therapy resistance in cancer. A study in lung tumors showed that while suppression of the iron–sulfur cluster biosynthetic enzyme NSF1 (nitrogen fixation gene 1) alone was not sufficient to induce ferroptosis, it however predisposed cancer cells to undergo ferroptosis [60]. This led the authors to hypothesize that suppression of NSF1 activates the iron-starvation response and triggers ferroptosis in combination with inhibition of glutathione biosynthesis [60].
The enzyme ACSL4 was found to be preferentially expressed in a subset of triple negative breast cancer cell lines and expression of ACSL4 was correlated with sensitivity to ferroptosis induction [23]. As this type of breast cancer is among the most difficult to manage, it stresses the urgency for developing new efficacious in vivo ferroptosis-inducing agents for the treatment of ACSL4 expressing tumors [23].
Signs of necroinflammation in ferroptotic tissue
Unlike the far better understood mechanisms of immunologically non-silent forms of regulated necrotic cell death routines, such as necroptosis and pyroptosis [61], our knowledge about the role of necroinflammation in ferroptosis is still in its infancy. This is undoubtedly owed to the fact that ferroptosis was introduced only in 2012 [1] and that research performed on oxytosis, a closely related—if not identical form of cell death—was mainly performed in cell culture/organoid systems [62], thus naturally lacking the immune compartment. Therefore, it remains to be investigated whether necroinflammation is modulating the death process per se or is activated in response to bursting of cells and concomitant release of DAMPs (alarmins).
Nonetheless, first experimental hints that markers of invading cells of the innate immune system including neutrophils, macrophages and microglial cells can be readily detected in various tissues of kidney and brain strongly suggested that ferroptotic cells indeed release potential immune-stimulating cellular components (Table 1). Perhaps the earliest study pointing towards an activation of the immune system was the report of the first conditional knockout mouse model for GPX4 [20]. Here, neuron-specific deletion of GPX4 (using the Camk2α-Cre transgenic mouse line) caused ataxia and epileptic seizures of newborn mice and a marked loss of pyramidal cells in the CA3 region of the hippocampus. This was accompanied by strong immunoreactivity of brain tissue against glial fibrillary acidic protein (GFAP), a marker for reactive astrogliosis due to dying neurons [20]. Strong GFAP staining in the cerebral cortex of two week old mice was also reported in a model of conditional deletion of the selenocysteine-specific tRNA using Camk2α-Cre and Trspfl/fl transgenic mice [38]. As the selenoenzyme GPX4 is nowadays accepted to be (one of) the main target(s) of selenium particularly in brain [63], one can infer that many of the phenotypes provoked by tissue-specific deletion of Trsp are, in fact, caused by impaired GPX4 expression and activity [64]. In two models of neuron-specific GPX4 ablation, Ran’s group then demonstrated the presence of neuroinflammation (as determined by staining against the microglia marker Iba1 (Ionized calcium binding adaptor molecule 1)) in the lumbar spinal cord region of dying motor neurons and in the CA1 region of the hippocampus [35, 36]. Iba1 positivity in the cerebral cortex was also recently reported in a mouse model expressing two hypomorphic GPX4 alleles [65]. In this model parvalbumin-positive GABAergic interneurons were identified as the specific neuronal subtype requiring selenium-containing GPX4, thus representing the limiting factor for mammalian life [66].
In kidney, using crystal (oxalate)- and folic acid-induced acute kidney injury (AKI) models as well as a genetic model of tamoxifen-inducible whole body GPX4 deletion, unequivocal evidence on the occurrence of necroinflammation in ferroptotic tissue has been provided (Table 1). In tamoxifen-induced Gpx4fl/fl_RosaCreERT2 mice, massive kidney tubular cell death caused acute renal failure and early death of knockout mice [15]. On the electron microscopy level rupturing cells can be observed releasing cell debris, mitochondria and even nuclei into the lumen of kidney tubules (Fig. 2). Although not formally shown for cells dying through ferroptosis, this is most likely linked to the release of classical DAMPs such as ATP, as known for other forms of AKI [67]. In the ferroptotic tissue, a marked activation of macrophages was noticeable as determined by F4/80 immunoreactivity [15]. In the crystal-induced mouse model of AKI, expression of proinflammatory cytokines and the infiltration of neutrophils into the damaged tissue were shown to be dampened by Fer1 [44]. Likewise, Fer1 suppressed the upregulation of pro-inflammatory markers, such as monocyte chemotactic protein 1 (MCP-1), TNFα, F4/80 and Fn14 (Tnfrsf12a, tumor necrosis factor receptor superfamily, member 12a), the latter belonging to the superfamily of TNF receptors [68]. And as detailed below, it seems that necroptosis, as evidenced by an upregulation of canonical players of this form of regulated necrosis, secondary to the primary damage inflicted by folic acid further aggravates the damage occurring during AKI [69].

Ferroptotic kidney tubular cells release cell debris and nuclei that in turn activate the innate immune system. Tamoxifen-inducible whole body GPX4 knockout mice die around two weeks after knockout induction due to massive cell death of kidney tubular cells and associated acute renal failure (see Ref. [15]). Bursting tubular cells release cellular debris including mitochondria (blue arrows) and nuclei into the lumen of kidney proximal tubules (red arrows). F4/80 immunostaining reveals massive activation of macrophages in diseased tissue. Micrographs adopted and modified from Friedmann Angeli et al. 2014 [15]. F4/80 adhesion G protein-coupled receptor E1, TEM transmission electron microscopy
The contribution of known features of ferroptosis including the presence of certain peroxidized lipids and perhaps products of arachidonate metabolism to the process of necroinflammation is conceivable but only little, if at all, studied to date [70]. While arachidonate metabolites are well known to modulate inflammatory events in both ways, peroxidized lipids are directly involved in the ferroptotic cell death per se. Yet, they might also play a non-canonical role in the activation of the innate immune system during the early phases of ferroptosis-sensitized cells as detailed in the following.
Arachidonate metabolism controlled by GPX4 and its potential role in ferroptosis
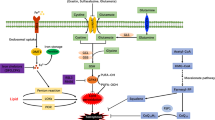
Actually long before the term ferroptosis was introduced and GPX4 established as the central regulator of this cell death paradigm, GPX4 was repeatedly shown to impact on inflammatory conditions by modulating LOX and PTGS activities [71]. Early studies performed from the eighties until the beginning of this century provided evidence that the activities of the arachidonic acid metabolizing enzymes LOX and PTGS are, in fact, controlled by the so-called “peroxide tone” (Fig. 3). The historical term “peroxide tone” describes the preexisting low level of peroxides and in particular lipid peroxides in cells [72], which had been recognized long time ago to increase the activities of the eicosanoid metabolizing enzymes PTGS [73] and LOX [74]. This is based on the fact that PTGS contains heme bound iron (Fe3+), whereas LOX contains non-heme bound iron (Fe2+) in their respective active sites. As both classes of enzymes require iron to be oxidized, in PTGS to yield a ferryl–oxo complex that oxidizes Tyr-385 in the oxygenase active site to produce a tyrosyl radical and in LOX to yield ferric iron, before they can catalyze the incorporation of molecular oxygen into arachidonic acid (PTGS and LOX) and other polyunsaturated fatty acids (LOX), this oxidation is mediated by cellular peroxides.

Arachidonate metabolizing enzymes are controlled by the peroxide tone. Oxidation of heme and non-heme containing enzymes PTGS and LOX enzymes, respectively, by cellular peroxides is essential for full activity. Since the GSH/GPX4 node controls cellular lipid peroxides, the entire PTGS/LOX axis is indirectly regulated by this system. GSH reduced glutathione, GSSG oxidized glutathione, LOX lipoxygenase, GPX4 glutathione peroxidase 4, H2O2 hydrogen peroxide, PGG2 prostaglandin G2, PTGS prostaglandin-endoperoxide synthase, HPETE hydroperoxyeicosatetraenoic acid, (P)LOOH (phospho)lipid hydroperoxide, PUFA polyunsaturated fatty acid
Since GPX4 dampens cellular peroxide levels, modulation of GPX4 expression levels and/or activity, experimentally or (patho)physiologically, indeed controls the levels of products generated by PTGS and LOX. For instance, the 5-LOX products leukotriene C4 (LTC4) and leukotriene B4 are strongly reduced in RBL-2H3 cells (a neoplastic rat basophile cell line) overexpressing GPX4 [75], an effect that is not due to a more efficient reduction of hydroperoxyeicosatetraenoic acids (HPETE) to hydroxyeicosatetraenoic acids (HETE) but due to a lowered activity of the 5-LOX enzyme. This phenotype is shared with the PTGS pathway as in the same cell line a strongly suppressed formation of PTGS2-derived prostaglandin D2 (PGD2) was reported [76]. Notably, PTGS2-dependent oxygenation of esterified arachidonic acid as 2-arachidonoylglycerol emerges to be by far more sensitive to the cellular peroxide tone than oxygenation of free arachidonic acids because higher levels of esterified PGE2 and PGF2α were observed in 3T3 mouse fibroblasts with suppressed GPX4 expression [77]. This is consistent with the fact that GPX4 preferably acts on lipid hydroperoxides in lipid bilayers. Conversely, depleting cells of intracellular GSH stimulated the synthesis of PTGS and LOX products in the human epidermoid carcinoma cell line A431 [78], while overexpression impeded arachidonate metabolism [79].
In the context of ferroptosis, PTGS2 was found to be upregulated in BJeLR cells treated with either RSL3 or erastin and in the corresponding xenograft tumors [16]. Yet, the pan-PTGS inhibitor indomethacin repeatedly failed to rescue from ferroptotic cell death, similar to what was seen in c-Myc/Ha-rasV12 transformed Gpx4 knockout cells [20, 80]. An analogous upregulation of PTGS2 mRNA/protein and its product PGE2 was detected in keratinocyte cultures and skin epithelium of keratinocyte-specific GPX4 null mice, and consequently celecoxib was shown to partially suppress impaired postnatal hair follicle morphogenesis of knockout mice [81]. Yet, the findings that PTGS inhibitors do not suppress ferroptosis elicited by either ferroptosis inducers or inhibition/genetic deletion of GPX4 indicates that upregulation of PTGS2 in ferroptotic conditions must be seen as a potential pharmacodynamic marker/biomarker for cells undergoing ferroptosis rather than a viable drug target to prevent ferroptosis.
In terms of LOX in the ferroptotic process, the picture seems to be rather complex or even obscure. Early studies suggested that GSH depletion causes 12-LOX and Ca2+ dependent cell death (now known as ferroptosis) in neurons, which, however, was reasoned just on the use of lipoxygenase inhibitors [82]. Nonetheless, the targeted knockout of 12/15-LOX in mice is protective in models of neurodegenerative diseases, such as ischemic stroke and traumatic brain injury [83, 84]. Later on, our laboratory reported that 12/15-LOX specific inhibitors rescue Tamoxifen-inducible Gpx4 knockout induced cell death in mouse fibroblasts (referred to as Pfa1 cells), and that 12/15-LOX deficient cells are accordingly more resistant to GSH depletion [20]. The inducible knockout of GPX4 in Pfa1 cells also led to higher levels of the LOX intermediary products 5-, 11-, 12- and 15-HETEs consistent with the results described in the foregoing [15]. However, Alox15 (the gene encoding 12/15-LOX) - Gpx4 double knockout mice failed to rescue acute renal failure of GPX4 null adult mice [15], embryonic lethality caused by the systemic loss of GPX4 [85], and the defects in the viability of GPX4 deficient T cells [41]. Just male subfertility of mice heterozygous for a non-functional GPX4 allele could be rescued by Alox15-deficiency by a yet not fully understood mechanism [86]. Additionally, concurrent silencing of six LOX isoforms (i.e. ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, ALOXE3) in the human renal carcinoma cell line G-401 conferred partial rescue against erastin-induced but, intriguingly, not against RSL3-induced ferroptosis [87]. In HT-1080 cells, knockdown of ALOX15B or ALOXE3 alone, however, was sufficient to render these cells resistant against erastin-induced ferroptosis. Thus, it seems that the preventive effects towards ferroptosis resulting from the knockout/knockdown of certain members of the LOX family might be restricted to the mode by which ferroptosis is induced, i.e. cysteine/glutathione depletion versus GPX4 inhibition/deletion.
Recently, phosphatidylethanolamine binding protein 1 (PEBP1), a scaffold protein known to interact with RAF1 and other kinases, was reported to interact with human ALOX15 and ALOX15B, thereby changing their substrate specificity to yield PE-OOH [70], which was identified earlier as proximate signal of ferroptotic cell death [21, 23]. The Pratt group has recently challenged a major contribution of LOX to the ferroptotic process. While overexpression of three selected LOX isoforms (ALOX5, ALOX12, and ALOX15) in HEK293 cells sensitized cells to erastin-induced cell death, incubation of cells with deuterated arachidonic acid (d-AA) at the respective positions (according to LOX specificity) did not rescue from ferroptosis [24]. Only when cells were incubated with d6-AA where all bis-allylic H-atoms were replaced with deuterium, cytoprotection was achieved in line with the large kinetic isotope effects shown for hydrocarbon autoxidation. Interestingly, many of the known “LOX-specific” inhibitors indeed rescued from RSL3-induced ferroptosis, but this could be solely attributed to their general radical-trapping antioxidant activity (RTA) and not to their “specific” inhibitory action towards LOX function. From this study one may conclude that LOX kickoff lipid peroxidation, although the bulk lipid peroxidation is derived from non-enzymatic lipid autoxidation [25].
It is tempting to speculate that LOX, beyond their role in oxygenating esterified PUFAs as signals of the ferroptotic death process, also indirectly contribute to ferroptosis by alerting innate immune cells by LOX-derived pro-inflammatory factor metabolites including leukotrienes (e.g. LTB4, LTB4, LTC4, LTD4, LTE4), HETEs and oxoeicosanoids (Fig. 4). This could be plausible as impaired GPX4 function for instance in response to moderate GSH depletion allows higher LOX activity due to the higher peroxide tone in cells. Innate immune cells, such as neutrophils and macrophages, attracted by this mechanism might then secrete pro-inflammatory factors including TNFα, which was shown to have an adverse impact on GPX4 stability [88]. In addition to this “priming” of the immune system, bursting ferroptotic cells release pro-inflammatory danger signals (i.e. DAMPs). Cells of the innate immune system then engage PTGS and/or LOX enzymes to perpetuate a state of inflammation by secreting leukotrienes, hepoxilins etc. and eventually factors that allow resolution of inflammation by secreting resolvins and lipoxins. Such an auto-amplification loop in tissue injury and organ failure has been proposed to occur also in the context of other immunologically non-silent forms of non-apoptotic cell death including necroptosis [5]. It thus follows that the spatiotemporal expression and activation of enzymes of the LOX family and PTGS in damaged tissues and in innate immune cells during sensitization towards ferroptosis and upon the release of DAMPs is likely multilayered involving many cell non-autonomous events, which, however, still need to be uncovered.

The GSH/GPX4 and LOX/PTGS systems may modulate the ferroptotic process on several levels. Since GPX4 and LOX (PTGS) are not only directly involved in the ferroptotic process by determining the levels of peroxides in ferroptosis-relevant phosphatidylethanolamine-containing arachidonic and adrenic acids (left), but also in the generation of HPETEs, HPODE (and its further downstream products) and prostaglandins (right), they may also contribute to the early process of innate immune cell activation. This in turn could spark a vicious cycle by negatively impacting on GPX4 expression levels. AA arachidonic acid, AdA adrenic acid, ACSL4 acyl-CoA synthetase long-chain family member 4, GPX4 glutathione peroxidase 4, H2O2 hydrogen peroxide, HPETE hydroperoxyeicosatetraenoic acid, HPODE hydroperoxylinoleic acid, LOX lipoxygenase, LPCAT lysophosphatidylcholine acyltransferase, (P)LOOH (phospho)lipid hydroperoxide, PE phosphatidylethanolamine, PTGS prostaglandin-endoperoxide synthase, TNFα tumor necrosis factor α
Conclusion
Accumulating evidence suggests that cells dying by ferroptosis secrete factors that strongly activate the innate immune system as shown in several pathological and genetic models of ferroptosis in vivo. So far, studies have mainly focused on two organs, i.e. kidney and brain, where ferroptosis emerges to be the root cause of tissue demise. Neuroinflammation determined by staining for reactive astrogliosis and activated microglia and necroinflammation in kidney by increased infiltration rates of neutrophils and macrophage activation have been repeatedly demonstrated, although additional factors including DAMPs and arachidonate products causing necroinflammation remain to be discovered and validated. A detailed knowledge would not only allow to better delineate the sequel of events of cell-autonomous versus non-autonomous effects during ferroptosis, but also might help to establish biomarkers for an improved study of ferroptosis in pathological contexts in humans. Moreover, recent data now indicate that different forms of cell death can be indeed triggered in the same tissue for instance in response to crystal- or folic acid induced AKI. In the latter, such a scenario was indeed shown recently: Ferroptosis induced by folic acid is the primary form of cell death causing secondary upregulation of components of the necroptosis machinery, which in turn further aggravates tissue detriment [69]. Since the activation of ferroptosis in the context of cancer is a highly attractive and hopefully powerful way to eradicate certain tumor entities, it remains to be shown whether cancer cells dying by this form of cell death elicit an adaptive immune response, as evidenced for instance for necroptotic cell death [89].
In conclusion, the study of necroinflammation in the context of ferroptotic cell death is still in its early phase unlike other forms of regulated cell death. Yet, a deeper molecular understanding of this largely untouched but obvious link will undoubtedly lead to novel therapeutic concepts not only by inhibiting necroinflammatory events to mitigate tissue damage in degenerative disease, but also to mount an adaptive immune response for the targeted elimination of residual, treatment-resistant cancer cells.
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–66.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–63.
Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–67.
Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255:2372–6.
Kobayashi S, Sato M, Kasakoshi T, Tsutsui T, Sugimoto M, Osaki M, et al. Cystathionine is a novel substrate of cystine/glutamate transporter: implications for immune function. J Biol Chem. 2015;290:8778–88.
Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256–63.
Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–40.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523.
Mandal PK, Seiler A, Perisic T, Kolle P, Banjac Canak A, Forster H, et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285:22244–53.
Griffith OW. Depletion of glutathione by inhibition of biosynthesis. Method Enzymol. 1981;77:59–63.
Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–8.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211.
Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett. 2012;22:1822–6.
Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162:441–51.
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–9.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4:387–96.
Friedmann Angeli JP, Conrad M. Lipoxygenases—killers against their will? ACS Cent Sci. 2018;4:312–4.
Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–43.
Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. 2017;38:489–98.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–15.
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–106.
Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17.
Wirth EK, Bharathi BS, Hatfield D, Conrad M, Brielmeier M, Schweizer U. Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol Trace Elem Res. 2014;158:203–10.
Wirth EK, Conrad M, Winterer J, Wozny C, Carlson BA, Roth S, et al. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 2010;24:844–52.
Schriever SC, Zimprich A, Pfuhlmann K, Baumann P, Giesert F, Klaus V, et al. Alterations in neuronal control of body weight and anxiety behavior by glutathione peroxidase 4 deficiency. Neuroscience. 2017;357:241–54.
Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–68.
Ueta T, Inoue T, Furukawa T, Tamaki Y, Nakagawa Y, Imai H, et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J Biol Chem. 2012;287:7675–82.
Wortmann M, Schneider M, Pircher J, Hellfritsch J, Aichler M, Vegi N, et al. Combined deficiency in glutathione peroxidase 4 and vitamin e causes multiorgan thrombus formation and early death in mice. Circ Res. 2013;113:408–17.
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111:16836–41.
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Liuyang ZY, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22:1520–30.
Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2:e90777.
Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–65.
Gascon S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, et al. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell. 2016;18:396–409.
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6.
Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–85.
Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23:120–7.
Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732–42.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–73.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30:918–30.
Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22:569–75.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33:890–904.
Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–43.
Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and consequences of necroinflammation. Physiol Rev. 2018;98:727–80.
Lewerenz L, Ates G, Methner A, Conrad M, Maher P. Oxytosis/ferroptosis—(Re-)emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front Neurosci. 2018;12:214.
Cardoso BR, Hare DJ, Bush AI, Roberts BR. Glutathione peroxidase 4: a new player in neurodegeneration? 63: Mol Psychiatry. 2017;22:328–335.
Friedmann Angeli JP, Conrad M. Selenium and GPX4, a vital symbiosis. Free Radic Biol Med. 2018; pii: S0891-5849(18)30104-7. https://doi.org/10.1016/j.freeradbiomed.2018.03.001. [Epub ahead of print]
Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409–22. e21
Green DR. An element of life. Cell. 2018;172:389–90.
Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol. 2016;27:27–39.
Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol JASN. 2017;28:218–29.
Martin-Sanchez D, Fontecha-Barriuso M, Carrasco S, Sanchez-Nino MD, Massenhausen AV, Linkermann A, et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci U S A. 2018;115:4182–4187.
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–41. e26
Huang HS, Chen CJ, Suzuki H, Yamamoto S, Chang WC. Inhibitory effect of phospholipid hydroperoxide glutathione peroxidase on the activity of lipoxygenases and cyclooxygenases. Prostaglandins Other Lipid Mediat. 1999;58:65–75.
Kulmacz RJ, Lands WEM . Peroxide tone in eicosanoid signaling. In: Forman HJ, Cadenas E, (eds.) Oxidative Stress and Signal Transduction. Boston, MA: Springer US: 1997. p 134–56. .
Kulmacz RJ, Lands WE. Requirements for hydroperoxide by the cyclooxygenase and peroxidase activities of prostaglandin H synthase. Prostaglandins. 1983;25:531–40.
Weitzel F, Wendel A. Selenoenzymes regulate the activity of leukocyte 5-lipoxygenase via the peroxide tone. J Biol Chem. 1993;268:6288–92.
Imai H, Narashima K, Arai M, Sakamoto H, Chiba N, Nakagawa Y. Suppression of leukotriene formation in RBL-2H3 cells that overexpressed phospholipid hydroperoxide glutathione peroxidase. J Biol Chem. 1998;273:1990–7.
Sakamoto H, Imai H, Nakagawa Y. Involvement of phospholipid hydroperoxide glutathione peroxidase in the modulation of prostaglandin D2 synthesis. J Biol Chem. 2000;275:40028–35.
Musee J, Marnett LJ. Prostaglandin H synthase-2-catalyzed oxygenation of 2-arachidonoylglycerol is more sensitive to peroxide tone than oxygenation of arachidonic acid. J Biol Chem. 2012;287:37383–94.
Chen CJ, Huang HS, Lin SB, Chang WC. Regulation of cyclooxygenase and 12-lipoxygenase catalysis by phospholipid hydroperoxide glutathione peroxidase in A431 cells. Prostaglandins Leukot Essent Fat Acids. 2000;62:261–8.
Chen CJ, Huang HS, Chang WC. Inhibition of arachidonate metabolism in human epidermoid carcinoma a431 cells overexpressing phospholipid hydroperoxide glutathione peroxidase. J Biomed Sci. 2002;9:453–9.
Schneider M, Wortmann M, Mandal PK, Arpornchayanon W, Jannasch K, Alves F, et al. Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia. 2010;12:254–63.
Sengupta A, Lichti UF, Carlson BA, Cataisson C, Ryscavage AO, Mikulec C, et al. Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of COX-2. J Invest Dermatol. 2013;133:1731–41.
Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–63.
Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH, et al. Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke. 2008;39:2538–43.
van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–8.
Brutsch SH, Wang CC, Li L, Stender H, Neziroglu N, Richter C, et al. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the alox15 gene does not rescue such knock-in mice. Antioxid Redox Signal. 2015;22:281–93.
Brutsch SH, Rademacher M, Roth SR, Muller K, Eder S, Viertel D, et al. Male subfertility induced by heterozygous expression of catalytically inactive glutathione peroxidase 4 is rescued in vivo by systemic inactivation of the Alox15 gene. J Biol Chem. 2016;291:23578–23588.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–75.
Latchoumycandane C, Marathe GK, Zhang R, McIntyre TM. Oxidatively truncated phospholipids are required agents of TNFalpha induced apoptosis. J Biol Chem. 2012.
Garg AD, Agostinis P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev. 2017;280:126–148.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG) CO 291/5-1, the Human Frontier Science Program (HFSP) RGP0013, the German Federal Ministry of Education and Research (BMBF) through the Joint Project Modelling ALS Disease In Vitro (MAIV, 01EK1611B) and the VIP + program NEUROPROTEKT (03VP04260).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by F. Pentimalli
Rights and permissions
About this article

Cite this article
Proneth, B., Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ 26, 14–24 (2019). https://doi.org/10.1038/s41418-018-0173-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0173-9
This article is cited by
-
Melanoma biology and treatment: a review of novel regulated cell death-based approaches
Cancer Cell International (2024)
-
NINJ1 induces plasma membrane rupture and release of damage-associated molecular pattern molecules during ferroptosis
The EMBO Journal (2024)
-
A New Perspective in the Treatment of Ischemic Stroke: Ferroptosis
Neurochemical Research (2024)
-
Knockdown of circSlc8a1 inhibited the ferroptosis in the angiotensin II treated H9c2 cells via miR-673-5p/TFRC axis
Journal of Bioenergetics and Biomembranes (2024)
-
Ferroptosis-related signature and immune infiltration characterization in acute lung injury/acute respiratory distress syndrome
Respiratory Research (2023)
