
Abstract
Pre-eclampsia, a syndrome of pregnant women, is one of the leading causes of maternal and fetal morbidity and mortality. Despite active research, the etiology of this disorder remains an enigma. Recent work has, however, provided promising explanations for the causation of the disorder and some of its phenotypes. Evidence indicates that the symptoms of hypertension and proteinuria, upon which the diagnosis of pre-eclampsia is based, have several underlying causes. Nevertheless, the treatment of pre-eclampsia has not changed significantly in over 50 years. This review describes the most recent insights into the pathophysiology of pre-eclampsia from both basic and clinical research, and attempts to provide a unifying hypothesis to reconcile the abnormalities at the feto–placental level and the clinical features of the maternal syndrome. The novel findings outlined in this review provide a rationale for potential future prophylactic and therapeutic interventions for pre-eclampsia.
Key Points
-
Pre-eclampsia results from placental hypoperfusion secondary to impaired establishment of uteroplacental vasculature
-
Abnormal biology of extravillous trophoblasts, decidua, uterine natural killer cells and maternal endothelium have pathogenic roles
-
Hypothesis: placental overexpression of arginase decreases L-arginine, reducing activity of the vasodilator nitric oxide
-
Hypothesis: hypoxia secondary to placental hypoperfusion induces release of soluble Fms-like tyrosine kinase 1 and reactive oxygen species into the maternal circulation, initiating endothelial dysfunction
-
The hypertension and proteinuria characteristic of pre-eclampsia might be ameliorated by supplementation with L-arginine or antioxidants, or by blocking Fms-like tyrosine kinase 1
Similar content being viewed by others

Introduction
Pre-eclampsia is a syndrome that is usually defined as the onset of hypertension and proteinuria after 20 weeks of gestation in previously normotensive non-proteinuric pregnant women.1 If left untreated, pre-eclampsia can progress to a convulsive state known as eclampsia.2 Pre-eclampsia is a leading cause of maternal and fetal morbidity and mortality.3,4 In the Western world, it affects between 2 and 7% of all pregnancies, but the incidence in other geographic areas with different ethnic or social characteristics can be up to three times greater.5,6 A worldwide incidence of 8,370,000 cases per year has been estimated.5,6 In developing countries, such as Colombia, 42% of maternal deaths are attributed to this disorder, which is also the major reason for premature delivery.7 The incidence of eclampsia is also high in developing countries (1.6 and 12 per 1,000 live births, respectively, in Colombia and India).8
Risk factors
Pre-eclampsia is primarily regarded as a disease of first pregnancy, although the protective effect of multiparity is said to be lost with change of partner.9 This supports the hypothesis that risk is reduced with repeated exposures to specific antigens from the same partner; however, recent evidence from the Medical Birth Registry of Norway indicates that the protective effect of previous pregnancy with the same partner is confounded by the time interval between births.10 These data, obtained from >1.8 million births over 31 years, showed that when the birth interval was >10 years, a multiparous woman had the same risk of developing pre-eclampsia as a primiparous woman. It has, however, been suggested that this 'birth-interval hypothesis' is not well founded.11
A systematic review of >1,000 controlled studies published from 1966 to 2002 found that a previous history of pre-eclampsia, multiple pregnancy, nulliparity, pre-existing diabetes, high BMI before pregnancy, maternal age ≥40 years, renal disease, hypertension, ≥10 years since previous pregnancy and presence of antiphospholipid antibodies all increased a woman's risk of developing pre-eclampsia.12 Increases in risk of more than ninefold, sevenfold and threefold, respectively, were documented for antiphospholipid antibodies, previous history of pre-eclampsia and diabetes. Based on this evidence, guidelines for risk assessment of pre-eclampsia have recently been issued.13
Other risk factors for pre-eclampsia are insulin resistance in concert with obesity14,15 and thrombophilia.16 In developing countries, protein-calorie undernutrition has been identified as an important risk factor;17 however, a negative correlation between calcium intake and incidence of pre-eclampsia has recently been detected in Guatemala, Colombia and India.7 This finding is supported by the observation that calcium supplementation is beneficial to women at high risk of gestational hypertension, and in communities with low dietary calcium intake.18,19,20
Genetic factors
The precise role of genetic factors in the development of pre-eclampsia is unclear, and no specific contributory gene has been identified. The inheritance pattern of the disease has been described as Mendelian (autosomal recessive and autosomal dominant with incomplete PENETRANCE), polygenic/multifactorial and mitochondrial. Small studies21,22,23,24 have indicated an association between pre-eclampsia and polymorphisms of genes that control blood pressure, coagulation or oxygen-free-radical metabolism—such as renin, angiotensinogen, endothelial nitric oxide synthase (eNOS), FACTOR V LEIDEN, METHYLTETRAHYDROFOLATE or lipoprotein lipase—but this has not been confirmed.25 Linkage analysis has identified three potential loci linked to susceptibility to pre-eclampsia: 2p13, 2p25 and 9p13.26,27 Notably, these loci account for only a small percentage of cases (other studies failed to confirm the associations).28,29 A fourth locus for pre-eclampsia was subsequently identified on 10q22. Maximal allele sharing between pre-eclamptic sisters at this locus was observed for maternal-derived but not paternal-derived alleles,30 indicating matrilineal inheritance.
The STOX1 gene in the 10q22 locus contains five different missense mutations. STOX1 is identical in affected sisters, co-segregates with the pre-eclamptic phenotype and is inherited matrilineally.31 A role has been proposed for the gene product (a DNA-binding protein) in controlling polyploidization of extravillous trophoblasts, although its precise biological function has not been clarified.31
In a large study by the British Genetics of Pre-eclampsia Consortium, 657 women affected by pre-eclampsia and their families were genotyped at sites of 28 SINGLE-NUCLEOTIDE POLYMORPHISMS in several genes, including those involved in angiotensin activity (angiotensinogen and angiotensin II type 1 receptor [AT1]) and oxidative stress (methylene tetrahydrofolate reductase).32 None of the genetic variants tested were found to confer a high risk of disease development, indicating that alterations of angiotensin activity and oxidative stress are not prime causes of pre-eclampsia.
Diagnosis and clinical features
The cardinal features of pre-eclampsia are hypertension and proteinuria. In the past, hypertension indicative of pre-eclampsia was defined as an elevation of >30 mmHg systolic pressure or >15 mmHg diastolic pressure above the patient's baseline blood pressure. This definition proved to be a poor indicator of outcome33 and, in 2000, the criteria were refined by the National High Blood Pressure Education Program (NHBPEP) Working Group.1 Since then, there has been considerable agreement between international working groups.34 The criteria define hypertension as a systolic blood pressure ≥140 mmHg or a diastolic level (Korotkoff 5) ≥90 mmHg on two or more occasions at least 4–6 h (but not more than 7 days) apart after 20 weeks of gestation in a woman with previously normal blood pressure. Nevertheless, the NHBPEP working group report1 also stated the collective opinion that 30 mmHg systolic and 15 mmHg diastolic increases, even at values <140/90 mmHg, should prompt patient monitoring for pre-eclampsia.
For most healthy, nulliparous women, the factor that differentiates pre-eclampsia from other gestational hypertensive disorders is concomitant proteinuria, defined as ≥0.3 g protein in a 24 h urine sample.1 When daily urine collection is not feasible, two random urine samples taken at least 4–6 h apart are usually acceptable.1 Under these sampling conditions, proteinuria is defined as ≥0.3 g/l protein or ≥1+ on a dipstick test-strip; however, as there is not yet consensus on the correlation between random protein concentration or urinary dipstick measurements and 24 h protein excretion in women with gestational hypertension,35,36,37 it is best to base diagnosis of proteinuria on quantitative measurement of 24 h urine samples.
The diagnostic criteria outlined above do not apply to women who have hypertension or proteinuria before 20 weeks of gestation.38,39 Thus, pre-eclampsia can also occur in women with pre-existing chronic hypertension, a condition that is often worsened by gestation (Box 1).1 More stringent diagnostic criteria are necessary, including a sudden exacerbation of hypertension, new onset or sudden exacerbation of proteinuria, thrombocytopenia and elevated levels of liver enzymes.1
Pre-eclampsia is regarded as severe if one or more of the criteria in Box 2 are fulfilled.40 As well as hypertension and proteinuria, pre-eclampsia can be associated with edema, visual disturbances, headache, and epigastric or right upper-quadrant pain with nausea and vomiting (Box 3). Hemolysis, elevated liver enzymes and thrombocytopenia are characteristic laboratory features of a severe variant of pre-eclampsia, the HELLP SYNDROME, which occurs in 5–8% of pre-eclamptic women.40 This multisystem, pregnancy-specific condition illustrates that pre-eclampsia can affect every maternal organ, predominantly the vascular, renal, hepatic, cerebral and coagulation systems (Box 3). Kidney function, with its reliance on adequate glomerular blood flow and selective permeability of glomerular capillaries, is particularly susceptible to the endothelial changes in pre-eclampsia that manifest functionally as vascular constriction, decline in renal blood flow and glomerular filtration rate, and clinically significant proteinuria.41 Oliguria or anuria can occur in patients with severe pre-eclampsia as a result of low cardiac output and high systemic vascular resistance,42 but acute renal failure is unusual.41
Pre-eclampsia can rapidly progress to a convulsive phase termed eclampsia, especially if untreated.2 The onset of eclampsia can be preceded by severe persistent headache and visual changes. The seizures and mental status changes of eclampsia are often, although not always, related to hypertensive encephalopathy. Maternal death is more likely in the presence of severe hypertension and eclampsia. Notably, the risk of cardiovascular complications later in life is increased in women who develop pre-eclampsia. This could be a function of the numerous risk factors and pathophysiological abnormalities that are shared by pre-eclampsia and coronary artery disease.43 Impaired uteroplacental blood flow or placental infarction in pre-eclampsia can affect the fetal–placental unit, causing intrauterine growth restriction, intrauterine fetal demise, oligohydramnios, or placental abruption. Generally, maternal and perinatal outcomes are better in mild pre-eclampsia that develops after 36 weeks of gestation than in cases that are symptomatic before 33 weeks of gestation.1,6,44,45 Maternal and perinatal morbidity and mortality are increased in women with pre-existing medical disorders36,39,46 and in developing countries.1,47
Screening of pregnant women for pre-eclampsia based on functional or biochemical tests, such as evaluation of placental perfusion and vascular resistance by Doppler scan, or measurement of markers of endothelial and oxidative stress, has been proposed.48 Unfortunately, the search for clinically useful screening tests has been disappointing, as shown in a recent analysis of 87 studies in 211,369 women.48 Other researchers assert that biochemical and/or hemodynamic markers are reliable predictive tools for pre-eclampsia.49 Uterine artery Doppler waveform analysis is used extensively in academic centers to identify women at risk of pre-eclampsia. Studies of Doppler waveform analysis as a screening test for adverse pregnancy outcomes have, however, generated conflicting results, probably due to differences in gestational age at time of examination and the lack of standardized analysis.50
Pathogenetic mechanisms: recent insights
Efforts to unravel the pathogenesis of pre-eclampsia have been hampered by the lack of clear diagnostic criteria for the disease and its subtypes. Consequently, several studies have included a variety of other conditions that do not necessarily reflect an adverse pregnancy outcome. More importantly, the definition of pre-eclampsia proposed in 2000 was designed for clinical use, to maximize sensitivity at the detriment of specificity (the risk of not detecting pre-eclampsia far outweighs the risks of overdiagnosis and overtreatment). In addition, most pathogenetic studies use specimens from patients with established pre-eclampsia rather than those obtained before clinical onset. This makes it difficult to define a clear relationship between specific mediators, mechanisms and clinical manifestations. Animal models have failed to provide definitive insights into the pathogenesis of pre-eclampsia because of their limited applicability to the human form of the disease. Despite these limitations, several theoretical mechanisms have been proposed that reconcile feto–placental abnormalities and clinical features of the maternal syndrome.
The feto–placental level
For pregnancy to proceed normally, the early blastocyst must adhere to and invade the uterine endometrium so that maternal blood can effectively bathe the PLACENTAL COTYLEDONS.51 During this process, extravillous CYTOTROPHOBLAST cells proliferate from the tips of anchoring chorionic villi to form a 'shell' lining the uterine cavity. Two populations of invasive extravillous trophoblasts have been identified. Interstitial trophoblasts invade the decidual stroma and reach the myometrium. At the end of their invasion path, these cells fuse to form multinuclear giant cells.52 Interstitial trophoblasts might release vasodilators (e.g. NO and carbon monoxide) that act on the SPIRAL ARTERIES before destruction by endovascular trophoblasts.52 Endovascular cytotrophoblasts invade the lumen of the uterine spiral arterioles, anchor to the vessel wall, and promote arteriole dilation. Thus, by the second trimester, the endometrial and superficial myometrial segments of spiral arteries are lined by cells of cytotrophoblastic origin, which changes them from small resistance vessels to flaccid, high-caliber capacitance vessels. This remodeling, which transforms the endometrium into a yet more developed tissue (the deciduas) is instrumental in ensuring adequate perfusion of the feto–placental unit.
Endovascular invasion does not proceed beyond the superficial portions of the uterine spiral arterioles, which retain their endothelial lining and muscular wall, and functionally remain relatively narrow-bore, high-resistance vessels.53 The net result is that blood flow into the intervillous space is dramatically reduced. Thus, it was thought that the principal defect in pre-eclampsia relates to endovascular trophoblast invasion and that interstitial trophoblast invasion is normal.54 Recent quantitative studies, however, of interstitial trophoblast invasion in uterine tissues taken from hysterectomies following cesarean section in women with pre-eclampsia, have revealed that both the invasive depth and numerical density of interstitial trophoblasts are significantly reduced.55 Failure of vascular invasion could, therefore, be preceded by impaired interstitial trophoblast invasion. Some patients with pre-eclampsia at term do not show severe growth restriction. Their placental histomorphometry is similar to that of normotensive gestational age-matched controls. Usually, however, pre-term placentas of women with pre-eclampsia are characterized by vascular and occlusive lesions that have been classified according to particular pathophysiological processes.56,57
Factors involved in abnormal placentation
Basic research and clinical data indicate that the maladaptation and inadequate invasion of uteroplacental arteries characteristic of pre-eclampsia result from intrinsic factors (abnormal biology of extravillous trophoblasts) acting in concert with extrinsic maternal uterine factors. These extrinsic factors, operating around the uteroplacental arteries, comprise impaired decidual remodeling, impaired function of uterine natural killer (NK) cells and maternal endothelial failure to express adhesion molecules. These factors might interact in a cascade-like fashion.
NK cells are the predominant population of decidual lymphoid cells. During early pregnancy they accumulate as a dense infiltrate around the invading cytotrophoblast cells. They cooperate with extravillous trophoblasts to remodel spiral arteries by producing cytokines that are involved in angiogenesis and vascular stability, such as vascular endothelial growth factor (VEGF), placental growth factor (PlGF) and ANGIOPOIETIN 2.58 Although the villous trophoblast cells exposed to maternal blood lack MHC class I and class II molecules, the invading cytotrophoblast expresses an unusual combination of the former: HLA-C, HLA-E and HLA-G. These molecules interact with receptors on the surface of NK cells, such as killer-cell immunoglobulin-like receptors (KIRs) and CD94. The ligand–receptor pairs might control the interaction of extravillous trophoblast cells and decidual NK cells. The human KIR locus consists of 7–15 closely packed genes on chromosome 19q, which encode both inhibitory and activating KIRs. These genes form numerous haplotypes that differ in both gene content and allele combinations.
Both HLA-C and KIR genes are polymorphic in humans, so every pregnancy has different combinations of paternally derived fetal HLA-C on trophoblasts and maternal KIRs on NK cells.59 Pre-eclampsia is more prevalent in women who are homozygous for group A KIR haplotypes (which contain mainly genes encoding inhibitory KIRs) than in women who are homozygous for group B haplotypes (which contain genes for activating KIRs) or those who are heterozygous.60 Moreover, it is the combination of fetal HLA-C2 genotype—the product of which binds more tightly to its cognate KIR than C1 product—and maternal AA KIR genotype that most frequently leads to pre-eclampsia.60 This combination is predicted to deliver the strongest inhibitory signals from extravillous trophoblasts to NK cells. It has therefore been proposed that inhibition of uterine NK cells has a role in defective trophoblast remodeling of maternal blood vessels; however, only a minority of pregnancies with C2 fetuses and AA mothers result in pre-eclampsia. This finding, along with the observation that not all pre-eclamptic pregnancies have the C2–AA genotype combination, indicates that additional factors are involved.
In normal pregnancy, as they invade the uterine wall, cytotrophoblasts lose the ability to divide and upregulate expression of matrix metalloproteinases, thereby promoting transformation of epithelial tissue to endothelium.61 Zhou et al.61 have shown that invasive cytotrophoblasts downregulate epithelial-like receptors such as endothelial (E)-cadherin and α6β4, which are replaced by endothelial adhesion molecules, such as platelet–endothelial cell-adhesion molecule (PECAM), vascular endothelial (VE)-cadherin, vascular cell-adhesion molecule 1 (VCAM1), and α4 and αvβ3 integrins.61 In pre-eclampsia, this transformation is impaired, and cytotrophoblasts do not invade and differentiate. Immunohistochemical staining of pre-eclamptic placental bed biopsies shows very low levels of VE-cadherin, VCAM1 and αvβ3 integrin,53 indicating that the cells fail to acquire an endothelial phenotype.
One hypothesis attributes this impaired transformation to insufficient local concentrations of angiogenic factors, including VEGF and PlGF, which are expressed at high levels during normal placentation.62 VEGF induces endothelial cell expression of αvβ3 integrin, a molecule associated with angiogenesis and cytotrophoblast invasion.61 Furthermore, cytotrophoblasts have VEGF receptors, including the Fms-like tyrosine kinase 1 (Flt1) receptor (also known as VEGF receptor-1).63 As high levels of Flt1 are found in invasive cytotrophoblasts as early as 6 weeks postfertilization,63 interaction of the receptor with VEGF and PlGF has been considered crucial for invasion and PSEUDO-VASCULOGENESIS. This possibility is supported by the fewer cell-to-cell interconnections and decreased expression of integrin α1. These accurate indices of defective cells' invasive capacity are observed in human cytotrophoblasts in vitro when an Flt1–Fc fusion protein that blocks binding of VEGF and PlGF to the Flt1 receptor is added to the culture medium.63 Pre-eclamptic placentas have decreased levels of VEGF messenger RNA,64 supporting the hypothesis that VEGF is involved in abnormal, shallow placentation.
Cytotrophoblast immunostaining for VEGFA and Flt163 is reduced in pre-eclampsia compared with normal pregnancy. Most Flt1 produced in the human placenta during later stages of gestation is a soluble form (sFlt1) generated by alternative splicing. sFlt1 lacks the transmembrane and cytoplasmic domains of Flt1, and is released in large amounts into the blood,65 where it reduces levels of free VEGF via soluble antagonism of both factors.65 Cytotrophoblasts isolated from pre-eclamptic placentas released more sFlt1 than those from normal placentas,63 and upregulation of sFlt1 messenger RNA has been detected in pre-eclamptic placentas.66 So, by preventing binding of VEGF to Flt1 receptors on cytotrophoblast cells, excessive production of sFlt1 in placental trophoblasts could be responsible for the defective cytotrophoblast differentiation and abnormal placentation of pre-eclampsia. However, chimeric placentas from tetraploid aggregates—comprising trophoblasts homozygous for a null allele of Flt1 and the normal Flt1+/+ maternal and fetal components—showed no defective formation of placental circulation or endovascular trophoblast invasion of maternal spiral arteries.67 The chimeric placentas supported normal fetal growth67 indicating that, in mice, expression of Flt1 in trophoblasts is not essential to normal placentation. Whether this observation is applicable to humans is not known; mouse placenta does not undergo the same degree of placental vascular remodeling and invasion as human placenta.
As insightful as these findings are, they do not answer the question of whether altered VEGF–Flt1 signaling has a role in the impaired placental vasculogenesis and maternal spiral uterine-artery remodeling of pre-eclampsia. Interestingly, the VEGF–Flt1 interaction induces release of NO,68 a potent vasodilator synthesized from the amino acid L-arginine (L-Arg) by the NOS family of isoenzymes69 (Figure 1). Human placenta does not form inducible or neuronal NOS,70 but eNOS is found in the endothelium of the umbilical cord, in the chorionic plate, and in villous cytotrophoblasts.70 Several reports have described intense eNOS expression in extravillous trophoblast cells.71,73,74 This finding could not, however, be confirmed by others.75 In trophoblast cells, NO upregulates expression and activity of the matrix-degrading proteases MMP2 and MMP9, which are required for invasion during embryo implantation.76 MMP2 also has a pivotal role in the relaxin–gelatinase pathways that are upregulated during pregnancy and contributes to reduced myogenic reactivity of small arteries through activation of the endothelial endothelin B receptor–NO pathways.77

Binding of vascular endothelial growth factor and placental growth factor to the Fms-like tyrosine kinase 1 receptor on cytotrophoblasts stimulates production of nitric oxide by endothelial nitric oxide synthase. Interaction of vascular endothelial growth factor and placental growth factor with the Fms-like tyrosine kinase 1 receptor is inhibited by soluble Fms-like tyrosine kinase 1. Normal placenta with sufficient tissue L-arginine sustains adequate generation of nitric oxide by endothelial nitric oxide synthase (left panel). By contrast, in pre-eclampsia, increased levels of soluble Fms-like tyrosine kinase 1 inhibit activation of endothelial nitric oxide synthase by the Fms-like tyrosine kinase 1 receptor. At the same time, excessive arginase II expression reduces the placental L-arginine concentration, causing endothelial nitric oxide synthase to preferentially facilitate superoxide anion production. Superoxide anion reacts with nitric oxide to form peroxynitrite, thus reducing the half-life of nitric oxide (right panel). This promotes abnormal placental perfusion and microvascular oxidative damage. eNOS, endothelial nitric oxide synthase; Flt1, Fms-like tyrosine kinase 1; L-Arg, L-arginine; NO, nitric oxide; O2−, superoxide anion; ONOO−, peroxynitrite; PlGF, placental growth factor; sFlt1, soluble Fms-like tyrosine kinase 1; VEGF, vascular endothelial growth factor.
NO release has been shown to mediate angiopoietin-induced growth and migration of extravillous trophoblast cell lines in vitro.78 NO is coupled through Flt1 signaling to cytotrophoblast invasion and motility induced by VEGF.79 It also mediates VEGF-induced upregulation of VE-cadherin in endovascular trophoblasts,80 all processes necessary for placental development. By virtue of these unique vasodilatory and angiogenic/vasculogenic properties, locally generated NO seems to be instrumental in promoting cytotrophoblast endovascular invasion during normal placentation.61 Thus, it is possible that the placental changes of pre-eclampsia reflect a reduced capacity of cytotrophoblast cells to form NO.
The diminished NO activity of pre-eclamptic placenta cannot be the consequence of a defect in NO-forming enzyme, because placentas of pre-eclamptic women express normal levels of eNOS and form normal amounts of NO.70,81 Instead, it results from reduced activity or half-life of NO. This translates into lower concentrations of cyclic GMP (the NO intracellular second messenger) in the placental circulation compared with normal pregnancy.70 A plausible explanation for reduced NO activity is that, in pre-eclamptic placenta, NO is rapidly degraded to peroxynitrite (ONOO−), a potent cytotoxic anion devoid of angiogenic properties.82 This explanation is supported by intense immunostaining for nitrotyrosine (a marker of ONOO− formation in vivo) observed in fetal blood vessels of floating villi, syncytiotrophoblasts and invasive cytotrophoblasts.83 Data indicate that NO interacts with reactive oxygen species (ROS), including superoxide anion (O2−), which are abundant in pre-eclamptic placenta compared with normal placenta.83
A biochemical explanation for excessive ROS production in pre-eclampsia has been proposed. In addition to synthesizing NO, NOS facilitates production of superoxide anion (O2−).84 The relative amounts of NO and O2− formed are tightly regulated by intracellular levels of L-Arg.85,86 In the presence of sufficient L-Arg, NOS in cultured mouse macrophages and human kidney cells85,87 generated only NO. Depletion of L-Arg led to production of both NO and O2−, and subsequent formation of ONOO−. It is reasonable to propose that a similar biochemical aberration in NOS function might occur in pre-eclamptic placenta, as L-Arg levels are lower than normal in pre-eclamptic villous tissue88 as a consequence of L-Arg consumption by arginase II. This enzyme, which degrades L-Arg to ornithine and urea,89 is markedly expressed in pre-eclamptic, but not normal, trophoblast cells.88 The mechanism of arginase II upregulation in pre-eclampsia is unknown, but testosterone—which stimulates arginase II activity in female mice and rats90 and is present at higher levels in pre-eclampsia than in normal pregnancy90,91—is a potential mediator (Figure 1).
The role of subnormal L-Arg/NO activity in sustaining deficient spiral artery invasion in pre-eclampsia is underscored by the recent finding that long-term L-Arg supplementation in women at risk of placentation disorders reduces uterine artery resistance, allows delivery at term and improves perinatal outcome.92 Conversely, chronic administration of the L-Arg antagonist nitro-L-Arg-methyl-ester to pregnant rats induces a pre-eclampsia-like syndrome characterized by sustained hypertension, proteinuria and intrauterine growth retardation.93 It also increases levels of blood and tissue markers of oxidative stress, further supporting the link between L-Arg deficiency and oxidative stress of pre-eclampsia. In this model, treatment with the antioxidants glutathione and quercetine normalized plasma levels of peroxidation products, and reduced proteinuria and the pup mortality rate.94
Defective NO-mediated vasodilatation conceivably translates into the relative vasoconstriction of feto–placental vessels and inadequate perfusion that characterize severe pre-eclampsia. The direct consequence of abnormal cytotrophoblast differentiation is placental hypoperfusion and ischemia, which trigger production of factors that might drive both the fetal and maternal syndromes. Hypoxia-inducible factor 1α (HIF1α), a subunit of HIF1, is overexpressed in trophoblast cells of pre-eclamptic placenta.52,95,96 HIF1 mediates cellular adaptations to low-oxygen conditions by activating transcription of several genes. In human villous explants under low-oxygen tension, HIF1 upregulates expression of transforming growth factor-β3 (TGF-β3),97 a potent inhibitor of early trophoblast differentiation.98 TGF-β3 is overexpressed in pre-eclamptic placenta.95 Its inhibition by antisense oligonucleotides in pre-eclamptic villous explant cultures restores the invasive capabilities of trophoblast cells,98 indicating that HIF1-mediated TGF-β3 expression could have a role in trophoblast dysfunction in pre-eclampsia.
In addition, placenta hypoperfusion and, particularly, intermittent perfusion, create a condition of hypoxia–reoxygenation that favors oxidative damage.99 Hypoxia increases activity of xanthine oxidase (XO),100 a member of the holoenzyme xanthine dehydrogenase (XDH)/XO family that generates ROS. XDH/XO expression and XO activity are increased in invasive cytotrophoblasts of pre-eclamptic women.83 These changes, together with the above-mentioned increased production of ONOO−, result in release of ROS from the placenta into the maternal circulation. These ROS eventually contribute to widespread maternal endothelial dysfunction.
The maternal level
The placental abnormalities described above have several maternal consequences (Figure 2). Although peripheral vascular resistance and blood pressure are substantially decreased in normal pregnancy,101 increased peripheral vascular resistance is universally present in pre-eclampsia as a result of generalized vasoconstriction. Angiotensin II (Ang II) infusion can elicit an increased pressor response compared with normal pregnancy,102 although this is not a universal finding.103 Plasma renin activity and Ang II levels are low,104 and platelet AT1 density is comparable to normal pregnancy, discounting the possibility that increased pressor response to Ang II in pre-eclampsia depends on increased receptor expression.

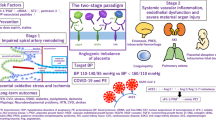
Abnormal placentation (stage 1), particularly lack of dilatation of the uterine spiral arterioles, is the common starting point in the genesis of pre-eclampsia, which compromises blood flow to the maternal–fetal interface. Reduced placental perfusion activates placental factors and induces systemic hemodynamic changes. The maternal syndrome (stage 2) is a function of the circulatory disturbance caused by systemic maternal endothelial cell dysfunction resulting in vascular reactivity, activation of coagulation cascade and loss of vascular integrity. Pre-eclampsia has effects on most maternal organ systems, but predominantly on the vasculature of the kidneys, liver and brain.
Most vasoactive substances have been investigated with respect to their involvement in the maternal syndrome of pre-eclampsia. The relevance of endothelin-1, a potent vasoconstrictor, remains unclear, as most studies (with some exceptions81) have detected increased levels in the plasma of pre-eclamptic women. The vasoconstrictor thromboxane A2 (TxA2), normally generated by activated platelets, macrophages and the kidney, is synthesized in excessive amounts in severe pre-eclampsia.105 Increased urinary excretion of TxA2 metabolites has been reported in some studies of pre-eclampsia but not in others.81 According to most reports, synthesis of vasodilatory mediators, such as prostacyclin (PGI2) and NO, is reduced in pre-eclampsia.81,106,107 Other authors have documented normal or even increased synthesis of NO.70 In most studies, NO synthesis was evaluated indirectly by measuring plasma and urinary levels of its metabolites (nitrites and nitrates), which are also present in food and water.70 So, variability in time, method of fluid sampling and dietary intake could explain discrepancies in the results of different studies. On the other hand, NO activity, measured as flow-mediated NO-dependent vasodilatation in resistance-sized, subcutaneous vessels, is severely reduced in pre-eclampsia compared with normal pregnancy.70 Increased plasma concentrations of the endogenous NO inhibitor asymmetric dimethylarginine have also been found in pre-eclampsia,108 although this was not confirmed.109 Perhaps all of the changes in levels of vasoactive substances in pre-eclampsia reflect a generalized inflammatory response that activates the endothelium and circulating leukocytes.
Two new findings have shed light on the pathophysiology of the maternal syndrome of pre-eclampsia. First, a correlation between reduced uteroplacental blood flow in early or mid-pregnancy and the subsequent onset of pre-eclampsia.110 Second, plasma and urinary levels of sFlt1 are higher than normal in patients with established pre-eclampsia, and begin to rise as early as 5 weeks before clinical onset.65,111 In a pilot cohort study, plasma levels of sFlt1 in women at 20 weeks of gestation with abnormal uterine perfusion were significant predictors of either pre-eclampsia or intrauterine growth retardation.112 Concurrent measurement of uterine perfusion and sFlt1 levels could therefore be an early screening test for severe complications of pregnancy, although lacking specificity for pre-eclampsia. Repeated evaluation during the first two pregnancies of 93 women showed that sFlt1 serum levels were higher in the first than in the second pregnancy, indicating that enhanced sFlt1 secretion might account for the increased risk of pre-eclampsia among nulliparous women.113
The way in which placental dysfunction relates to placental sFlt1 production remains unknown. Because hypoxia upregulates production of Flt1 in placental trophoblasts,114,115 restricted placental blood flow might increase placental production of sFlt1 to pathological levels, thus initiating the maternal syndrome. In normal primiparous women, plasma sFlt1 tends to increase during the last month of pregnancy,65 despite normal uteroplacental blood flow. Therefore, what could differentiate pre-eclampsia from normal pregnancy is abnormal, early placental production and eventual high circulating concentrations of sFlt1, possibly secondary to placental vasculature constriction and hypoperfusion. Consistent with the scavenger effects of sFlt1, concentrations of unbound plasma VEGF and PlGF, and urinary PlGF, are decreased in pre-eclampsia.65,66,116,117 Evidence is emerging that lower than normal urinary PlGF levels mid-gestation might help predict development of pre-eclampsia.117
So, circulating sFlt1 deprives the vasculature of the kidney, liver, brain and other organs of essential survival and maintenance signals, ultimately triggering the maternal vascular dysfunction of pre-eclampsia (Figure 3). There is evidence that, in addition to angiogenesis, VEGF induces vasodilation by enhancing endothelial synthesis of NO68 and PGI2. As a consequence, low circulating VEGF levels will increase vascular tone and cause hypertension, a hallmark of the maternal syndrome. The organs most affected by pre-eclampsia have fenestrated vascular endothelium, which is needed for physiological diffusion of water and solutes. Animal models have shown that VEGF is important in maintaining the health of fenestrated endothelium.118 This implies that organs with fenestrated endothelium could be prone to damage by sFlt1-mediated VEGF blockade.

Most of the Fms-like tyrosine kinase 1 produced by human placenta is a soluble form generated by alternative splicing. Soluble Fms-like tyrosine kinase 1 is released in large amounts into the blood. Soluble Fms-like tyrosine kinase 1 binds both vascular endothelial growth factor and placental growth factor, reducing their free levels in the blood by working as a soluble antagonist of both factors, and maintaining normal endothelial function of maternal vasculature (left panel). Pre-eclamptic placenta releases higher amounts of soluble Fms-like tyrosine kinase 1 than normal placenta (right panel), depriving the vasculature of kidney, liver, brain and other organs of essential maintenance signals, thereby triggering the maternal vascular dysfunction of pre-eclampsia. PlGF, placental growth factor; sFlt1, soluble Fms-like tyrosine kinase 1; VEGF, vascular endothelial growth factor.
Pregnant and non-pregnant rats receiving sFlt1 via adenoviral transfection developed hypertension, proteinuria and endothelial abnormalities66 reminiscent of those in pre-eclamptic women. Observed glomerular enlargement with occlusion of capillary loops by swelling and hypertrophy of endocapillary cells66 mimics lesions of human pre-eclampsia. In the rat model, administration of VEGF reverses hypertension and proteinuria and ameliorates glomerular changes.144 Administration of sFlt1 or VEGF-neutralizing antibodies to normal mice caused severe proteinuria via glomerular endothelial cell detachment and damage, and downregulation of nephrin (a key protein of the glomerular filtration apparatus).119 Similarly, neutralizing antibodies and VEGF-receptor inhibitors administered to cancer patients as antiangiogenic therapy can cause hypertension, proteinuria and coagulopathy.120 Moreover, podocyte-specific heterozygous deletion of the VegfA gene in mice caused a renal disease characterized by endotheliosis and proteinuria, the renal lesions of pre-eclampsia.121 Together, these observations indicate that increased production of sFlt1 in pre-eclampsia contributes to the maternal symptoms. The gene encoding Flt1 in humans is on chromosome 13. Patients with trisomy 13 are at increased risk of developing pre-eclampsia compared with those with other trisomies and normal pregnant women.122,123
The contribution of the renin–angiotensin system to the maternal syndrome of pre-eclampsia is intriguing. As mentioned earlier, responsiveness to Ang II is enhanced in pre-eclampsia, but not as a result of increased AT1 levels. In omental vessels and platelets from pre-eclamptic women, there is a significant increase in heterodimerization between the AT1 and the B2 receptor of the vasodepressor bradykinin.124 This does not occur in normal pregnancy. A fourfold to fivefold increase in B2 receptor protein levels was also documented, correlated with the increase in receptor heterodimerization. Expression of AT1–B2 heterodimer increased responsiveness to Ang II and conferred resistance on AT1 to inactivation by oxidative stress of pre-eclampsia. The mechanism by which the AT1–B2 heterodimer forms is speculative. It has been proposed that inflammation and ischemia—features of pre-eclampsia—increase expression of B2 receptors, which promotes heterodimerization with the normally expressed AT1. This observation provides a plausible but not conclusive explanation for the enhanced vascular Ang-II-responsiveness of pre-eclampsia. The plasma of pre-eclamptic women also contains agonistic antibodies that target vascular smooth muscle AT177,125 and disappear after pregnancy; however, the origin of these antibodies and whether their disappearance is consistent with the time course of the resolution of pre-eclampsia is unknown. An intriguing possibility is that the AT1–B2 receptor heterodimer acts as a neoantigen, eventually activating an immune response to generate the above-mentioned agonistic autoantibodies.
In addition to enhancing vascular tone, activation of AT1 (by Ang II or agonistic antibodies) and AT1–B2 receptor heterodimers generates ROS through long-term stimulation of endothelial and vascular smooth muscle NADH/NADPH OXIDASE.77 AT1-agonistic antibodies can also target vascular smooth muscle angiotensin receptors to activate tissue factor,125 and mesangial cells to release interleukin-6 and plasminogen-activator-inhibitor-1.126 These findings might account for activation of coagulation and initiation of renal inflammation, other prominent features of the maternal disease. Furthermore, AT1-agonistic antibodies caused stimulation of plasminogen-activator-inhibitor-1 in human trophoblasts in vitro and perturbed trophoblast invasiveness, indicating that the antibodies might also have a role in impaired trophoblast invasion.127 The importance of AT1–B2 heterodimerization and agonistic AT1 antibodies for the pathogenesis of the maternal syndrome depends to some extent on whether these factors are specific for pre-eclampsia or operate in other hypertensive and vascular disorders.
It has been postulated that oxidative stress resulting from placental ischemia is the fundamental abnormality leading to endothelial damage in pre-eclampsia. Formation of oxidized LDLs is thought to initiate endothelial damage in atherosclerosis. In pre-eclampsia, fibrinoid necrosis of vessel walls, with accumulation of lipid-laden foam cells, is the hallmark of oxidized LDL.128 Moreover, increased concentrations of IgG antibodies directed against oxidized LDL have been documented in pre-eclampsia, although not by all investigators. The possibility that oxidative stress has a role in determining endothelial dysfunction of pre-eclamptic maternal syndrome has sound biochemical underpinnings. ROS alter endothelial metabolism by blocking mitochondrial electron transport, oxidizing proteins and initiating lipid peroxidation.82 Lipid peroxides and ONOO− activate cyclooxygenase-1 and cyclooxygenase-2,129 thus increasing TxA2 synthesis. As mentioned earlier, levels of this vasoconstrictive prostanoid are elevated in pre-eclampsia. At the same time, lipid peroxides and ONOO− inhibit endothelial PGI2 synthase,129 thereby ultimately contributing to the TxA2–PGI2 imbalance that precedes the onset of clinical symptoms of pre-eclampsia.
Unifying hypothesis
Although our understanding of the pathophysiology of pre-eclampsia is still incomplete, the recently published studies reviewed above provide elements for a unifying hypothesis (Figure 4). We propose that pre-eclampsia might be a disease of placentation triggered by reduced activity or half-life of NO, secondary to a low tissue L-Arg concentration that is a consequence of excessive arginase expression. In response to the hypoxia that results from abnormal placentation and placental hypoperfusion, the placenta releases several factors into the maternal circulation, including sFlt1 and ROS, which initiate endothelial dysfunction and subsequent multisystem organ defects.

We propose that pre-eclampsia is a disease of placentation triggered by reduced activity or half-life of nitric oxide secondary to low placental L-arginine concentration, which is in turn related to excessive arginase II expression. Low L-arginine levels stimulate endothelial nitric oxide synthase to generate reactive oxygen species (e.g. peroxynitrite and hydroxyl radical) and locally exacerbate oxidative stress. Reduced flow through the placental L-arginine–nitric oxide pathway is also consistent with the higher resistance and hypoperfusion of the fetal–placental circulation. In response to the restricted placental blood flow and consequent hypoxia, the placenta releases into the maternal circulation a number of factors including soluble Fms-like tyrosine kinase 1 and reactive oxygen species, which initiate the vascular dysfunction characteristic of the maternal syndrome. Soluble Fms-like tyrosine kinase 1 is present at high concentrations in the blood of pre-eclamptic women, and administration of soluble Fms-like tyrosine kinase 1 to animals produces a maternal syndrome resembling that of pre-eclampsia. In a further attempt to ameliorate perfusion of the placenta as well as of the maternal organs involved in pre-eclampsia, upregulation of the B2 receptor for the vasodilator bradykinin occurs. The B2 receptors heterodimerize with the angiotensin II type 1 receptor, thereby increasing vascular and inflammatory responsiveness to angiotensin II, paradoxically reducing systemic organ perfusion and promoting generation of reactive oxygen species. Concomitant production of agonist angiotensin II type 1 receptor autoantibodies also contributes to oxidative stress. A positive-feedback loop is initiated that eventually results in the full-blown clinical syndrome of pre-eclampsia. Abs, antibodies; Ang II, angiotensin II; AT1, angiotensin II type 1 receptor; eNOS, endothelial nitric oxide synthase; L-Arg, L-arginine; MMP2, matrix metalloproteinase 2; MMP9, matrix metalloproteinase 9; NO, nitric oxide; O2−, superoxide anion; OH˙, hydroxyl radical; PGI2, prostacyclin; sFlt1, soluble Fms-like tyrosine kinase 1; TxA2, thromboxane A2; VEGF, vascular endothelial growth factor.
Therapeutic perspectives
Novel findings on the pathophysiology of the placental and maternal abnormalities of pre-eclampsia provide a rationale for potential prophylactic or therapeutic intervention. L-Arg or antioxidant supplementation,130 aimed at providing more substrate to eNOS and lowering the rate of NO degradation to ONOO−, respectively, could help correct placental and systemic vascular dysfunction. Administration of L-Arg could theoretically counterbalance the excessive degradation of this amino acid in the placenta by arginase II, and stimulate eNOS to release more NO and less superoxide anion. Preliminary data from a recent trial in 17 pre-eclamptic women showed that intravenous infusion of L-Arg was associated with a significant reduction of blood pressure.131 In a larger placebo-controlled study, 3 weeks of L-Arg supplementation in pre-eclamptic women increased NO synthesis and lowered blood pressure compared with those given placebo.132 In a preliminary study in women at high risk of placentation disorders (assessed as BILATERAL NOTCHING and high uterine artery resistance) chronic oral L-Arg supplementation from 10 weeks of gestation decreased mean arterial pressure and uterine artery resistance, improved endothelium-mediated vasodilatation and ameliorated perinatal outcome.92
A combination of the antioxidant vitamins C and E given to pregnant women at high risk of pre-eclampsia reduced circulating levels of plasminogen-activator-inhibitor-1 (a marker of endothelial activation), lowered indices of oxidative stress and decreased the frequency of pre-eclampsia compared with placebo.130,133 These results indicate that oxidative stress might be a target for prevention of pre-eclampsia. Larger trials have begun in the US, Canada, Mexico, England and several developing nations.134 The NIH has funded two antioxidant trials; the first is under way in Brazil and the second is being carried out in nulliparous women in the National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network.135 These trials are expected to elucidate the effectiveness of antioxidants in prevention of pre-eclampsia.
Strategies to normalize circulating levels of free VEGF and PlGF by reducing the plasma concentration of sFlt1 or blocking its effects might be expected to ameliorate the systemic vascular and inflammatory response of the maternal syndrome. In mutant mice, Flt1 is not required for establishment of the maternal–fetal interface in the placenta, indicating that suppression or binding of sFlt1 to cure the maternal syndrome might not have adverse effects on the placenta. Clinical trials with VEGF are also being planned.144
Conclusions
The lack of a specific animal model of pre-eclampsia hinders experimental evaluation of pathogenetic mechanisms and effectiveness of potential new therapeutic approaches to this disorder. The model obtained by administering an L-Arg inhibitor to pregnant rats is the most commonly used. It might be a suitable background in which to test the effect of arginine deprivation on placental development, and to evaluate strategies to overcome such a defect with compounds that either prevent ONOO− formation (such as arginase inhibitors and antioxidants) or block sFlt1 release from the placenta. The pre-eclampsia-like rat model induced by adenoviral-mediated administration of sFlt166 could also be used to test potential therapeutics.
Ongoing clinical studies are assessing the potential beneficial effect of L-Arg in women with pre-eclampsia; in particular, whether increasing L-Arg bioavailability might restore physiological NO production and inhibit ONOO− formation in pre-eclamptic placenta. This takes the form of a case–control pilot study of women given oral L-Arg supplementation from the time of diagnosis of pre-eclampsia to delivery, compared with placebo-treated pre-eclamptic women. Results could form the basis of a large, multicenter trial aimed at exploring whether prophylactic L-Arg supplementation reduces the incidence of pre-eclampsia in women at high risk of this disease, which is one of the leading causes of maternal and fetal morbidity and mortality.
Review criteria
We searched PubMed for articles published between 1970 and 2005. The keywords used were “pre-eclampsia”, “gestational hypertension”, “clinical features”, “pathophysiology” and “treatment”. We also searched the bibliographies of the articles retrieved for further relevant references. Because of the large number of articles identified, the decision on which to include was based on personal judgment. Moreover, as the diagnostic criteria for pre-eclampsia were refined in 2000 by the National High Blood Pressure Education Program Working Group,1 this review focuses on studies published during the past 5 years.
References
National High Blood Pressure Education Program Working Group on High Pressure in Pregnancy (2000) Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 183: S1–S22
Mahmoudi N et al. (1999) Eclampsia: A 13-year experience at a United States tertiary care center. J Womens Health Gender Based Med 8: 495–500
de Swiet M (2000) Maternal mortality: confidential enquiries into maternal deaths in the United Kingdom. Am J Obstet Gynecol 182: 760–766
Sibai BM et al. (1993) Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome). Am J Obstet Gynecol 169: 1000–1006
Villar K et al. (2003) Eclampsia and pre-eclampsia: a health problem for 2000 years. In Preeclampsia, 189–207 (Eds Critchley H et al.) London: RCOG Press
Sibai BM (2003) Diagnosis and management of gestational hypertension and preeclampsia. Obstet Gynecol 102: 181–192
Lòpez-Jaramillo P et al. (2001) Preeclamplsia: from epidemiological observations to molecular mechanisms. Braz J Med Biol Res 34: 1227–1235
Belizàn JM and Villar J (1980) The relationship between calcium intake and edema-proteinuria and hypertension-gestosis: a hypothesis. Am J Clin Nutr 33: 2202–2210
Li D and Wi S (2000) Changing paternity and the risk of preeclampsia in the subsequent pregnancy. Am J Epidemiol 151: 57–62
Skjaerven R et al. (2002) The interval between pregnancies and the risk of preeclampsia. N Engl J Med 346: 33–38
Dekker G and Robillard PY (2003) The birth interval hypothesis: does it really indicate the end of the primipaternity hypothesis? J Reprod Immunol 59: 245–251
Duckitt K and Harrington D (2005) Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. BMJ 330: 565
Milne F et al. (2005) The pre-eclampsia community guideline (PRECOG): how to screen for and detect onset of pre-eclampsia in the community. BMJ 330: 549–550
Walker JJ (2000) Pre-eclampsia. Lancet 356: 1260–1265
Wolf M et al. (2002) First trimester insulin resistance and subsequent preeclampsia: a prospective study. J Clin Endocrinol Metab 87: 1563–1568
Kupferminc M et al. (1999) Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 340: 9–13
Brewer T (1976) Role of malnutrition in pre-eclampsia and eclampsia. Am J Obstet Gynecol 125: 281–282
Hofmeyr GJ et al. (2003) Calcium supplementation to prevent pre-eclampsia—a systemic review. S Afr Med J 93: 224–228
Atallah AN et al. (2002) Calcium supplementation during pregnancy for preventing hypertensive disorders and related problems. Cochrane Database Syst Rev 2002: CD001059
Villar J and Belizan JM (2000) Same nutrient, different hypotheses: disparities in trials of calcium supplementation during pregnancy. Am J Clin Nutr 71 (Suppl 5): S1375–S1379
Ward K et al. (1993) A molecular variant of angiotensinogen associated with preeclampsia. Nat Genet 4: 59–61
Arngrimsson R et al. (1997) Evidence for a familial pregnancy-induced hypertension locus in the eNOS-gene region. Am J Hum Genet 61: 354–362
Dizon-Townson D et al. (1996) The factor V Leiden mutation may predispose women to severe preeclampsia. Am J Obstet Gynecol 175: 902–905
Sohda S et al. (1997) Methylenetetrahydrofolate reductase polymorphism and pre-eclampsia. J Med Genet 34: 525–526
O'Shaughnessy K et al. (1999) Factor V Leiden and thermolabile methylenetetrahydrofolate reductase gene variants in an East Anglian preeclampsia cohort. Hypertension 33: 1338–1341
Arngrimsson R et al. (1999) A genome-wide scan reveals a maternal susceptibility locus for preeclampsia on chromosome 2p13. Hum Mol Genet 8: 1799–1805
Laivuori H et al. (2003) Susceptibility loci for preeclampsia on chromosomes 2p25 and 9p13 in Finnish families. Am J Hum Genet 72: 168–177
Lachmeijer AM et al. (2001) A genome-wide scan for preeclampsia in the Netherlands. Eur J Hum Genet 9: 758–764
Moses EK et al. (2000) A genome scan in families from Australia and New Zealand confirms the presence of a maternal susceptibility locus for pre-eclampsia, on chromosome 2. Am J Hum Genet 67: 1581–1585
Oudejans CB et al. (2004) The parent-of-origin-effect of 10q22 in pre-eclamptic females coincides with two regions clustered for genes with down-regulated expression in androgenetic placentas. Mol Hum Reprod 10: 589–598
van Dijk M et al.: Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet, in press
GOPEC Consortium (2005) Disentangling fetal and maternal susceptibility for pre-eclampsia: a British multicenter candidate-gene study. Am J Hum Genet 77: 127–131
Levine RJ et al. (2000) Should the definition of preeclampsia include a rise in diastolic blood pressure of >/= 15 mm Hg in association with proteinuria? Am J Obstet Gynecol 183: 787–792
Brown MA et al. (2001) The classification and diagnosis of the hypertensive disorders of pregnancy: a statement from the International Society for the Study of Hypertension in Pregnancy (ISSHP). Hypertens Pregn 20: IX–XIV
Waugh JJ et al. (2004) Accuracy of urinalysis dipstick techniques in predicting significant proteinuria in pregnancy. Obstet Gynecol 103: 769–777
Sibai BM (2004) Diagnosis, controversies, and management of HELLP syndrome. Obstet Gynecol 103: 981–991
Yamasmit W et al. (2004) Random urinary protein-to-creatinine ratio for prediction of significant proteinuria in women with preeclampsia. J Matern Fetal Neonatal Med 16: 275–279
Brown MA et al. (2000) The detection, investigation and management of hypertension in pregnancy: executive summary. Aust NZ J Obstet Gynecol 40: 133–138
Sibai BM (2002) Chronic hypertension in pregnancy. Obstet Gynecol 100: 369–377
Gilstrap LC III and Ramin SM on behalf of the ACOG Committee on Practice Bulletin (2002) Diagnosis and management of preeclampsia and eclampsia. Int J Obstet Gynecol 77: 67–75
Moran P et al. (2004) The renal response to preeclampsia. Semin Nephrol 24: 588–595
Hibbard JU et al. (2004) Cardiovascular changes in preeclampsia. Semin Nephrol 24: 580–587
Haukkamaa L et al. (2004) Risk for subsequent coronary artery disease after preeclampsia. Am J Cardiol 93: 805–808
Zhang J et al. (2003) Severe maternal morbidity associated with hypertensive disorders in pregnancy in the United States. Hypertens Pregn 22: 203–212
Hauth JC et al. (2000) Pregnancy outcomes in healthy nulliparous women who subsequently developed hypertension. Obstet Gynecol 95: 24–28
Buchbinder A et al. (2002) Adverse perinatal outcomes are significantly higher in severe gestational hypertension than in mild preeclampsia. Am J Obstet Gynecol 186: 66–71
Duley L (2003) Pre-eclampsia and the hypertensive disorders of pregnancy. Br Med Bull 67: 161–176
Conde-Agudelo A et al. (2004) World Health Organization systematic review of screening tests for preeclampsia. Obstet Gynecol 104: 1367–1391
North RA (2003) Can we predict pre-eclampsia? In Preeclampsia, 257–275 (Eds Critchley H et al.) London: RCOG Press
Axt-Fliedner R (2004) Second trimester uterine artery Doppler ultrasound as a screening test for adverse pregnancy outcome. Clin Exp Obstet Gynecol 31: 9–11
Red-Horse K et al. (2004) Trophoblast differentiation during embryo implantation and formation of the maternal–fetal interface. J Clin Invest 114: 744–754
Lyall F (2005) Priming and remodelling of human placental bed spiral arteries during pregnancy—a review. Placenta 26 (Suppl A): S31–S36
Zhou Y et al. (1997) Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype: One cause of defective endovascular invasion in this syndrome? J Clin Invest 99: 2152–2164
Pijnenborg R et al. (1991) Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br J Obstet Gynaecol 98: 648–655
Kadyrov M et al. (2003) Preeclampsia and maternal anemia display reduced apoptosis and opposite invasive phenotypes of extravillous trophoblast. Placenta 24: 540–548
Moldenhauer JS et al. (2003) The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am J Obstet Gynecol 189: 1173–1177
Teasdale F (1985) Histomorphometry of the human placenta in maternal preeclampsia. Am J Obstet Gynecol 152: 25–31
Moffett-King A (2002) Natural killer cells and pregnancy. Nat Rev Immunol 2: 656–663
Parham P (2004) NK cells and trophoblasts: partners in pregnancy. J Exp Med 200: 951–955
Hiby SE et al. (2004) Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med 200: 957–965
Zhou Y et al. (1997) Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest 99: 2139–2151
Sharkey A et al. (1993) Expression of vascular endothelial growth factor in human placenta. J Reprod Fertil 99: 609–615
Zhou Y et al. (2002) Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol 160: 1405–1423
Cooper J et al. (1996) VEGF mRNA levels in placentae from pregnancies complicated by preeclampsia. Br J Obstet Gynaecol 103: 1191–1196
Levine R et al. (2004) Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350: 672–683
Maynard S et al. (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658
Hirashima M et al. (2003) Trophoblast expression of fms-like tyrosine kinase 1 is not required for the establishment of the maternal-fetal interface in the mouse placenta. Proc Natl Acad Sci USA 100: 15637–15642
Papapetropoulos A et al. (1997) Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 100: 3131–3139
Moncada S and Higgs A (1993) The L-arginine-nitric oxide pathway. N Engl J Med 329: 2002–2012
Baylis C et al. (1998) Recent insights into the roles of nitric oxide and renin-angiotensin in the pathophysiology of preeclamptic pregnancy. Semin Nephrol 18: 208–230
Thomson AJ et al. (1997) Nitric oxide synthase activity and localization do not change in uterus and placenta during human parturition. Hum Reprod 12: 2546–2552
Martin D and Conrad KP (2000) Expression of endothelial nitric oxide synthase by extravillous trophoblast cells in the human placenta. Placenta 21: 23–31
Eis AL et al. (1995) Immunohistochemical localization of endothelial nitric oxide synthase in human villous and extravillous trophoblast populations and expression during syncytiotrophoblast formation in vitro. Placenta 16: 113–126
Orange SJ et al. (2003) Placental endothelial nitric oxide synthase localization and expression in normal human pregnancy and pre-eclampsia. Clin Exp Pharmacol Physiol 30: 376–381
Lyall F et al. (1999) Human trophoblast invasion and spiral artery transformation. The role of nitric oxide. Am J Pathol 154: 1105–1114
Novaro V et al. (2001) Regulation of metalloproteinases by nitric oxide in human trophoblast cells in culture. Reprod Fertil Dev 13: 411–420
Davison JM et al. (2004) New aspects in the pathophysiology of preeclampsia. J Am Soc Nephrol 15: 2440–2448
Dunk C et al. (2000) Angiopoietin-1 and angiopoietin-2 activate trophoblast Tie-2 to promote growth and migration during placental development. Am J Pathol 156: 2185–2199
Ahmed A et al. (1997) Role of VEGF receptor-1 (Flt-1) in mediating calcium-dependent nitric oxide release and limiting DNA synthesis in human trophoblast cells. Lab Invest 76: 779–791
Chang CC et al. (2005) Induction of VE-cadherin in rat placental trophoblasts by VEGF through a NO-dependent pathway. Placenta 26: 234–241
Noris M et al. (1996) The role of vasoactive molecules of endothelial origin in the pathophysiology of normal pregnancy and pregnancy-induced hypertension. Curr Opin Nephrol Hypertens 5: 347–352
Radi R et al. (1991) Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 288: 481–487
Many A et al. (2000) Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am J Pathol 156: 321–331
Wang W et al. (2000) Superoxide production and reactive oxygen species signaling by endothelial nitric-oxide synthase. J Biol Chem 275: 16899–16903
Xia Y et al. (1996) Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA 93: 6770–6774
Vasquez-Vivar J et al. (1998) Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA 95: 9220–9225
Xia Y and Zweier JL (1997) Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA 94: 6954–6958
Noris M et al. (2004) L-arginine depletion in preeclampsia orients nitric oxide synthase toward oxidant species. Hypertension 43: 614–622
Li H et al. (2001) Regulatory role of arginase I and II in nitric oxide, polyamine, and proline synthases in endothelial cells. Am J Physiol 280: E75–E82
Lowe D (2000) Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric Oxide 4: 441–458
Carlsen SM et al. (2005) Early second-trimester maternal hyperandrogenemia and subsequent preeclampsia: a prospective study. Acta Obstet Gynecol Scand 84: 117–121
Germain A et al. (2004) Evidence supporting a beneficial role for long term L-arginine supplementation in high risk pregnancies. Hypertension 44: e1
Molnar M et al. (1994) Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol 170: 1458–1466
Tanir HM et al. (2005) Effect of quercetine and glutathione on the level of superoxide dismutase, catalase, malonyldialdehyde, blood pressure and neonatal outcome in a rat model of pre-eclampsia induced by NG-nitro-L-arginine-methyl ester. Eur J Obstet Gynecol Reprod Biol 118: 190–195
Caniggia I and Winter JL (2002) Hypoxia inducible factor-1: oxygen regulation of trophoblast differentiation in normal and pre-eclamptic pregnancies—A review. Placenta 23 (Suppl A): S47–S57
Rajakumar A et al. (2004) Evidence of the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 25: 763–769
Caniggia I et al. (2000) Hypoxia-inducible factor-1 mediates biological effects of oxygen in human trophoblast differentiation through TGFβ3 . J Clin Invest 105: 577–587
Caniggia I et al. (1999) Inhibition of TGFβ3 restores the invasive capability of extravillous trophoblast in preeclamptic pregnancies. J Clin Invest 103: 1641–1650
Hung T-H et al. (2002) Hypoxia-reoxygenation. A potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res 90: 1274–1281
Parks D et al. (1988) Conversion of xanthine dehydrogenase to oxidase in ischemic rat intestine: a reevaluation. Am J Physiol 254: G768–G774
McLaughlin MK and Roberts JM (1999) Hemodynamic changes in pregnancy. In Hypertensive Disorders in Pregnancy, 92–95 (Eds Lindheimer MD et al.) Stamford: Appleton & Lange
Shah D (2005) Role of the renin–angiotensin system in the pathogenesis of preeclampsia. Am J Renal Physiol 288: F614–F625
Bowyer L et al. (2003) Forearm blood flow in pre-eclampsia. BJOG 110: 383–391
Elsheikh A et al. (2001) The renin-aldosterone system during normal and hypertensive pregnancy. Arch Gynecol Obstet 264: 182–185
Walsh S (2004) Eicosanoids in preeclampsia. Prostaglandins Leukot Essent Fatty Acids 70: 223–232
Seligman SP et al. (1994) The role of nitric oxide in the pathogenesis of preeclampsia. Am J Obstet Gynecol 171: 944–948
Garmendia JV et al. (1997) Nitric oxide in different types of hypertension during pregnancy. Clin Sci 93: 413–421
Fickling SA et al. (1993) Plasma concentrations of endogenous inhibitor of nitric oxide synthesis in normal pregnancy and pre-eclampsia. Lancet 34: 242–243
Lopez-Jaramillo P et al. (1996) Cyclic guanosidine 3,5 monophosphate concentrations in pre-eclampsia: effects of hydralazine. Br J Obstet Gynaecol 103: 33–38
Pridjian G and Puschett JB (2002) Preeclampsia. Part 1: Clinical and pathophysiologic considerations. Obstet Gynecol Surv 57: 598–618
Buhimschi CS et al. (2005) Urinary angiogenic factors cluster hypertensive disorders and identify women with severe preeclampsia. Am J Obstet Gynecol 192: 734–741
Stepan H et al. (2004) Soluble fms-like tyrosine kinase 1. N Engl J Med 351: 2241–2242
Wolf M et al. (2005) Circulating levels of the antiangiogenic marker sFLT-1 are increased in first versus second pregnancies. Am J Obstet Gynecol 193: 16–22
Luttun A and Carmeliet P (2003) Soluble VEGF receptor Flt1: the elusive preeclampsia factor discovered? J Clin Invest 111: 600–602
Li H et al. (2005) Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta 26: 210–217
Livingston JC et al. (2000) Reductions of vascular endothelial growth factor and placental growth factor concentrations in severe preeclampsia. Am J Obstet Gynecol 183: 1554–1557
Levine RJ et al. (2005) Urinary placental growth factor and risk of preeclampsia. JAMA 293: 77–85
Masuda Y et al. (2001) Vascular endothelial growth factor enhances glomerular capillary repair and accelerates resolution of experimentally induced glomerulonephritis. Am J Pathol 159: 599–608
Sugimoto H et al. (2003) Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem 278: 12605–12608
Yang J et al. (2003) A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 349: 427–434
Eremina V et al. (2003) Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111: 707–716
Boyd P et al. (1987) Pre-eclampsia and trisomy 13: a possible association. Lancet 2: 425–427
Bdolah Y et al. (2005) Trisomy 13 and preeclampsia: an old link with a new explanation. J Soc Gynecol Invest 12 (Suppl 2): S196A
AbdAlla S et al. (2001) Increased AT1 receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med 7: 1003–1009
Dechend R et al. (2000) AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation 101: 2382–2387
Bobst SM et al. (2005) Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin-6 and plasminogen activator inhibitor-1 secretion. Am J Hypertens 18: 330–336
Xia Y et al. (2003) Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. J Soc Gynecol Investig 10: 82–93
Hubel C et al. (1989) Lipid peroxidation in pregnancy: New perspectives on preeclampsia. Am J Obstet Gynecol 161: 1025–1034
Cosentino F et al. (2003) High glucose causes upregulation of cyclooxygenase-2 and all prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation 107: 1017–1023
Chappell L et al. (1999) Effect of antioxidants on the occurrence of pre-eclampsia in women at increased risk: a randomized trial. Lancet 354: 810–816
Facchinetti F et al. (1999) L-Arginine infusion reduces blood pressure in preeclamptic women through nitric oxide release. J Soc Gynecol Invest 6: 202–207
Rytlewski K et al. (2005) Effects of prolonged oral supplementation with L-arginine on blood pressure and nitric oxide synthesis in preeclampsia. Eur J Clin Invest 35: 32–37
Chappell L et al. (2002) Vitamin C and E supplementation in women at risk of preeclampsia is associated with changes in indices of oxidative stress and placental function. Am J Obstet Gynecol 187: 777–784
Roberts JM and Speer P (2004) Antioxidant therapy to prevent preeclampsia. Semin Nephrol 24: 557–564
Fraser WD et al. (2005) The vitamin E debate: implications for ongoing trials of pre-eclampsia prevention. BJOG 112: 684–688
Sibai B et al. (2005) Pre-eclampsia. Lancet 365: 785–799
Mautner W et al. (1962) Preeclamptic nephropathy. An electron microscopic study. Lab Invest 11: 518–530
Gallery E and Lindheimer M (1999) Pathology and pathophysiology of preeclampsia: Alterations in volume homeostasis. In Chesley's Hypertensive Disorders in Pregnancy, 327–347 (Eds Lindheimer M et al.) Stamford: Appleton & Lange
Hayashi T (1956) Uric acid and endogenous creatinine clearance studies in normal pregnancy and toxemias of pregnancy. Am J Obstet Gynecol 71: 859–870
Zissin R et al. (1999) Hepatic infarction in preeclampsia as part of the HELLP syndrome: CT appearance. Abdom Imaging 24: 594–596
Port J and Beauchamp NJ (1998) Reversible intracerebral pathologic entities mediated by vascular autoregulatory dysfunction. Radiographics 18: 353–367
Saleh A et al. (1992) Markers for endothelial injury, clotting and platelet activation in preeclampsia. Arch Gynecol Obstet 251: 105–110
Stubbs T et al. (1986) Evidence of accelerated platelet production and consumption in non thrombocytopenic preeclampsia. Am J Obstet Gynecol 155: 263–265
Lindheimer MD (2005) Unraveling the mysteries of preeclampsia. Am J Obstet Gynecol 193: 3–4
Acknowledgements
We are indebted to Professor Franklin H Epstein (Senior Physician, Renal Division, Department of Medicine, Beth Israel Deaconess Medical Center, Boston) and William Applebaum (Professor of Medicine, Harvard Medical School, Boston) for their critical reading of the manuscript and insightful discussion. We also thank Dr Prudence Hill (Department of Anatomical Pathology, St Vincent's Hospital, Melbourne) for her critical reading.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Noris, M., Perico, N. & Remuzzi, G. Mechanisms of Disease: pre-eclampsia. Nat Rev Nephrol 1, 98–114 (2005). https://doi.org/10.1038/ncpneph0035
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/ncpneph0035
This article is cited by
-
Dietary supplements and vascular function in hypertensive disorders of pregnancy
Pflügers Archiv - European Journal of Physiology (2023)
-
Drug Safety in Episodic Migraine Management in Adults. Part 2: Preventive Treatments
Current Pain and Headache Reports (2022)
-
The Evidence of Aspirin Use in Prevention of Adverse Pregnancy Outcomes (APOs): Should It Be Continued Long Term After an APO?
Current Treatment Options in Cardiovascular Medicine (2021)
-
Lipoxin A4 suppresses angiotensin II type 1 receptor autoantibody in preeclampsia via modulating caspase-1
Cell Death & Disease (2020)
-
Changes of lipoxin levels during pregnancy and the monthly-cycle, condition the normal course of pregnancy or pathology
Inflammation Research (2020)
