
Abstract
Regulatory CD4+ T cells (Tr cells), the development of which is critically dependent on X-linked transcription factor Foxp3 (forkhead box P3), prevent self-destructive immune responses1. Despite its important role, molecular and functional features conferred by Foxp3 to Tr precursor cells remain unknown. It has been suggested that Foxp3 expression is required for both survival of Tr precursors as well as their inability to produce interleukin (IL)-2 and independently proliferate after T-cell-receptor engagement, raising the possibility that such ‘anergy’ and Tr suppressive capacity are intimately linked2,3,4. Here we show, by dissociating Foxp3-dependent features from those induced by the signals preceding and promoting its expression in mice, that the latter signals include several functional and transcriptional hallmarks of Tr cells. Although its function is required for Tr cell suppressor activity, Foxp3 to a large extent amplifies and fixes pre-established molecular features of Tr cells, including anergy and dependence on paracrine IL-2. Furthermore, Foxp3 solidifies Tr cell lineage stability through modification of cell surface and signalling molecules, resulting in adaptation to the signals required to induce and maintain Tr cells. This adaptation includes Foxp3-dependent repression of cyclic nucleotide phosphodiesterase 3B, affecting genes responsible for Tr cell homeostasis.
Similar content being viewed by others


Main
In males, Foxp3 deficiency results in fatal early-onset systemic autoimmune disease5. In heterozygote Foxp3wt/null females only one-half of T cells harbours the mutant Foxp3 allele due to random X-chromosome inactivation, whereas autoimmunity is controlled by a normal Tr population expressing the Foxp3 wild-type allele. Thus, we were able to genetically mark cells actively transcribing a Foxp3null allele, yet lacking Foxp3 protein (hereafter called Tfn for Foxp3null-expressing T cells), through an in-frame insertion of GFP into a stop-codon-disrupted Foxp3 locus (Foxp3gfpko) and investigate their features in mice (Fig. 1a; see also Supplementary Figs 1 and 2a). Female Foxp3gfpko/wt mice were healthy, whereas male Foxp3gfpko mice developed the same severity of autoimmunity as Foxp3 knockout (Foxp3null) mice6, resulting in death at ∼4 weeks of age. Thymocyte and peripheral lymphoid organ cellularity did not differ between Foxp3gfpko/wt and Foxp3gfp/gfp mice, nor did the proportion of Foxp3+ Tr cells and Foxp3- CD4+ T cells (data not shown). As our main focus was to characterize Tfn cells in healthy Foxp3gfpko/wt mice, analysis of autoimmune male Foxp3gfpko mice is included as Supplementary Fig. 2.

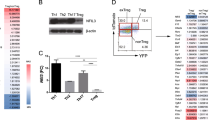
a, We modified our previously described GFP–Foxp3 fusion protein targeting construct8 by inserting stop codon/frameshift cassettes downstream of GFP and also into Foxp3 exon 5. In this manner, potentially important regulatory sequences were retained without the possibility that cryptic re-initiation of Foxp3 translation would generate full-length protein. b, Flow cytometric analysis of CD4+CD8-HSAlow thymocytes or CD4+ lymph node cells from female Foxp3gfp/gfp or Foxp3gfpko/wt mice. c, Frequency of GFP+ cells among either CD4+CD8-HSAlow thymocytes (Thy, circles) or CD4+ lymph node cells (LN, diamonds) for individual mice. d, Flow cytometric analysis of CD4+ lymph node cells for the indicated markers. The four cell subsets are demarcated in b as follows: GFP–CD25– Tn cells (grey), GFP–CD25+Foxp3-CD4+ T cells (green), GFP+ Tr cells (red) from Foxp3gfp/gfp mice, and GFP+ Tfn cells from Foxp3gfpko/wt mice (blue). e, Ki67 staining of lymph node cells from the indicated mice. f, Proliferation of sorted T cells in response to the indicated reagents. Results are representative of three independent experiments. Data represent the mean and standard deviation for triplicate wells. g, Cytokine production by sorted cells from Foxp3gfp/gfp (Tn, CD25+Foxp3-CD4+, Tr cells) or Foxp3gfpko/wt (Tfn cells) mice in response to plate-bound anti-CD3 and anti-CD28 assayed by enzyme-linked immunosorbent assay. Results are representative of three separate experiments; data represent mean and maximum/minimum for duplicate wells and colours are as described in d.
Tfn cells constituted ∼1–3% of mature CD4+ thymocytes and peripheral CD4+ T cells, indicating that Foxp3 is not required to rescue Tr precursors from negative selection (Fig. 1b, c). This is consistent with a reported abundance of T-cell receptors (TCRs) characteristic of Tr cells in Foxp3null mice7. As ectopic expression of Foxp3 has been shown to induce a state of hyporesponsiveness in CD4+ T cells3, we expected Tfn cells to appear highly activated, similar to CD25+Foxp3-CD4+ T cells (Fig. 1b), which exhibit characteristics of activated, pro-inflammatory T cells8. Tfn cells, however, were small and CD62Lhigh, more similar to Tr cells and naive CD4+ cells than to CD25+Foxp3-CD4+ T cells (Fig. 1b, d). Tfn cells expressed intermediate levels of CD25, CD44, CTLA4, GITR and ICOS in comparison to Tr cells (Fig. 1b, d; see also Supplementary Fig. 3a). Notably, reduced IL-7R expression, which is considered to be a distinguishing feature of Tr cells, was lowest on Tfn cells in comparison to the other T-cell subsets (Fig. 1d). In contrast to Tr cells, Tfn cells exhibited negligible proliferative activity in vivo based on proliferation-associated antigen Ki67 expression (Fig. 1e).
In vitro, both Tfn and Tr cells showed an anergic phenotype, which was reversed in both Tfn and Tr cells by provision of IL-2; however, proliferation was more readily restored in Tfn cells by limited CD28 co-stimulation (Fig. 1f). Thus, anergy in Tfn cells is less stable in comparison to Tr cells. Tfn cells were also similar to Tr cells—and distinct from Tn (GFP-CD25-CD4+ T cells from Foxp3gfp/gfp mice) and CD25+Foxp3-CD4+ T cells—in their inability to generate IL-2 and T-helper 1 and 2 (Th1 and Th2) cytokines (Fig. 1g). Thus, IL-2 expression may be blocked transcriptionally during Tr cell differentiation in a Foxp3-independent manner. Lack of IL-2 production and decreased IL-7R make both Tfn and Tr cells reliant on exocrine IL-2, but Tfn cells are probably less competitive due to intermediate CD25 levels. Indeed, we found that Tfn cells were hyper-responsive to elevated IL-2 levels in vivo (Supplementary Fig. 7). Together, our analyses show that Tr cell development coincides with Foxp3-independent acquisition of several key Tr cell characteristics.
Unlike TR cells, some Tfn cells produced either IL-10 or IL-17, resembling Tr1 (IL-10-producing Foxp3-CD4+ T cells9) and Th17 cells, respectively (Fig. 1g; see also Supplementary Fig. 2g). Consistent with observed IL-17 production, both Tfn and CD25+Foxp3-CD4+ T cells were found to express orphan nuclear receptor RORγt (Rorc), which has been shown to both promote IL-17 and block IL-2 production in Th17 cells10 (Supplementary Fig. 3b). Notably, Rorc transcription was reduced only fourfold in TR relative to Tfn cells but Il17 transcription was suppressed 7,500-fold, suggesting an ability of Foxp3 to over-ride RORγt activity.
The defining feature of Tr cells is their ability to suppress pro-inflammatory immune function. Tfn cells were incapable of suppressing CD4+CD25- T-cell proliferation in vitro (Fig. 2a) and controlling effector T-cell expansion and ensuing splenomegaly and lymphadenopathy after adoptive co-transfer into T-cell-deficient recipient mice (Fig. 2b, c). Spleens and lymph nodes of mice that had received CD45RBhighCD25-CD4+ effector T cells with Tfn cells were similar in cellularity to those of mice that had received effector T-cells alone, whereas the co-transfer of Tr cells with effector T cells significantly limited effector T-cell expansion. Similarly, effector T cells were increased in size in the presence of Tfn cells, but not Tr cells, likely reflecting increased proliferation (Fig. 2c). Lack of suppression was not due to loss of Tfn cells because donor Tfn cells competed efficiently with effector T cells during homeostatic expansion, manifested by an increase in their proportion and number (Fig. 2d).

a, FACS purified Tr or Tfn cells were compared for their ability to suppress CD25-CD4+ T-cell proliferation in the presence of concavalin A and T-cell-depleted splenic antigen presenting cells. Data represent mean and standard deviation for triplicate wells; results are representative of four separate experiments. b–d, To examine suppressor activity in vivo, Tr or Tfn cells were co-transferred with allelically marked effector T cells into lymphopenic animals. Sorted Ly5.1+CD45RBhighCD25-CD4+ effector T cells (Teff, 3 × 105) were transferred into B6.SCID mice alone or with sorted TR or Tfn cells (105). Five weeks after transfer, splenocytes were enumerated (b) and lymph node and spleen cells were analysed by flow cytometry. Cell size of the effector T-cell population (c) and abundance and GFP expression of the donor Tr or Tfn cell populations (d) are shown. Data are representative of two independent experiments. e, Quantification of Foxp3 cDNA prepared from the indicated cell populations. For peripheral cells, error bars represent mean and maximum/minimum for biological duplicates.
Tfn cells also differed from Tr cells in their ability to retain Foxp3 transcription. Whereas ∼90% of donor Tr cells from Foxp3gfp/gfp mice retained expression of GFP-tagged Foxp3 protein, two-thirds of the expanded Tfn population lost GFP expression (Fig. 2d). Thus, in the course of homeostatic proliferation, Foxp3 transcription in the absence of Foxp3 function was not efficiently retained. Furthermore, quantification of Foxp3 messenger RNA in both thymic and peripheral Tfn and Tr cells supported the model that Foxp3 promotes its own transcription (Fig. 2e). Although we cannot rule out the possibility that the premature stop codons destabilize Foxp3gfpko mRNA, we have found that conditional deletion of Foxp3 exons 1–5 in mature Tr cells also results in an identical reduction of Foxp3 mRNA levels11. Together, these findings demonstrate that stability of Foxp3 expression and suppressor activity is dependent upon Foxp3 function rather than coinciding with its expression.
To characterize the Foxp3-dependent transcriptional programme, global gene expression profiling was performed. Automated gene clustering based on similarity in fold-change values between Tn cells and CD25+Foxp3-CD4+ T cells, Tfn cells or Tr cells revealed that a distinct set of genes with moderate fold-change values in Tfn cells was further amplified in Tr cells. These genes encoded many of the cell surface markers currently used to identify Tr cells, such as CD25, GITR and CTLA4. To incorporate this expression pattern, gene clusters were then hand-curated to generate eight clusters with ‘Foxp3-amplified’ genes residing in clusters T4 and P4 (Fig. 3a, b; see also Supplementary Fig. 5). For both thymocytes and peripheral cells, Foxp3-dependent genes constituted the largest cluster, reflecting a dominant role for Foxp3 in broadly effecting cellular physiology and phenotype (Fig. 3a, b; clusters T3, P3). Notably, ‘Foxp3-dependent’ (T3/P3) and ‘Foxp3-amplified’ genes (T4/P4) most faithfully retained the same expression pattern in thymic and peripheral cells, with Foxp3-amplified genes containing the highest proportion of shared genes (Supplementary Fig. 6a). These findings corroborate our functional studies in elucidating a novel facet of Tr cell differentiation whereby transcriptional and functional characteristics induced concurrently or before Foxp3 transcription are enforced by Foxp3 to become principal features of Tr cell biology.

a, b, Genes differentially expressed in CD25+Foxp3-CD4+ (T25), Tr and Tfn cells relative to Tn cells were identified for both thymic (a) and peripheral (b) T-cell subsets. For each cluster, values and line thicknesses represent the number of upregulated or downregulated genes, and error bars represent the full range of log2 expression ratios. c, Expression patterns of genes that may have a role, or have been demonstrated to have a role, in Tr cell effector function. Foxp3-mediated repression of genes encoding pro-inflammatory cytokines and chemokines is also shown.
To ascribe functional meaning to the defined gene clusters, comparisons with gene annotation databases were performed (Supplementary Fig. 6b, c), revealing that the Foxp3-dependent genes shared between thymic and peripheral data sets were significantly enriched for genes encoding cell surface and extracellular proteins. In contrast, the most enriched functional category for peripheral Tr cells was cell-cycle-associated genes (P6), demonstrating that Foxp3 rescues the inferior proliferative activity of Tfn cells in agreement with the analyses of cell size and Ki67 expression (Fig. 1d, e). Also for peripheral cells, the gene cluster specifically regulated by Foxp3 (P3) was enriched for genes involved in intercellular communication. Together, these data suggest that a second major role for Foxp3 is to adapt developing Tr cells by altering how environmental cues are integrated into cellular processes.
Because our experiments showed that suppressor function was Foxp3-dependent, we examined clusters T3 and P3 for genes defining potential Tr effector mechanisms (Fig. 3c). This analysis suggested several such mechanisms, including suppressive soluble factors12,13,14, generation of extracellular adenosine15,16,17,18, and release of reactive oxygen19, and a possible, previously unanticipated role for Tr cells in regulating wound repair.
Among Foxp3-dependent genes, Pde3b (cyclic nucleotide phosphodiesterase 3B, cGMP-inhibited) was the most repressed (Fig. 4a, b). As cyclic AMP–protein kinase A pathways are well characterized attenuators of T-cell function20, reduced PDE3B levels implied cAMP-mediated adaptation in Tr cells to chronic TCR and IL-2 signalling. Notably, naive T cells fail to downregulate PDE3B upon TCR engagement (Fig. 4b), and we found that Foxp3 binds a highly conserved region in the first intron of Pde3b (ref. 21). Thus, reduced PDE3B expression represents the first unique marker of Tr cells and may be considered more definitive then Foxp3 itself as it reports Foxp3 function. Hypothesizing that increased expression of PDE3B in Tr cells would be deleterious to Tr cell homeostasis, we introduced PDE3B or a catalytically inactive mutant, PDE3B(H801A)22, into Tr cells by retroviral gene transfer (Supplementary Fig. 8a, f, g). After transfer into T-cell-deficient recipients, PDE3B-expressing Tr cells were reduced in number by ∼60% in comparison to empty vector controls, whereas ectopic PDE3B expression in Foxp3-CD4+ T cells (Th) had no effect on their homeostasis (Fig. 4c). Tr cells expressing PDE3B(H801A) also exhibited reduced numbers, although to a lesser degree. Loss of PDE3B-transduced Tr cells was due to cell death or inefficient expansion rather than loss of Tr phenotype, because enumeration of expanded donor Tr and Th cells based on differing Ly5 congenic markers rather than Tr phenotype revealed the same reduction of Tr cell numbers (Fig. 4c).

a, b, Pde3b gene expression was analysed by real-time polymerase chain reaction (a) and by flow cytometry (b). Error bars represent mean and maximum/minimum for biological duplicates. Expression in GFP–CD4+ and CD8+ gated cells after 3 days stimulation of total Foxp3gfp/gfp splenocytes with ConA is also shown (dashed lines). T25, CD25+Foxp3-CD4+ T cells. c, Frequency in recipient mice of transduced (GFP+) Tr cells (Ly5.1-CD4+GITR+) or Tn cells (Ly5.1+CD4+GITR-), as well as the frequency of transduced Ly5.1-CD4+ cells (see Supplementary Fig. 8a). Results are representative of five separate experiments. d, The effect of ectopic PDE3B (PDE3B/Vector) on expression of the listed cell-cycle-associated genes in cluster P6 was added to Tr/Tn values for the same genes (Fig. 3b) and plotted as described (Supplementary Fig. 8c). The effect of PDE3B(H801A) and PDE3B on the same genes is also shown. e, f, Proliferative responses of Pde3b+/+ or Pde3b-/- CD4+ T cells to anti-CD3 in the presence of irradiated APC (e) and to allogeneic (BALB/c) stimulator cells (f). Results are representative of two separate experiments; data represent mean and standard deviation for triplicate wells. g, h, Pre-activated CD4+CD25- T cells were cultured with IL-2 alone or with the PDE3 inhibitor cilostamide or the PDE4 inhibitor rolipram as described in Supplementary Methods. Viable cell counts (g) and percentage live cells (h) are shown. Results are representative of three separate experiments.
Transcriptional profiling of recovered cells suggested mechanistic underpinnings for reduced homeostatic fitness of PDE3B-transduced Tr cells, including reduced expression of genes encoding mitochondrial and biosynthetic proteins (Supplementary Fig. 8b). Remarkably, cell cycle genes shared by CD25+Foxp3-CD4+ T cells and Tr cells (cluster P6) reverted to Tfn-like expression levels in PDE3B-transduced Tr cells in a manner that was largely dependent on PDE3B catalytic activity (Fig. 4d). This reversion to Tfn-like gene expression was also observed for many other Foxp3-dependent genes (Supplementary Fig. 8c). In apparent contrast to the deleterious effect of ectopic PDE3B on Tr cell homeostasis, we found no difference in the proliferative responses of PDE3B-deficient and -sufficient CD4+ T cells23, and proliferation of wild-type T cells was not altered by PDE3 inhibitors (Fig. 4e, f and data not shown). However, we observed a substantial increase in viability and numbers of pre-activated Tn cells after IL-2-driven expansion in the presence of a PDE3, but not PDE4, inhibitor, suggesting that apoptosis associated with chronic IL-2-induced proliferation may be facilitated by PDE3B activity (Fig. 4g, h). Thus, we have identified Pde3b repression as a central component of Foxp3-dependent Tr cell maintenance, as its re-expression resulted in a reduction of biosynthetic processes, the attenuation of Foxp3-dependent proliferative fitness and the loss of some Foxp3-dependent gene expression.
Our study suggests that rather than initiating a de novo developmental programme in self-reactive T cells, Foxp3 takes advantage of preceding and coincidental features of Tr precursor cells probably facilitated by TCR signalling. This is accomplished through consolidation of the state of anergy and paracrine IL-2 dependence. Furthermore, Foxp3 amplifies and stabilizes expression of genes encoding cell surface or secreted molecules—like Fgl2, CD73, CD39, TRAIL or CTLA4—normally elaborated by conventional T cells upon TCR stimulation and capable of negative feedback regulation of T-cell activation ‘in trans’. At the same time, Foxp3 enforces repression of TCR-activation-dependent immune response effector cytokines including IL-4, interferon-γ, tumour-necrosis factor-α, IL-17 and IL-21. These observations might explain failure to identify a single non-redundant mechanism of suppression mediated by Tr cells. Finally, Foxp3 alters how Tr cells respond to environmental cues by modulating cell surface and signalling molecules to promote both Tr homeostasis and lineage stability. Essential adaptation to these signals results from Foxp3-dependent downregulation of PDE3B to support normal homeostasis and metabolic function and maintain a part of the Tr cell transcriptional programme. Our results suggest that, in resemblance of evolutionary processes, a lineage commitment factor may act in both an opportunistic and adaptive fashion during cellular differentiation by amplifying beneficial and correcting disabling features of precursor cells.
Methods
Mice
B6.SCID, TCRα-/- and TCRβδ-/- mice were obtained from The Jackson Laboratory, and C57BL/6 mice were from the Charles River Breeding Laboratories. All mice were maintained at the University of Washington specific pathogen-free facility. Generation of Foxp3gfpko is described in Supplementary Methods.
Flow cytometry and cell sorting
Staining and sorting of cells and intracellular staining for Ki67 and PDE3B are described in Supplementary Methods.
In vitro assays
T-cell stimulation, suppression and cytokine release assays were performed according to conventional methods as described in Supplementary Methods. To examine cell survival in the presence of PDE inhibitors, CD25-CD4+ T cells were stimulated with plate-bound anti-CD3 and anti-CD28 antibodies. On day 2 cells were removed from antibody-coated plates and on day 4 cells were washed and plated at 2 × 105 cells ml-1 with IL-2 (100 U ml-1) or IL-2 plus cilostamide or rolipram (each at 5 mM). On day 7 cells were split 1:4 into the same conditions and on day 13 cells were counted and analysed by flow cytometry.
Gene expression analysis
Gene expression was determined with Affymetrix mouse 430 2.0 microarrays and clusters were generated by Pearson squared K-means clustering with MultiExperiment Viewer (TIGR) and by hand, as described (Supplementary Methods).
PDE3B expression
Mixtures of transduced and non-transduced CD25+CD4+ cells from B6 mice and CD25-CD4+ cells from B6.SJL mice (Ly5.1+) were transferred into TCRα-/- recipients. Three weeks after transfer cells were analysed by flow cytometry and isolated by fluorescence-activated cell sorting (FACS) for gene expression analysis (Supplementary Methods and Supplementary Fig. 8).
References
Fontenot, J. D. & Rudensky, A. Y. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nature Immunol. 6, 331–337 (2005)
van Santen, H. M., Benoist, C. & Mathis, D. Number of T reg cells that differentiate does not increase upon encounter of agonist ligand on thymic epithelial cells. J. Exp. Med. 200, 1221–1230 (2004)
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003)
Khattri, R., Cox, T., Yasayko, S. A. & Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nature Immunol. 4, 337–342 (2003)
Wildin, R. S. & Freitas, A. IPEX and FOXP3: clinical and research perspectives. J. Autoimmun. 25, (suppl.)56–62 (2005)
Fontenot, J. D., Gavin, M. A. & Rudensky, A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunol. 4, 330–336 (2003)
Hsieh, C. S., Zheng, Y., Liang, Y., Fontenot, J. D. & Rudensky, A. Y. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nature Immunol. 7, 401–410 (2006)
Fontenot, J. D. et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341 (2005)
Roncarolo, M. G. et al. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 212, 28–50 (2006)
Ivanov, I. I. et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17(+) T helper cells. Cell 126, 1121–1133 (2006)
Williams, L. M. & Rudensky, A. Y. Maintenance of the Foxp3 dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nature Immunol. (in the press).
Chan, C. W. et al. Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. J. Immunol. 170, 4036–4044 (2003)
Stumhofer, J. S. et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nature Immunol. 7, 937–945 (2006)
Batten, M. et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nature Immunol. 7, 929–936 (2006)
Yegutkin, G. G., Henttinen, T., Samburski, S. S., Spychala, J. & Jalkanen, S. The evidence for two opposite, ATP-generating and ATP-consuming, extracellular pathways on endothelial and lymphoid cells. Biochem. J. 367, 121–128 (2002)
Yegutkin, G. G., Samburski, S. S., Jalkanen, S. & Novak, I. ATP-consuming and ATP-generating enzymes secreted by pancreas. J. Biol. Chem. 281, 29441–29447 (2006)
Ohta, A. & Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920 (2001)
Erdmann, A. A. et al. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood 105, 4707–4714 (2005)
Sheppard, F. R. et al. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 78, 1025–1042 (2005)
Torgersen, K. M., Vang, T., Abrahamsen, H., Yaqub, S. & Tasken, K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell. Signal. 14, 1–9 (2002)
Zheng, Y. et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature doi: 10.1038/nature05563 (in the press)
Zhang, W. & Colman, R. W. Conserved amino acids in metal-binding motifs of PDE3A are involved in substrate and inhibitor binding. Blood 95, 3380–3386 (2000)
Choi, Y. H. et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J. Clin. Invest. 116, 3240–3251 (2006)
Acknowledgements
We are grateful to all members of the Rudensky laboratory for discussions. We also thank K. Forbush, L. Karpik and T. Chu for help. This work was supported by grants from the Arthritis Foundation and the Leukemia and Lymphoma Society (M.A.G.) and grants from the National Institutes of Health (A.Y.R.). A.Y.R. is a Howard Hughes Medical Institute Investigator.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Figures 1-8 with legends and additional references. (PDF 587 kb)
Rights and permissions
About this article
Cite this article
Gavin, M., Rasmussen, J., Fontenot, J. et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775 (2007). https://doi.org/10.1038/nature05543
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature05543
This article is cited by
-
A promising target for breast cancer: B7-H3
BMC Cancer (2024)
-
The regulation and differentiation of regulatory T cells and their dysfunction in autoimmune diseases
Nature Reviews Immunology (2024)
-
Anti-CTLA-4 nanobody as a promising approach in cancer immunotherapy
Cell Death & Disease (2024)
-
Genetic engineering of regulatory T cells for treatment of autoimmune disorders including type 1 diabetes
Diabetologia (2024)
-
Regulatory T cells in the face of the intestinal microbiota
Nature Reviews Immunology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
