
Abstract
Few data are available concerning the role of risk markers for Alzheimer’s disease (AD) in progression to AD dementia among subjects with mild cognitive impairment (MCI). We therefore investigated the role of well-known AD-associated single-nucleotide polymorphism (SNP) in the progression from MCI to AD dementia. Four independent MCI data sets were included in the analysis: (a) the German study on Aging, Cognition and Dementia in primary care patients (n=853); (b) the German Dementia Competence Network (n=812); (c) the Fundació ACE from Barcelona, Spain (n=1245); and (d) the MCI data set of the Amsterdam Dementia Cohort (n=306). The effects of single markers and combined polygenic scores were measured using Cox proportional hazards models and meta-analyses. The clusterin (CLU) locus was an independent genetic risk factor for MCI to AD progression (CLU rs9331888: hazard ratio (HR)=1.187 (1.054–1.32); P=0.0035). A polygenic score (PGS1) comprising nine established genome-wide AD risk loci predicted a small effect on the risk of MCI to AD progression in APOE-ɛ4 (apolipoprotein E-ɛ4) carriers (HR=1.746 (1.029–2.965); P=0.038). The novel AD loci reported by the International Genomics of Alzheimer's Project were not implicated in MCI to AD dementia progression. SNP-based polygenic risk scores comprising currently available AD genetic markers did not predict MCI to AD progression. We conclude that SNPs in CLU are potential markers for MCI to AD progression.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most common form of neurodegenerative dementia, representing 50–60% of all dementia cases. AD pathology commences years, or even decades, before the appearance of clinical symptoms, and current consensus among scientists is that prevention should be started at an early phase in individuals at increased risk. Patients with mild cognitive impairment (MCI) are at increased risk of developing AD dementia. However, the MCI group is heterogeneous, and wide variation in the annual progression to AD dementia rate has been reported, with estimates ranging from 4 to 31%. In a recent study, which involved the follow-up of 550 MCI subjects for an average of 26.6 months,1 the present authors found that the majority (45.5%) of those MCI individuals who subsequently developed dementia displayed the AD dementia phenotype. Thus, predicting which MCI cases will actually progress to AD dementia is an important challenge. Several clinical measures and biomarkers have been proposed for this purpose, including neuroimaging, cerebrospinal levels of amyloid-β and phosphorylated and total tau. However, the predictive value of these biomarkers is low.2, 3 Accordingly, research conducted in recent decades has tended to focus on identifying factors that render MCI patients more susceptible to AD dementia.4 This research is important as the early detection of AD will be essential once an efficacious method of preventing or delaying the disease becomes available.
Individual risk for AD is determined by genetic, environmental and demographic factors, as well as interactions between them. The estimated genetic component of AD, that is, the so-called heritability, is as high as 79%. Hence in AD, the majority of pathophysiological pathways are likely to be driven by, or include, genetic determinants. Recent genome-wide association studies (GWAS) and whole-exam sequencing approaches have indeed identified several common and rare low-penetrance risk variants.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16
Within routine clinical practice, the implementation and evaluation of AD risk markers in the prediction of MCI to AD dementia progression is in its inception. To date, the APOE (apolipoprotein E) locus is the only marker to have shown a consistent association with MCI to AD progression.17 For other reported AD genetic markers, studies of MCI to AD dementia progression using single-nucleotide polymorphisms (SNPs), or combinations of SNPs in polygenic scores (PGS), have generated conflicting results.18, 19
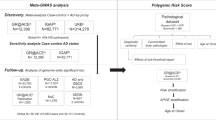
The aim of the present study was to investigate the role of established AD genetic markers in the progression of MCI to AD using follow-up data from four independent MCI data sets (n=3216 subjects).
Materials and methods
Patients
The present cohort comprised MCI patients from Germany, Spain and the Netherlands. These individuals were drawn from the following cohorts: (a) the German study on Aging, Cognition and Dementia in primary care patients (AgeCoDe; n=853);20 (b) the German Dementia Competence Network (DCN; n=812);21 (c) the Fundació ACE from Barcelona (ACE, n=1245);1 and (d) the MCI data sets of the Amsterdam Dementia Cohort (ADC, n=306).22 Effective sample size varied depending on phenotype analyses and covariation matrices (Table 1). Clinical characteristics, neuropsychological assessment, behavioral and functional scales, and progression to AD dementia rates for each MCI data set are shown in Table 1, Supplementary Table 1 and at http://detritus.fundacioace.com/pgs. The study was approved by the respective ethics committees, and all participants provided written informed consent before inclusion.
DNA extraction, SNP selection and genotyping
DNA from 3216 MCI samples was extracted using commercial methods. SNP selection was based on a review of the literature. Here, only those SNPs in loci identified by GWAS or meta-GWAS efforts were selected. To avoid missing loci, for all of the loci selected for PGS construction, whenever possible, alternative SNPs in linkage disequilibrium were also selected (i.e. linkage disequilibrium proxies). This additional SNP thus served as a backup in the event that the primary selected SNP failed in the sequenom assay. Further details on the references used to select SNPs, the genotyping procedures and genotyping quality control are provided in Supplementary Table 2 and in the Genotyping procedures section of the Supplementary Data file. The sequenom technology genotyping methods are described elsewhere.16
Statistical analysis
To investigate the influence of genetic markers, demographic factors and PGS on MCI to AD dementia progression, methods from survival analysis were used. For the 40 individual SNPs and the three PGS of interest, hazard ratios (HRs) were calculated using the following three models: (i) crude (model 0); (ii) age- and gender-adjusted (model 1); and (iii) age-, gender-, APOE- and education-adjusted (model 2) (for details see Statistical Analysis in the Supplementary Data file). Unless otherwise specified, the subsequent text refers to model 1 only.
PGS construction and evaluation
PGS were calculated in accordance with Purcell et al.23 (for details, see Polygenic Score Construction and Polygenic Score Evaluation in the Supplementary Data file). PGS were constructed using sets of AD-associated loci identified in recent GWAS. Inclusion of SNPs in the PGS was based on definitive evidence of association in large meta-GWAS reported by the International Genomics of Alzheimer’s project (IGAP).15 Since the established association between APOE ɛ4 and AD is also present in our four cohorts, the APOE region was excluded from the PGS calculation. PGS1 comprised the nine established AD-associated SNPs reported before publication of the IGAP consortium results (see Supplementary Table 3 and Part A in the Supplementary Data file). PGS2 comprised 9 of the 11 novel AD-associated SNPs identified by IGAP (Supplementary Table 3 and Part B in the Supplementary Data file).15 PGS3 comprised all SNPs from PGS1 and 2. Each of the three calculated PGS was used as a dose, and the proportional hazards model was employed using the three models applied for the analysis of single SNPs. Meta-analysis techniques were used to estimate the global effects of SNPs and PGS. The meta-analysis was conducted using the standard fixed effect approach implemented in the YAMAS software. YAMAS implements standard fixed and random-effects meta-analysis, and operates on beta and standard error.24
Results
Univariate analyses
The demographic characteristics of the cohorts are summarized in Table 1. The results obtained for each analyzed SNP are shown in Table 2.
In the meta-analysis, the APOE-ɛ4 allele (rs429358 C allele) showed an association with the rate of MCI to AD dementia progression in all cohorts, with a homogeneous effect being observed across data sets (HR=1.84 (1.64–2.04), heterogeneity index (I2)=0, P=1.35 × 10−27) (Figures 1a and 2a). Interestingly, the relative risk was ~50% of that reported in GWAS.6, 7, 8, 9 Furthermore, the ɛ4 effect increased with age, reaching its most pronounced effect between 65 and 80 years. In contrast, the APOE-ɛ2 allele conferred a protective effect against MCI to AD dementia progression to other APOE genotypes (Figure 1a). As with ɛ4, the effect of APOE-ɛ2 was dose dependent and homogeneous across data sets. The meta-analysis confirmed the protective effect of APOE-ɛ2 (HR=0.69 (0.51–0.86), I2=0, P=0.004; Table 2). Six MCI subjects carrying the APOE-ɛ2 allele in a homozygous state did not progress to AD dementia during the observational time period.

Cox proportional hazard model multivariate dementia-free survival analyses for APOE (apolipoprotein E) genotypic score (a) and clusterin (CLU) rs111360000 (b). Hazard ratio meta-analyses were adjusted according to data set, age and gender.

Effect size of the APOE (apolipoprotein E) (a) and clusterin (CLU) (b) loci in mild cognitive impairment (MCI) to Alzheimer’s disease (AD) dementia progression following stratification for age. Notes: Meta-analysis of hazard ratio (HR) for progression to AD dementia in APOE-ε4 carriers following stratification for age. The progression rate for each age stratum is shown in the secondary Y2 axis.
An additional association signal was observed in SNPs at the CLU locus (rs9331888, rs11136000). For these variants, a nominally significant result was obtained in the AgeCoDe cohort, and a consistent trend towards association was observed in the DCN, ACE and ADC cohorts (Table 2). The meta-analysis yielded a significant association for both CLU SNPs (P=0.003 and 0.01, respectively). Although rs11136000 showed a heterogeneous HR across the series, the HR for rs9331888 was homogeneous across the four cohorts (Table 2). Association findings for the CLU SNPs withstood all adjustments (Table 2, Figure 1b and Supplementary Data files). No major difference in the effect sizes of the CLU SNPs was observed following stratification for APOE status, gender or age (Table 4 and Figure 2b; P>0.71). Stratification for these variables confirmed the orthogonality of CLU markers with key covariates.
Of the remaining SNPs genotyped in the present study, only rs641120 (located at the SORL1 locus) showed nominal significance with MCI to AD dementia progression (HR=0.89, P=0.043, model 0). However, this finding did not withstand adjustment.
PGS in MCI to AD dementia progression
The results of the hazard models analysis of PGS are shown in Table 3. In the meta-analysis of PGS1, a trend towards association was observed (HR=1.31, P=0.1). Interestingly, stratification according to APOE genotype revealed a consistently higher effect size for PGS1 in APOE-ɛ4 carriers (Table 4). The meta-analysis of PGS1 showed that the effect in APOE-?4 carriers was nominally significant (HR=1.74 (1.03–2.97), P=0.04). However, combined analysis revealed no statistically significant interaction between PGS1 and the APOE locus in any of the four data sets (P=0.14). In contrast, PGS2 did not contribute to MCI to AD dementia progression. The effect size for PGS2 observed in the meta-analyses indicated a nonsignificant protective effect. This suggests that the accumulation of risk alleles was implicated in protection from MCI to AD dementia progression in the present series.
The analysis of PGS3 yielded an intermediate and nonsignificant result (HR=1.03, P=0.96). The PGS3 results reflect the findings of PGS1, as biased by the noise from PGS2. No significant interaction was found between PGS3 and age, gender, APOE-ɛ4 status or cohort (Tables 3 and 4).
Discussion
For several years, intensive research has attempted to identify the role of genetic factors in the progression of MCI to AD dementia. To date, however, only the APOE locus has shown a consistent association. Elias-Sonnenschein et al.17 performed a meta-analysis of 35 prospective MCI studies, which comprised a total of 6095 subjects. Of these, 1236 individuals progressed to AD dementia within a 2.9-year period of follow-up. For MCI subjects carrying the APOE-ɛ4 allele, the authors reported an odds ratio of 2.29 (1.88–2.80) for progression to AD dementia. The present findings support the hypothesis that the APOE-ɛ4 allele is implicated in MCI to AD dementia progression (HR=2.20 (1.88–2.53) for subjects carrying APOE-ɛ4 allele). However, we cannot exclude the possibility that additional loci around APOE may also modulate the age at onset for AD, as has been suggested for TOMM40, a gene adjacent to APOE.25, 26 Detailed mapping data of the linkage disequilibrium region around APOE are now available. These have identified a poly-T length polymorphism in an intron of TOMM40. Interestingly, research has demonstrated that the allele distribution of the poly-T polymorphism explains a larger proportion of the observed survival curves of age at onset in AD than is the case for APOE-ɛ4 containing haplotypes alone.25, 26 To confirm the role of TOMM40 poly-T in AD progression, genotyping of this poly-T is currently being scheduled in our large MCI data set.
In the present study, the MCI to AD dementia progression rate increased continuously with age, whereas the effect of the allele APOE-ɛ4 on AD dementia progression decreased after the age of 80 years (Figure 2a). However, previous research has shown that both the incidence of AD and the AD risk effect of APOE-ɛ4 decrease in the elderly.27, 28 The observation of a reduced association between APOE-ɛ4 and MCI to AD dementia progression is consistent with the survivor effect, as APOE-ɛ4 is a risk factor for both a shorter lifespan and dementia.29 A plausible hypothesis therefore is that most APOE-ɛ4-carrying MCI patients from the present cohorts had converted to dementia or died at an earlier age, thereby causing an enrichment of survivor APOE-ɛ4 MCI carriers among our elderly MCI subjects. This latter group is protected against the progression risk effect conferred by APOE-ɛ4, and this may have led to the observed reduction in the association between APOE-ɛ4 and progression to AD dementia in the present study. This hypothesis is supported by the fact that a reduced APOE-ɛ4 allele frequency was found within this age group compared with younger individuals (Figure 2a).
Besides APOE, no other SNP or PGS combination reached study-wise statistical significance (Bonferroni-corrected P-value=0.00125). However, for some of these markers (i.e. SNPs contributing to PGS1), definitive evidence of association AD has been reported. Hence, the application of Bonferroni correction in this context could be considered overconservative, as our study was based on validated AD susceptibility loci.
The univariate analyses identified a consistent effect on MCI to AD dementia progression for two SNPs (rs11136000 and rs9331888) in the CLU gene (P=0.0035). For both SNPs, a small but consistent effect was observed in all four series, as well as in the meta-analysis. The effect sizes and allele directions of both SNPs are consistent with those reported in previous AD case control GWAS.7 Rodriguez-Rodriguez et al.19 also obtained a significant result for rs11136000 allele T in MCI to AD dementia progression in a small data set. The effect size observed in the Rodriguez-Rodriguez series9 was inflated compared with both the present data and previous results on the role of CLU markers in AD risk.6, 7, 15 Nonetheless, the reported confidence interval overlaps with our evaluation. Therefore, the present CLU results represent an independent replication of a previous report, and have confirmed, in a much larger sample size, the involvement of CLU in MCI to AD dementia progression.
Interestingly, other research has shown that the rate of cognitive decline among individuals who were cognitively normal at study baseline, but who subsequently developed MCI or AD, was significantly faster in those carrying the C allele of rs11136000 compared with non-carriers.30 Furthermore, cognitively normal carriers of the risk allele C of rs11136000 have been reported to show a significant increase in regional cerebral blood flow in brain areas intrinsic to memory processes.30 Overall, the genetic evidence supports the hypothesis that the CLU locus makes an independent contribution to MCI to AD dementia progression.
Along the same lines, the gene product of the CLU gene, clusterin/apolipoprotein J, has been proposed as a potential biomarker for AD. In this regard, the plasma concentration of apolipoprotein J has been associated with the severity and speed of disease progression in AD patients, as well as with atrophy of the entorhinal cortex and the hippocampus in AD.31, 32 In the prodromal stages of AD, for example, MCI elevated plasma levels of apoliprotein J have also been associated with lower rate of brain atrophy.33 This atrophy involved the hippocampus and the entorhinal cortex, that is, brain regions affected in the early stages of AD pathogenesis. Together, these findings suggest that clusterin levels respond in a selective manner along the cascade of events occurring in AD, and that this commences during the prodromal stages. This protective plasma response may modulate, at least in part, the progression of MCI to AD dementia. Our data provide additional support for this hypothesis, as they demonstrate an association between genetic variability in CLU and MCI to AD dementia progression. Although the precise molecular mechanism through which genetic variability in CLU modulates plasma clusterin levels remains unclear, research suggests the potential involvement of genetic variability in CLU in the modulation of gene expression. Hence, CLU appears a promising therapeutic target for AD.
The lack of association for most of the investigated SNPs in the present study may suggest that AD susceptibility loci have only small effects in terms of MCI to AD dementia progression risk. If this is the case, the present MCI data sets would have limited statistical power to detect them. Another power-reducing factor may have been the inclusion in MCI subjects who will never develop AD dementia or who will convert to other unrelated forms of dementia. In support of this, the effect sizes of true AD susceptibility genes in the present MCI series were low compared with conventional AD case–control data sets (OR=3.5 vs HR=2.2 for APOE), and the progression rate for elderly MCI subjects was higher compared with that in the case–control context. An alternative explanation is that the relative risk in GWAS studies was obtained from analysis of progression from healthy control status to AD dementia. In this case, only part of this relative risk was examined in the present study, as our series comprised individuals who were already diagnosed with MCI, many of whom had not yet converted to AD dementia, yet but would do so in the near future.1 Hence, ‘missing’ relative risk in MCI studies may be found when analyzing progression of healthy individuals to MCI or by extending the period of follow-up. Alternatively, the lack of association observed for most SNPs in the present study may suggest that many genuine genetic risk factors for AD exert their pathological effects earlier, that is, during pathological processes that occur during the pre-MCI stages of the disease. This hypothesis would imply that differing genetic factors contribute to AD susceptibility as compared with AD progression. In fact, selecting intermediate phenotypes such as MCI, which are more proximal to a specific event along the causal chain of AD, may capture more variations in the underlying heritable traits and further enhance the statistical power of the study. Interestingly, a number of previous studies selected intermediate AD phenotypes for genetic association analyses, which included neuropsychiatric test measures,34 magnetic resonance imaging data,35, 36 biomarkers from blood and cerebrospinal fluid37, 38 and direct measurements of AD pathology.39 Most of the association signals identified by these studies do not overlap with known genetic susceptibility genes.
On the other hand, genetic studies based on longitudinal samples provide new insight into the pathways related to disease progression. A recent GWAS (based on 18F-florbetapir PET) of time-dependent amyloid accumulation in AD implicated the microglial activation-associated gene, IL1RAP.40 Furthermore, the authors also found that APOE and CLU affect amyloid accumulation, which is consistent with the known effects of these molecules on disease susceptibility. The IL1RAP gene was also associated with a greater likelihood of progression from MCI to AD dementia. Interestingly, the interleukin-1 proinflammatory pathway, to which IL1RAP belongs, is involved in plaque-associated activation of microglia and amyloid burden.41 This inflammatory pathway is shared with CLU because clusterin modulates neuroinflammation by inhibiting the inflammatory response associated with complement activation.42 In the case of APOE, research has linked the gene product, apoE, to innate inflammatory responses induced via the TLR4 and interleukin-4 R-receptor pathways.43 Furthermore, apoE and clusterin cooperate to regulate the clearance and deposition of amyloid-β in the brain.44 Notably, clusterin and apoE promote the clearance of amyloid-β by interacting with several receptors located on microglia cells, including TREM2.42, 45, 46 These findings, together with our own, suggest that immune responses and microglial clearance of amyloid-β have a role in disease progression from MCI to AD dementia. Interestingly, a recent study on AD identified a significant association with signals in SNPs located in genes involved in immune response pathways,47 suggesting a partial overlap between disease progression pathways and those that increase susceptibility to AD.
Previous AD studies have investigated the predictive value of PGS constructed using the effects sizes of multiple SNPs. For example, Verhaaren et al.48 constructed a genetic risk score (GRS) that was similar to the present PGS1. By using this GRS in 5171 non-dementia cases from Rotterdam, the authors demonstrated that although the GRS without APOE was associated with the development of AD (P=0.010), it provided only a marginal improvement in the prediction of AD dementia beyond that provided by age, sex and APOE status (area under the curve: 0.8159 vs 0.8148, respectively). Using a similar strategy, Rodriguez-Rodriguez et al.19 used a GRS based on eight non-APOE genetic AD risk variants to study its effect on MCI to AD dementia progression, and on rapid progression from MCI to AD dementia. Although the authors observed no association between GRS and progression risk, they found that AD converters harboring six or more risk alleles progressed twice as rapidly to AD compared with individuals with less than six risk alleles. Thus, the present findings for PGS1 are consistent with these previous studies, and support the hypothesis that the first identified AD susceptibility locus has only a limited role in MCI to AD dementia progression. Interestingly, whereas PGS1 achieved nominal significance in APOE-?4 carriers in the present study, this was not observed by Rodriguez-Rodriguez et al.19 (Table 4). This may have been due to an enrichment of truly prodromal AD within APOE-?4 carriers, who were therefore likely to progress to AD dementia within our observational time. Nevertheless, this observation with PGS1 suggests that AD susceptibility genes other than APOE also contribute to disease progression. However, the predictive value of the PGS1 composite effect for diagnosis is too small to improve prediction, and this precludes its use in routine clinical practice.
The markers included in PGS1 are the best AD-associated SNP set reported to date, as—with the exception of CD33—all were reconfirmed in the large replication data set included in the IGAP effort. Many of the SNPs discovered by IGAP only reached GWAS significance during the last round of replication. Consequently, many of these SNPs still await an extensive independent replication effort to confirm genuine loci and remove false positives.16 The existence of some false positives among the IGAP results cannot be excluded, and this would affect PGS results.
The present study had several limitations. First, the sample size may have been too small to detect certain associations. Unexpectedly, we observed a worsening of PGS risk prediction following the addition of the novel SNPs identified by the IGAP. In fact, a nonsignificant protective effect was observed for PGS2 in our meta-analyses (P=0.25). A possible explanation for this finding is that novel loci included in PGS2 have even smaller effect sizes than the SNPs included in PGS1, in which case our sample would have been too small to detect association. Alternatively, the effect of the IGAP-SNPs may be restricted to very late or early onset AD, or to an undetermined and very specific subgroup of AD patients that was poorly represented in our MCI data sets. Second, only 9 of 11 novel risk loci found by IGAP were represented in PGS2 and PGS3. Unfortunately, the genotyping method failed for rs9271192 at HLA-DRB5–HLA-DRB1, and for rs10838725 at CELF1, and no additional backup SNPs were available for either locus. Thus, the conclusions drawn for PGS2 and PGS3 should be viewed with caution. Notwithstanding, the small effect sizes of the two markers are unlikely to have made a strong contribution to the overall effect of PGS2 or, more particularly, PGS3. Nevertheless, future efforts are necessary to investigate the potential implications of these missing markers (either by themselves or in combination with other loci) in terms of the progression of MCI to AD dementia.
In summary, the present data support the hypothesis that CLU has an independent role in MCI to AD progression. As in previous studies, the data also confirm the role of APOE in this process. Furthermore, our longitudinal data suggest that the genetic effect of AD risk factors on MCI progression may be age-dependent. Finally, our findings confirm the poor predictive value of the current genome-wide AD risk loci for MCI to AD dementia progression. Further studies in larger longitudinal MCI samples are now warranted to replicate these data, and to disentangle the genetic factors that influence the progression of MCI to AD dementia. Information on loci acting in the prodromal stages of AD, that is, in patients with MCI, will be of relevance for drug target selection in secondary prevention trials.
References
Espinosa A, Alegret M, Valero S, Vinyes-Junqué G, Hernández I, Mauleón A et al. A longitudinal follow-up of 550 mild cognitive impairment patients: evidence for large conversion to dementia rates and detection of major risk factors involved. J Alzheimers Dis 2013; 34: 769–780.
Gainotti G . Origins, controversies and recent developments of the MCI construct. Curr Alzheimer Res 2010; 7: 271–279.
Drago V, Babiloni C, Bartrés-Faz D, Caroli A, Bosch B, Hensch T et al. Disease tracking markers for Alzheimer’s disease at the prodromal (MCI) stage. Adv Alzheimer’s Dis 2011; 26 (Suppl 3):159–199.
Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC et al. The Alzheimer’s disease neuroimaging initiative: a review of papers published since its inception. Alzheimer’s Dement J Alzheimer's Assoc 2013; 9: e111–e194.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 1977–1981.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009; 41: 1088–1093.
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010; 303: 1832–1840.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 2011; 43: 429–435.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011; 43: 436–441.
Antúnez C, Boada M, Gonzalez-Perez A, Gayán J, Ramírez-Lorca R, Marín J et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med 2011; 3: 33.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonson PV, Snaedal J et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013; 368: 107–116.
Guerreiro R, Wotjas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013; 368: 117–127.
Boada M, Antúnez C, Ramírez-Lorca R, DeStefano AL, González-Pérez A, Gayán J et al. ATP5H/KCTD2 locus is associated with Alzheimer's disease risk. Mol Psychiatry 2014; 19: 682–687.
Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron 2013; 78: 256–268.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45: 1452–1458.
Ruiz A, Heilmann S, Becker T, Hernández I, Wagner H, Thelen M et al. Follow-up of loci from the International Genomics of Alzheimer's Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry 2014; 4: e358.
Elias-Sonnenschein LS, Viechtbauer W, Ramakers IH, Verhey FR, Visser PJ . Predictive value of APOE-ɛ4 allele for progression from MCI to AD-type dementia: a meta-analysis. J Neurol Neurosurg Psychiatry 2011; 82: 1149–1156.
Hu X, Pickering E, Liu YC, Hall S, Fournier H, Katz E et al. Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer’s disease. PLoS One 2011; 6: e16616.
Rodríguez-Rodríguez E, Sánchez-Juan P, Vázquez-Higuera JL, Mateo I, Pozueta A, Berciano J et al. Genetic risk score predicting accelerated progression from mild cognitive impairment to Alzheimer's disease. J Neural Transm 2013; 120: 807–812.
Kornhuber J, Schmidtke K, Frölich L, Perneczky R, Wolf S, Hampel H et al. Early and differential diagnosis of dementia and mild cognitive impairment: design and cohort baseline characteristics of the German Dementia Competence Network. Dement Geriatr Cogn Disord 2009; 27: 404–417.
Jessen F, Wiese B, Bickel H, Eiffländer-Gorfer S, Fuchs A, Kaduszkiewicz H et al. Prediction of dementia in primary care patients. PLoS One 2011; 6: e16852.
Van der Flier WM, Pijnenburg YAL, Prins N, Lemstra AW, Bouwman FH, Teunissen CE et al. Optimizing patient care and research: the Amsterdam Dementia Cohort. J Alzheimers Dis 2014; 41: 313–327.
Purcell SM, Wray NR, Stone JL, Visscher PM, O' Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Meesters C, Leber M, Herold C, Angisch M, Mattheisen M, Drichel D et al. Quick, 'Imputation-free' meta-analysis with proxy-SNPs. BMC Bioinform 2012; 13: 231.
Roses AD . An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Arch Neurol 2010; 67: 536–541.
Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenom J 2010; 10: 375–384.
Miech RA, Breitner JCS, Zandi PP, Khachaturian AS, Anthony JC, Mayer L . Incidence of AD may decline in the early 90 s for men, later for women: The Cache County study. Neurology 2002; 58: 209–218.
Valerio D, Raventos H, Schmeidler J, Beeri MS, Villalobos LM, Bolaños-Palmieri P et al. Association of apolipoprotein E-e4 and dementia declines with age. Am J Geriatr Psychiatry 2014; 22: 957–960.
Kulminski AM, Arbeev KG, Culminskaya I, Arbeeva L, Ukraintseva SV, Stallard E et al. Age, gender, and cancer but not neurodegenerative and cardiovascular diseases strongly modulate systemic effect of the apolipoprotein E4 allele on lifespan. PLoS Genet 2014; 10: e1004141.
Thambisetty M, Beason-Held LL, An Y, Kraut M, Nalls M, Hernandez DG et al. Alzheimer risk variant CLU and brain function during aging. Biol Psychiatry 2013; 73: 399–405.
Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 2010; 67: 739–748.
Hardy J, Guerreiro R, Lovestone S . Clusterin as an Alzheimer biomarker. Arch Neurol 2011; 68: 1459–1460.
Thambisetty M, An Y, Kinsey A, Koka D, Saleem M, Guntert A et al. Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. NeuroImage 2012; 59: 212–217.
McQueen MB, Bertram L, Lange C, Becker KD, Albert MS, Tanzi RE et al. Exploring candidate gene associations with neuropsychological performance. Am J Med Genet Part B 2007; 144B: 987–991.
Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s Disease. PLoS One 2009; 4: e6501.
Seshadri S, DeStefano AL, Au R, Massaro JM, Beiser AS, Kelly-Hayes M et al. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham Study. BMC Med Genet 2007; 8: S15.
Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, Nicosia F et al. Increased brain beta-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol 2003; 60: 29–35.
Peskind ER, Li G, Shofer J, Quinn JF, Kaye JA, Clark CM et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol 2006; 63: 936–939.
Bennett DA, De Jager PL, Leurgans SE, Schneider JA . Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology 2009; 72: 1495–1503.
Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI). GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer's disease implicates microglial activation gene IL1RAP. Brain 2015; 138: 3076–3088.
Prinz M, Priller J, Sisodia SS, Ransohoff RM . Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 2011; 14: 1227–1235.
Nuutinen T, Suuronen T, Kauppinen A, Salminen A . Clusterin: a forgotten player in Alzheimer's disease. Brain Res Rev 2009; 61: 89–104.
Tai LM, Ghura S, Koster KP, Liakaite V, Maienschein-Cline M, Kanabar P et al. APOE-modulated Aβ-induced neuroinflammation in Alzheimer's disease: current landscape, novel data, and future perspective. J Neurochem 2015; 133: 465–488.
DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 2004; 41: 193–202.
Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 2015; 290: 26043–26050.
Bailey CC, DeVaux LB, Farzan M . The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem 2015; 290: 26033–26042.
Jones L, Lambert JC, Wang LS, Choi SH, Harold D, Vedernikov A et al. Convergent genetic and expression data implicate immunity in Alzheimer's disease. International Genomics of Alzheimer's Disease Consortium (IGAP). Alzheimers Dement 2015; 11: 658–671.
Verhaaren BF, Vernooij MW, Koudstaal PJ, Uitterlinden AG, van Duijn CM, Hofman A et al. Alzheimer's disease genes and cognition in the nondemented general population. Biol Psychiatry 2013; 73: 429–434.
Acknowledgements
We thank all patients for their participation in this project. We are obliged to Trinitat Port-Carbó and her family for their support of the Fundació ACE research programs. Fundació ACE collaborates with the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED, Spain), and is one of the participating centers of the Dementia Genetics Spanish Consortium (DEGESCO). CIBERNED is an Instituto de Salud Carlos III ISCIII Project. AR is supported by Grant PI13/02434 (Acción Estratégica en Salud, Instituto de Salud Carlos III (ISCIII), Ministerio de Economía y Competitividad, Spain) and Obra Social ‘La Caixa’ (Barcelona, Spain). Part of this work will be included in the doctoral thesis of AE from the University of Barcelona. TB and MMN are members of the German Research Council (DFG)-funded Excellence Cluster ImmunoSensation. This publication was funded, in part, by the German Federal Ministry of Education and Research (Grants KND: 01GI0102, 01GI0420, 01GI0422, 01GI0423, 01GI0429, 01GI0431, 01GI0433, 01GI0434; Grants KNDD: 01GI0710, 01GI0711, 01GI0712, 01GI0713, 01GI0714, 01GI0715, 01GI0716, 01ET1006B). Research performed at the VUmc Alzheimer center is part of the neurodegeneration research program of the Neuroscience Campus Amsterdam, the Netherlands. The VUmc Alzheimer Center is supported by Stichting Alzheimer Nederland and Stichting VUmc Fonds. The clinical database structure was developed using funding from Stichting Dioraphte. EL was supported by a research fellowship from Alzheimer Nederland (WE 15-2014-04). We thank Christine Schmäl for her important contribution. This article is dedicated to Prof. Hanns Hippius, MD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Lacour, A., Espinosa, A., Louwersheimer, E. et al. Genome-wide significant risk factors for Alzheimer’s disease: role in progression to dementia due to Alzheimer's disease among subjects with mild cognitive impairment. Mol Psychiatry 22, 153–160 (2017). https://doi.org/10.1038/mp.2016.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.18
This article is cited by
-
Step by step: towards a better understanding of the genetic architecture of Alzheimer’s disease
Molecular Psychiatry (2023)
-
Genome-wide variants and polygenic risk scores for cognitive impairment following blood or marrow transplantation
Bone Marrow Transplantation (2022)
-
Identification of genetic loci shared between Alzheimer’s disease and hypertension
Molecular Genetics and Genomics (2022)
-
New insights into the genetic etiology of Alzheimer’s disease and related dementias
Nature Genetics (2022)
-
Predictability of polygenic risk score for progression to dementia and its interaction with APOE ε4 in mild cognitive impairment
Translational Neurodegeneration (2021)
