
Abstract
Quantitative trait locus (QTL) analysis detects regions of a genome that are linked to a complex trait. Once a QTL is detected, the region is narrowed by positional cloning in the hope of determining the underlying candidate gene—methods used include creating congenic strains, comparative genomics and gene expression analysis. Combined cross analysis may also be used for species such as the mouse, if the QTL is detected in multiple crosses. This process involves the recoding of QTL data on a per-chromosome basis, with the genotype recoded on the basis of high- and low-allele status. The data are then combined and analyzed; a successful analysis results in a narrowed and more significant QTL. Using parallel methods, we show that it is possible to narrow a QTL by combining data from two different species, the rat and the mouse. We combined standardized high-density lipoprotein phenotype values and genotype data for the rat and mouse using information from one rat cross and two mouse crosses. We successfully combined data within homologous regions from rat Chr 6 onto mouse Chr 12, and from rat Chr 10 onto mouse Chr 11. The combinations and analyses resulted in QTL with smaller confidence intervals and increased logarithm of the odds ratio scores. The numbers of candidate genes encompassed by the QTL on mouse Chr 11 and 12 were reduced from 1343 to 761 genes and from 613 to 304 genes, respectively. This is the first time that QTL data from different species were successfully combined; this method promises to be a useful tool for narrowing QTL intervals.
Similar content being viewed by others


Introduction
Although quantitative trait locus (QTL) mapping is a very useful approach for identifying regions on the genome associated with a phenotype of interest, the mapped intervals are often very broad and contain many genes. The number of genes within the QTL must then be narrowed using genetic and bioinformatic approaches (DiPetrillo et al., 2005). These methods yield promising results, substantially reducing the candidate gene number. However, the steps are tedious and time consuming, and the work required is commensurate with the starting number of genes. Thus, any method that narrows the QTL confidence interval, thereby excluding many less likely candidate genes, saves time and money. A recent advance in QTL mapping involves combining QTL cross data; in this process, the data from different crosses are combined when a QTL is identified in the same chromosomal region and it is believed that the same gene underlies the QTL.
In a QTL located using genotype data from F2 mice from a cross between inbred strains, a parental strain is determined to be the ‘high-allele strain’ for that QTL by the effect plot for the peak marker; the homozygous genotype with the higher average phenotype carries the high allele. When genotype data from different crosses are combined, the high alleles are coded identically on a per-chromosome basis and the original cross is used as a covariate in analysis. Combining the data sets adds power to the QTL analysis, and if the underlying gene is the same, the resulting confidence interval is reduced. This method has been used in other species such as the pig (Uleberg et al., 2005), and it complements other recent advancements in QTL mapping, such as the meta-analysis of QTL logarithm of the odds ratio (LOD) values (Wuschke et al., 2007; Schmidt et al., 2008) or the pooling of assigned P-values along an entire QTL on the basis of localized LOD scores (Peirce et al., 2007).
The level of homology between genomes of different species is variable at both the species and genomic level, depending both on the overall relatedness of the two species at hand and on the underlying physiology and genomic architecture that define the species. Closely related species may share a similar genomic structure with contiguous patches of nearly identical sequence and shared clusters of genes. In the last decade, the sequencing of the mouse (Waterston et al., 2002) and the rat (Gibbs et al., 2004) has allowed for a detailed analysis of the genomic divergence of the two species, revealing areas of both high- and low-sequence conservation (Hancock, 2004).
For many phenotypes, QTLs are concordant among different species. High-density lipoprotein (HDL) cholesterol QTLs are homologous between humans and mice (Korstanje and DiPetrillo, 2004; Wang and Paigen, 2005), and kidney disease and hypertension QTLs are homologous among rat, mouse and humans (Herrera et al., 2006; Garrett et al., 2010). This QTL concordance suggests an underlying shared contiguity of genetically mapped loci, which opens up the possibility of expanding the combining of crosses beyond the use of one species. A successful combination is perhaps most likely the use of data from the mouse and the rat, as the two species are closely related, and research involving both in the laboratory uses inbred strains and similar crossing strategies.
We explored the possibility of narrowing HDL cholesterol QTL by combining data from one rat cross (Kovacs et al., 2000; Kloting et al., 2001) and two mouse crosses (Drake et al., 2001; Cervino et al., 2005; Mehrabian et al., 2005; Wittenburg et al., 2005). Our results show that in parallel with the combination of QTL data sets from the same species, it is possible to both increase the statistical significance of a QTL and narrow the confidence interval of the homologous QTL region using combined data from two different species.
Materials and methods
QTL data sets
The WxDA data set
WOKW and DA rats were reciprocally crossed to produce two F2 crosses of 76 (WOKW × DA) F2 and 74 (DA × WOKW) F2 male and 72 (WOKW × DA) F2 and 68 (DA × WOKW) F2 female rats. Animals received a standard chow diet. Blood samples were taken at 28, 30 and 32 weeks, and HDL cholesterol levels were determined using a Roche Cobas Mira Plus auto analyzer (Roche, Basel, Switzerland). Values did not differ between the two crosses, and the crosses were therefore combined. Details of this data set were previously published by Kovacs et al. (2000) and Klöting et al. (2001). In these papers, 126 microsatellite markers were used for genome-wide genotyping; since the publication, 19 additional markers have been genotyped for a total of 145 markers.
The PxD2 data set
PERA/EiJ and DBA/2J mice were reciprocally crossed to produce 324 F2 progeny (166 females and 158 males). All animals were fed a chow diet until 6–8 weeks of age, followed by an 8-week atherogenic diet (Nishina et al., 1990). In all, 97 microsatellite markers were used for genome-wide genotyping. Details of this data set were previously published by Wittenburg et al. (2005).
The BxD2 data set
This data set has been described by Drake et al. (2001), Cervino et al. (2005) and by Mehrabian et al. (2005) and is publicly available at http://www.diabetesgenome.org/thirdpartydata/lusis_060424/. C57BL/6J female mice were crossed with DBA/2J males, and F1 mice were crossed to produce 111 female F2 progeny. The F2 females were fed a chow diet for 12 months and then fed an atherogenic diet for 16 weeks before phenotypic measurements were taken. The mice were genotyped using 139 microsatellite markers.
Placing rat markers on the mouse genome
To determine the version 3.4 (November 2004 update) base-pair positions of the microsatellite markers used in the WxD cross, marker IDs were used as input using the batch version of University of California, Santa Cruz (UCSC) genome browser's Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables?command=start). Markers that were not available through the UCSC genome browser were looked up individually in Ensembl (http://www.ensembl.org); markers without base-pair positions at that point were discarded from the data set. Of the 145 markers, 138 were assigned updated positions. UCSC's web-based version of the batch coordinate conversion tool LiftOver (http://genome.ucsc.edu/cgi-bin/hgLiftOver) was used to convert the rat genome positions to homologous mouse genome positions (National Center for Biotechnology Information build 37). Using this method, all but 10 of the 138 markers were converted; the remaining markers were positioned in Ensembl, and using Ensembl's Comparative Genomics tool, genes adjacent to the rat marker and both homologous and contiguous to the mouse genome were determined. The mouse position of the nearest homologous gene in a contiguous sequence of genes was used as the rat marker's homologous position. For example, using Liftover, base-pair positions for rat marker D10Mgh12 are not homologous to the mouse genome. However, the gene Lcp2 is adjacent to D10Mgh12 at 19 019 978–19 066 754 bp and contiguous to the other homologous mouse positions on Chr 11 at 33 947 144–33 992 281. Using the above methods, all but three rat markers, namely, D2Wox32, D8Mit6 and D10Mgh2, were aligned to the mouse genome.
Assigning genetic map positions to the rat markers
Rat genome genetic positions for the rat markers were determined by interpolation to the single-nucleotide polymorphism map recently published by the STAR Consortium (Saar et al., 2008); the version 3.4 base-pair positions and STAR map cM positions are publicly available online at http://www.well.ox.ac.uk/rat_mapping_resources/SNPmaps.html. Marker base-pair positions were interpolated to the STAR map using MATLAB (Natick, MA, USA). The homologous mouse positions of the rat markers were interpolated to the Revised Shifman genetic map of the mouse (Cox et al., 2009) using the Center for Genome Dynamic's online Mouse Map Converter (http://cgd.jax.org/mousemapconverter/). Mouse chromosome and base-pair positions were used as input, and sex-averaged cM positions were selected as output.
Single data set QTL analysis
Individual QTL analyses were carried out on the WxD rat data set using rat positions, and then on each single mouse data set using mouse positions. QTL analyses were carried out using R version 2.8.1 and R/qtl version 1.11–12 (Broman et al., 2003). X-chromosome genotyping data were omitted. Genome scans were carried out using the expectation-maximization algorithm (Lander and Botstein, 1989) with 2 cM resolution, and significance thresholds were determined by permutation testing (1000 permutations). To determine the sex that contributed more to a QTL, sex versus HDL effect plots were created. Thereafter, the data sets were separated by sex and reanalyzed to determine the adjusted QTL peak and confidence interval positions. For all analyses, 95% confidence intervals were determined by Bayesian analysis using the bayesint function in R/qtl, which calculates an approximate interval (end points around the maximum LOD) for a given chromosome using the genome scan output. Allele effects were determined using the effect plot function in R/qtl using the QTL peak marker or marker nearest to the peak as the reference marker.
Combining the rat and mouse QTL data sets and multispecies QTL analysis
The rat marker names were listed in chromosome and base-pair order in an Excel spreadsheet, along with the rat genetic position, the homologous mouse position and the mouse genetic position. In the spreadsheet, the QTL peaks, confidence intervals and allele assignments for both the rat and mouse individual crosses were shaded. This allowed for the visualization of homologous and contiguous rat and mouse markers within a mouse QTL. Data were chosen for combination if the following criteria were met: (1) a set of markers within a rat HDL QTL significantly overlapped a mouse QTL on the basis of homology, with at least half of the markers from each species-specific QTL overlapping; (2) the markers were contiguous in both genomes; and (3) the peaks of the homologous QTL were close enough to suggest that the QTLs from each species were caused by the same gene. In each case, the homologous peak positions were either adjacent or aligned in the same row in the Excel spreadsheet. In some cases, markers within a rat QTL were not included in the combined-species analysis. For example, a rat QTL marker was excluded if it was within a rat QTL but outside of a homologous mouse QTL.
The original rat HDL cholesterol values were converted to the same unit as was used for mice (mg/dl). Before combining data sets, the HDL phenotype was log transformed and then standardized by a Z-score in R within each cross. Data sets were combined on the basis of mouse chromosome, as described in Wittenburg et al. (2005), by coding high- and low-allele strains the same and then combining the data sets into one file.
The data were combined and analyzed one chromosome at a time. We followed the linear models and analyses described in Li et al. (2005) (see equations 1–5) for all QTL scans. R/qtl version 1.11-12 was used for QTL analyses, and the expectation-maximization algorithm with a resolution of 2 cM was used for the scan and for determining significance thresholds with 1000 permutations. For the combined plots, significance thresholds were based on the additive model. Only one covariate was used in the analysis, representing either cross, species or sex, as there were no nested factors to consider within each covariate.
Determining the number of genes within the QTL confidence interval
The National Center for Biotechnology Information build 37 gene lists for Mus Musculus for Chr 11 and 12 were downloaded by Ensembl's BioMart (http://www.ensembl.org/biomart/martview); gene attributes selected for download were Ensembl Gene ID, Ensembl Transcript ID, Associated Gene Name, Gene Start (bp), Gene End (bp) and Description. The list of genes was downloaded as a CSV file and then opened in MS Excel. The gene list was sorted by Gene Start base-pair position and all gene repeats were removed (reflecting multiple transcripts per gene); such repeats were detected as duplicate Ensembl Gene IDs. To determine the number of genes within a QTL confidence interval, any gene with a starting base-pair position within the confidence interval was counted. To convert from cM to build 37 base-pair positions, we used the Mouse Map Converter available on The Jackson Laboratory's Center for Genome Dynamics Website (http://cgd.jax.org/mousemapconverter). We chose the sex-averaged cM position as input. On comparing the gene lists within the QTL confidence intervals before and after species combination, the confidence interval resulting from analysis with the species-additive covariate model was chosen for the postanalysis interval.
Results
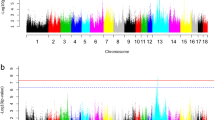
Results of the rat WxDA genome scan are shown in Figure 1. Running the first genome scan with sex as a covariate revealed the presence of multiple sex-specific QTLs on Chr 2, 4, 6, 7 and 9. Plotting genome scans using male and female data separately revealed the sex that contributes to each sex-specific QTL detected in the covariate analysis and also unveiled any sex effects not detected in the first scan. Figures 1c and d show additional sex differences on Chrs 1, 3, 5, 8, 10 through 14, and 17. Owing to the high incidence of sex-specific or sex-influenced QTL, the male and female data from the rat and mouse data sets were separated for individual species analysis; only QTLs from these analyses were considered for multispecies combination. The separation of sexes also facilitates the analysis of the combined data, as the influence of sex, species and cross covariates may be assessed as one covariate. The genome scan plots for the separate mouse crosses are not shown, as they were published previously (Drake et al., 2001; Cervino et al., 2005; Mehrabian et al., 2005; Wittenburg et al., 2005).

QTL scans for the rat WxDA genome scan with (a) sex as an additive covariate, (b) sex as an interactive covariate, (c) males only and (d) females only. The horizontal dashed lines represent suggestive (P=0.63) and significant (P=0.05) levels as determined by 1000 permutation tests.
The markers listed for rat Chr 6 and 10 from the WxDA data set, along with their rat genetic map positions and mouse genetic map positions on Chr 12 and 11, are shown in Figure 2. The following rat–mouse QTL HDL data combinations were tested:
-
1
Rat WxDA males (Chr 6; W, high allele)+mouse BxD2 females (Chr 12; B, high allele)
-
2
Rat WxDA females (Chr 10; W, high allele)+mouse PxD2 females (Chr 11; P, high allele)

Rat–mouse concordance between cholesterol QTLs on two different chromosomes. Genetic maps for rat chromosomes are shown on the left and for mouse chromosomes on the right. Rat marker names are shown in alignment with their genetic positions on the rat genome, and with their homologous positions on the mouse genome. Gray boxes show the original (precombination) QTL confidence interval for each species; the black bar represents the QTL peak. The LOD score for each QTL is shown adjacent to the peak. (a) Rat WxDA Chr 6 male (most left) and female QTLs are aligned with mouse BxD2 Chr 12 QTL. The most proximal rat marker (D6Pas1) is homologous to mouse Chr 17. The remaining markers align to mouse Chr 12 contiguously. The female rat data was not chosen for this combination, as the rat peak positions are not within the rat/mouse overlapping regions. (b) Rat WxDA Chr 10 female QTL is aligned with the mouse PxD2 Chr 11 female QTL. The region immediately surrounding the most distal rat marker, D10Mgh2, is not homologous to the mouse genome; the other markers align to mouse Chr 11 contiguously.
Of these individual QTLs, the PxD2 Chr 11 QTL is the only one that was previously published (Li et al.., 2005, Wittenburg et al.., 2005). Rat Chr 6 and 10 QTLs were suggestive (LOD=2.05 and LOD=2.2, respectively) and not published previously.
Phenotypes were standardized by Z-score in the individual QTL data sets and combined by identically coding the high- and low-allele strains. The results of the QTL analyses for the combinations are shown in Figures 3 and 4. Table 1 lists the peaks, confidence intervals and LOD scores for the original mouse QTL, and for the additive and interactive QTL resulting from the combination of data from different species. The numbers of genes within the original and species-additive confidence intervals are shown in Table 1.

Combining rat Chr 6 data with mouse Chr 12 data. All cM positions are with respect to mouse Chr 12. Confidence intervals for the original and species-additive QTLs are depicted by black boxes; the box closest to the bottom represents the original 95% confidence interval in the mouse. The interactive and additive plots are indistinguishable, suggesting that QTL is caused by the same gene in both the rat and the mouse.

Combining rat Chr 10 data with mouse Chr 11 data. All cM positions are with respect to mouse Chr 11. Confidence intervals for the original and species-additive QTLs are depicted by black boxes; the box closest to the bottom represents the original 95% confidence interval in the mouse. The interactive plot has a higher LOD score than the additive plot, but not by a significant amount (ΔLOD=0.8). Overall, the LOD score is increased and the QTL is substantially narrowed, suggesting that the underlying gene is shared between the rat and the mouse.
Figure 3 shows the result of combining rat Chr 6 data with mouse Chr 12 data. The interactive, additive and noncovariate plots are all identical and overlaid, indicating that the QTL is probably not species specific. The LOD is substantially higher than the original mouse QTL and the confidence interval is narrowed from 44 to 22 cM. The number of genes in the confidence interval is reduced from 613 to 304 genes.
Figure 4 shows that combining rat Chr 10 data with the mouse PxD Chr 11 female QTL data resulted in a narrowing of the confidence interval and an increase in LOD from 2.3 to 3.7 (additive model). The additive and noncovariate plots are identical and overlaid. Although the interaction LOD score is somewhat higher than the additive model, the difference between them is not significant (ΔLOD=0.8). Thus, we conclude that QTL is not species specific. The confidence interval was reduced from 41 to 19 cM and the number of genes in the confidence interval was reduced from 1343 to 761 genes.
Discussion
Previously, genotype data from multiple mouse QTL data sets were successfully combined; if the QTL genes are the same, or close to each other with the same mode of inheritance and the same direction of the allele effect, the combined analysis results in higher LOD scores and narrower QTLs. Using the same methodology, we combined HDL QTL data sets from two different species, the rat and the mouse. Both combinations resulted in a successful increase in statistical significance and a narrowing of the QTL confidence interval. More important than the positional effects, however, this narrowing reduced the number of underlying candidate genes. The mouse QTL on Chr 11 was narrowed from a confidence interval spanning 41 cM to one spanning 17 cM, and the QTL on Chr 12 was reduced from 44 to 22 cM. The numbers of underlying candidate genes were reduced by 43 and 54 %, respectively.
These results are the first report of a successful combination of QTL data from two different species. Although the methodology involved in combining cross data was not novel, the steps required to prepare the data sets for combination were thoroughly researched and tested by us and may now be repeated. It is notable that in clarifying the steps required for aligning rat and mouse QTL at the marker level, several factors normally considered in QTL analysis were omitted for simplicity and for the value of testing by assumption. We designed the data combinations so that sex, diet or any other condition was a cofactor only in addition to species, that is, the data sets may have been from different species and different sexes, but one data set never included both male and female mice. Diet significantly contributes to lipid metabolism, and the rats and mice from the different crosses were not fed identical diets or for the same time time periods before phenotyping (see Materials and methods); however, its effects were not considered in this study. In implementing this procedure for actual positional cloning, a model with multiple cofactors should be used.
If QTLs for the same trait are found in more than one species, it is common when narrowing the QTL to eliminate regions within the QTL that are not homologous to QTL regions for the same trait in the other species. This is especially true when the two species in comparison are extensively studied, as in humans and mice, or when the two species are very closely related, as among rodents. We recommend that in parallel or in conjunction with combining QTL data from multiple crosses within a species, combining QTL data from different species may be used along with all other QTL narrowing techniques, including cross-specific haplotyping and genome-wide association studies based on all strains. As previously suggested (Peirce et al., 2007), combining crosses complements the meta-analysis of QTL significance values.
Although rat and mouse are closely related rodents, this study represents a useful approach for combining data from other species, even from less related species such as mouse and human—if the species are extensively studied and if there is an appropriate arrangement of contiguous loci, as required in the selection of the data sets combined in this analysis. In attempting a mouse–human study, we would need to address the differences in overall metabolism in theoretical speculation and in data analysis. For example, in humans, females on an average have higher HDL cholesterol than do males, but in mice the opposite relationship is true. Such sex or other covariate differences in physiology are usually considered after one-species analyses are complete, when individual candidate genes are investigated. In combining QTL data within a species, we assume that allelic effects are parallel, which allows us to combine the data. A successful combination, that is, the achievement of a higher LOD and narrower confidence interval, provides substantial evidence that the two QTLs share the same underlying gene. We have shown that such success is possible with two closely related species, and because the rat and mouse are both closely related and physiologically similar, it is not a far stretch to draw the same conclusion. If the two species were different regarding HDL metabolism, as in mice and humans, narrowing the QTL by combined data analysis suggests the same underlying gene. However, because the physiological differences are not considered in the combined QTL analysis, any successful combination is based on a multidimensional assumption. The underlying differences in physiology are so complex in their polygenicity and dependent pathways that the data analysis would possibly need to coincide with physiological modeling. It may be possible to quantify metabolic differences between species on the basis of such modeling and then standardize the phenotype before combining QTL data accordingly.
In addition to metabolic differences, genomic function at the level of recombination must be considered. In the analysis presented here, homologous QTL with an underlying contiguous sequence of loci were combined. As differences in recombination rate between the rat and mouse were not taken into account during the analysis, a likely result is the skewing of the genetic map created in the context of the mouse. Recombination rates should be consulted as data from more divergent species are combined.
In summary, sex, diet, species, physiology and genomic differences are all factors to be considered when analyzing combined QTL data sets. In the examples that we present, we use a simplified one-covariate model, encompassing all possible differences between the two data sets. Most important, we have described detailed methods for combining data sets from two different species. The successful combinations we achieved reveal a promising addition to the process of QTL narrowing. Had a combination not been successful, we would not have been able to declare that the individual QTL were species –specific, as they could also be diet, physiology, sex (in one case) or genome structure specific. We recommend using a multicovariate model when analyzing combined data sets for QTL narrowing.
Advances in comparative genomics contribute greatly to the understanding of animal development and physiology. Incorporating such knowledge is essential for the construction of genomes, proteomes and metabolic networks, and for theorizing and elucidating the mechanisms of molecular and phenotypic evolution. The near completion of entire genome homology maps, such as the one available between the rat and the mouse, allows for the ability to combine data in a way that is useful both intellectually and statistically. The methods introduced here recognize the decade-long explosion of empirical advancements and the resulting field of bioinformatics.
Beyond the alignment of homologous markers and genes, the integration of QTL data for analysis adds a new level to species homology, because theories of shared physiology may be tested mathematically. Although statistical significance is both arbitrary and necessary for experimental validity, the increase in statistical significance (LOD) seen in these combinations is overshadowed by the potential insight gained into the underlying shared genome organization. The candidate genes underlie the QTL, but the replication and expression of those genes are dictated by the underlying DNA sequence and chromosome mechanics.
References
Broman KW, Wu H, Sen S, Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890.
Cervino AC, Li G, Edwards S, Zhu J, Laurie C, Tokiwa G et al. (2005). Integrating QTL and high-density SNP analyses in mice to identify Insig2 as a susceptibility gene for plasma cholesterol levels. Genomics 86: 505–517.
Cox A, Ackert-Bicknell CL, Dumont BL, Ding Y, Bell JT, Brockmann GA et al. (2009). A new standard genetic map for the laboratory mouse. Genetics 182: 1335–1344.
DiPetrillo K, Wang X, Stylianou IM, Paigen B (2005). Bioinformatics toolbox for narrowing rodent quantitative trait loci. Trends Genet 21: 683–692.
Drake TA, Schadt E, Hannani K, Kabo JM, Krass K, Colinayo V et al. (2001). Genetic loci determining bone density in mice with diet-induced atherosclerosis. Physiol Genomics 5: 205–215.
Garrett MR, Pezzolesi MG, Korstanje R (2010). Integrating human and rodent data to identify the genetic factors involved in chronic kidney disease. J Am Soc Nephrol 21: 398–405.
Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S et al. (2004). Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521.
Hancock JM (2004). A bigger mouse? The rat genome unveiled. Bioessays 26: 1039–1042.
Herrera VL, Tsikoudakis A, Ponce LR, Matsubara Y, Ruiz-Opazo N (2006). Sex-specific QTLs and interacting loci underlie salt-sensitive hypertension and target organ complications in Dahl S/jrHS hypertensive rats. Physiol Genomics 26: 172–179.
Klöting I, Kovacs P, van den Brandt J (2001). Sex-specific and sex-independent quantitative trait loci for facets of the metabolic syndrome in WOKW rats. Biochem Biophys Res Commun 284: 150–156.
Korstanje R, DiPetrillo K (2004). Unraveling the genetics of chronic kidney disease using animal models. Am J Physiol Renal Physiol 287: F347–F352.
Kovacs P, van den Brandt J, Klöting I (2000). Genetic dissection of the syndrome X in the rat. Biochem Biophys Res Commun 269: 660–665.
Lander ES, Botstein D (1989). Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199.
Li R, Lyons MA, Wittenburg H, Paigen B, Churchill GA (2005). Combining data from multiple inbred line crosses improves the power and resolution of quantitative trait loci mapping. Genetics 169: 1699–1709.
Mehrabian M, Allayee H, Stockton J, Lum PY, Drake TA, Castellani LW et al. (2005). Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet 37: 1224–1233.
Nishina PM, Verstuyft J, Paigen B (1990). Synthetic low and high fat diets for the study of atherosclerosis in the mouse. J Lipid Res 31: 859–869.
Peirce JL, Broman KW, Lu L, Williams RW (2007). A simple method for combining genetic mapping data from multiple crosses and experimental designs. PLoS One 2: e1036.
Saar K, Beck A, Bihoreau MT, Birney E, Brocklebank D, Chen Y et al. (2008). SNP and haplotype mapping for genetic analysis in the rat. Nat Genet 40: 560–566.
Schmidt C, Gonzaludo NP, Strunk S, Dahm S, Schuchhardt J, Kleinjung F et al. (2008). A meta-analysis of QTL for diabetes-related traits in rodents. Physiol Genomics 34: 42–53.
Uleberg E, Wideroe IS, Grindflek E, Szyda J, Lien S, Meuwissen TH (2005). Fine mapping of a QTL for intramuscular fat on porcine chromosome 6 using combined linkage and linkage disequilibrium mapping. J Anim Breed Genet 122: 1–6.
Wang X, Paigen B (2005). Genetics of variation in HDL cholesterol in humans and mice. Circ Res 96: 27–42.
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562.
Wittenburg H, Lyons MA, Li R, Kurtz U, Mossner J, Churchill GA et al. (2005). Association of a lithogenic Abcg5/Abcg8 allele on chromosome 17 (Lith9) with cholesterol gallstone formation in PERA/EiJ mice. Mamm Genome 16: 495–504.
Wuschke S, Dahm S, Schmidt C, Joost HG, Al-Hasani H (2007). A meta-analysis of quantitative trait loci associated with body weight and adiposity in mice. Int J Obes (Lond) 31: 829–841.
Acknowledgements
We thank Sharon Tsaih for comments, Joanne Currer for writing assistance and Jesse Hammer for preparation of figures. This work was funded by HL081162, HL095668 and R37 HL077796 from the National Heart, Lung, and Blood Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Cox, A., Sheehan, S., Klöting, I. et al. Combining QTL data for HDL cholesterol levels from two different species leads to smaller confidence intervals. Heredity 105, 426–432 (2010). https://doi.org/10.1038/hdy.2010.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.75
