
Abstract
Background:
Mipsagargin (G-202; (8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin)-Asp-γ-Glu-γ-Glu-γ-GluGluOH)) is a novel thapsigargin-based targeted prodrug that is activated by PSMA-mediated cleavage of an inert masking peptide. The active moiety is an inhibitor of the sarcoplasmic/endoplasmic reticulum calcium adenosine triphosphatase (SERCA) pump protein that is necessary for cellular viability. We evaluated the safety of mipsagargin in patients with advanced solid tumours and established a recommended phase II dosing (RP2D) regimen.
Methods:
Patients with advanced solid tumours received mipsagargin by intravenous infusion on days 1, 2 and 3 of 28-day cycles and were allowed to continue participation in the absence of disease progression or unacceptable toxicity. The dosing began at 1.2 mg m−2 and was escalated using a modified Fibonacci schema to determine maximally tolerated dose (MTD) with an expansion cohort at the RP2D. Plasma was analysed for mipsagargin pharmacokinetics and response was assessed using RECIST criteria.
Results:
A total of 44 patients were treated at doses ranging from 1.2 to 88 mg m−2, including 28 patients in the dose escalation phase and 16 patients in an expansion cohort. One dose-limiting toxicity (DLT; Grade 3 rash) was observed in the dose escalation portion of the study. At 88 mg m−2, observations of Grade 2 infusion-related reaction (IRR, 2 patients) and Grade 2 creatinine elevation (1 patient) led to declaration of 66.8 mg m−2 as the recommended phase II dose (RP2D). Across the study, the most common treatment-related adverse events (AEs) were fatigue, rash, nausea, pyrexia and IRR. Two patients developed treatment-related Grade 3 acute renal failure that was reversible during the treatment-free portion of the cycle. To help ameliorate the IRR and creatinine elevations, a RP2D of 40 mg m−2 on day 1 and 66.8 mg m−2 on days 2 and 3 with prophylactic premedications and hydration on each day of infusion was established. Clinical response was not observed, but prolonged disease stabilisation was observed in a subset of patients.
Conclusions:
Mipsagargin demonstrated an acceptable tolerability and favourable pharmacokinetic profile in patients with solid tumours.
Similar content being viewed by others


Main
Proper function of the sarcoplasmic/endoplasmic reticulum calcium adenosine triphosphatase (SERCA) pump protein, which transfers Ca2+ from the cytosolic compartment of the cell to the lumen of the sarcoplasmic and endoplasmic reticulum, is necessary for normal cellular viability (Furuya et al, 1994, 1996; Lin et al, 1997; Denmeade et al, 1999; Tombal et al, 2000, 2002; Denmeade and Isaacs, 2005). The natural product thapsigargin (TG) binds tightly to the transmembrane portion of the SERCA pump (Toyoshima et al, 2003; Søhoel et al, 2006; Winther et al, 2010), inhibits its function Rasmussen et al, 1978; Thastrup et al, 1990) and induces cell death in both normal and malignant cell types (Denmeade et al, 2003). Sustained TG-driven inhibition of the SERCA pump depletes endoplasmic reticulum calcium stores, triggering the opening of plasma membrane calcium channels and the resulting rapid elevation in cytoplasmic calcium (Thastrup et al, 1990; Randriamampita and Tsien, 1993; Tombal et al, 1999; Deniaud et al, 2008; Winther et al, 2010). This elevation triggers the endoplasmic reticulum stress/unfolded protein response, caspase activation, release of apoptotic factors from the mitochondria and direct activation of calcium-dependent endonucleases that cleave cellular DNA resulting in cell death (Rao et al, 2002; Breckenridge et al, 2003; Deniaud et al, 2008; Tabas and Ron, 2011). Unfortunately, despite the known cytotoxic effects of TGs, the clinical application of TG has been hindered by its nonspecificity in harming nonproliferating cells.
To direct the cytotoxic activity of TG to tumour tissue and spare normal tissue, we have developed a protease-activated prodrug strategy in which a highly potent primary amine-containing TG analogue is coupled to a protease-specific peptide carrier (Denmeade and Isaacs, 1998; Denmeade et al, 2003) (Figure 1). The TG analogue, 8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin (12ADT), retains the capacity to bind the SERCA pump and maintains TG cytotoxic potency (Jakobsen et al, 2001; Singh et al, 2005). To target 12ADT to the SERCA pump specifically to tumour cells, we focussed on the unique carboxypeptidase activity and restricted expression of the prostate-specific membrane antigen (PSMA; folate hydrolase 1, glutamate carboxypeptidase II), a type II membrane protein first characterised in prostate cancer but subsequently shown to be expressed in the endothelial cells (ECs) of a variety of solid tumours types (Liu et al, 1997; Chang et al, 1999a, 1999b; Haffner et al, 2009; Samplaski et al, 2011). The only normal tissue known to express significant levels of PSMA is prostatic epithelium, with some evidence of expression in duodenal mucosa and a subset of proximal renal tubules (Silver et al, 1997). Our previous analysis of 340 human tumour samples and 32 control tissues from commercially available microarrays showed that nearly 95% of hepatocellular and renal cancers, ∼75% of ovarian and breast cancers, 57% of melanoma samples and 43% of bladder cancer specimens stained positive for PSMA; in contrast, we and others have shown a lack of expression in normal vascular endothelium (Liu et al, 1997; Silver et al, 1997; Chang et al, 1999a, 1999b; Denmeade et al, 2012).

Structure of mipsagargin. Sequential PSMA-mediated hydrolysis of mipsagargin ((8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin)-Asp-γ-Glu-γ-Glu-γ-GluGluOH) yields the cytotoxic thapsigargin analogue 12ADT-Asp.
Mipsagargin (G-202) was developed as a PSMA-targeted TG-based prodrug. Specifically, we coupled a PSMA-specific peptide substrate to 12ADT to produce a PSMA prodrug that upon PSMA-mediated proteolytic cleavage of the peptide selectively kills PSMA-expressing cells in vitro and produces marked regression of a variety of human tumour xenografts in mice (Denmeade et al, 2012). These preclinical studies further reveal that 3 days of treatment was superior to 1 or 2 days of treatment in inducing tumour regression in animal models, but extended treatment periods, including 5- and 10-day treatment regimens, were not more effective. In addition, studies in animals suggested that a 3-day regimen was effective and tolerated at higher doses without evidence of acquired resistance or sensitivity in several animal models of human tumours (Denmeade et al, 2012).
We hypothesised that mipsagargin would selectively target PSMA-expressing tumours and/or tumour neovasculature ECs resulting in clinical efficacy and, given the lack of expression of PSMA in normal cells, have a good safety profile in humans. A phase I trial was initiated to evaluate the safety, tolerability and pharmacokinetic profile of intravenous administration of mipsagargin in patients with locally advanced or metastatic solid tumours, refractory to standard therapy, or for whom no standard therapy was available.
Materials and methods
Study design and dose escalation schema
This was a multicentre, open-label phase I study of escalating doses of mipsagargin administered to cohorts of 3–6 patients with advanced solid tumours at three sites in the United States. Mipsagargin was administered by intravenous infusion over 1 h on three consecutive days (days 1, 2 and 3) of each 28-day cycle. The protocol was reviewed and approved by the Institutional Review Board (IRB) at each institution before initiation and described on the clinicaltrials.gov registry (NCT01056029). In the absence of prohibitive toxicity, patients were allowed to continue treatment until development of progressive disease. The primary objectives of the study were to determine the maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs), establish the recommended phase II dose and determine the pharmacokinetics of G-202 administered under this schedule. Secondary objectives were to investigate the safety profile of G-202 and document any evidence of antitumour activity in patients with advanced solid tumours.
As described in Table 1, the study initially followed a modified Fibonacci dose escalation to minimise the number of patients treated with potentially subtherapeutic doses. At each dose level during stage 1, 3-patient cohorts were treated, with 100% dose escalation between cohorts. Dosing increments were reduced and stage 2 escalation was implemented when a patient experienced two equivalent ⩾Grade 2 drug-related adverse events (AEs) or DLT. Dose escalation was allowed to continue until the MTD was established. The MTD was defined as the highest dose at which no more than one of six patients experienced a DLT. If two or more patients experienced a DLT, a total of six patients were treated at the previous dose level. The following drug-related AEs (National Cancer Institute Common Terminology Criteria for Adverse Events; NCI CTCAE version 4.03) occurring during the first cycle of treatment were prospectively defined as DLTs: Grade 4 neutropenia lasting ⩾5 days, Grade 3 or higher febrile neutropenia, Grade 4 thrombocytopenia or Grade 3 or 4 thrombocytopenia with bleeding, Grade 3 or higher nausea, vomiting or diarrhoea despite optimal management and Grade 3 nonhaematologic toxicities lasting >5 days or any Grade 4 or higher nonhaematologic toxicities. Treatment was held for patients who experienced a DLT, but could be resumed at the prior dose level if the AE recovered to ⩽Grade 2 within 1 week; only one dose reduction was allowed per patient. Dose reduction was also made for patients who had a delay in treatment for >1 week because of lack of recovery of any toxicity. Subsequent retreatment of such patient was allowed at the investigator’s decision and after full consideration of potential benefit and risk. Intrapatient dose escalation was not allowed. Once the MTD was identified, the RP2D regimen was established and explored in an expansion cohort, planned to include up to 18 patients, with the goal of gaining further experience at the RP2D and/or in particular patient or tumour types.
Derivation of starting dose
The starting dose was derived from repeat-dose GLP toxicology studies conducted in Cynomolgus monkeys and Sprague-Dawley rats. In monkeys, intravenous infusions of 1, 5 or 10 mg kg−1 over a 1-h period on 3 consecutive days and repeated following a 4-week treatment-free interval were associated with nephrotoxicity. At the 1 mg kg−1 dose, signs of nephrotoxicity (elevations in blood urea nitrogen, creatinine and albumin) were minimal to mild and showed clear reversibility by the end of the treatment-free period. Based on these observations, the dose of 1.2 mg m−2 given by intravenous infusion on days 1, 2 and 3 of a 28-day cycle was selected as the starting dose for the phase I clinical trial.
Eligibility
All patients were at least 18 years of age, and had histologically confirmed malignancy that was metastatic or unresectable and for which standard curative measures did not exist or were no longer effective; the malignancy was measurable or nonmeasurable according to Response Evaluation Criteria in Solid Tumours (RECIST) 1.1 criteria. Patients had Eastern Cooperative Oncology Group performance status (ECOG PS) of ⩽2 and life expectancy estimated to be at least 3 months. Women of child-bearing potential had documentation of a negative pregnancy test. Mipsagargin treatment did not begin until >4 weeks after the patient’s last therapy or major surgical procedure. Acceptable hepatic, renal, haematologic and coagulation profile status, as well as urinalysis with no evidence of +2 or higher proteinuria, was required of all patients. Patients with prostate cancer were to continue androgen deprivation therapy with LHRH agonists, unless such therapy was discontinued 6 months before first dosing with mipsagargin. Exclusion criteria included known and untreated CNS metastases, a prolonged QTc interval (>450 ms) or any medication known to prolong the QT interval, active uncontrolled infection, history of hepatitis B or C in the absence of hepatocellular carcinoma, requirement for anticoagulants, pre-existing cardiac conditions, pregnancy or lactation or other conditions that would contraindicate investigational drug use in the opinion of the investigator.
Data collection
In the first 28-day cycle, patients underwent weekly physical examinations, along with monitoring of vital signs, ECOG PS, haematology, serum chemistry and urinalysis. In subsequent cycles, safety assessments were performed after week 1 of each cycle and before initiation of each new cycle. Data concerning AEs were collected for up to 30 days after the last study treatment for all patients who received any amount of mipsagargin. Per protocol, any deaths reported within 30 days of discontinuation of study drug treatment were to be reported as AEs, including deaths because of disease progression. For patients with measurable disease, tumour assessment was performed at baseline, after 8 weeks (2 cycles) of treatment and every 8 weeks thereafter using the RECIST (version 1.1) to determine stable disease, progressive disease and best overall response. A complete or partial response required confirmation after 4 weeks. Tumour response was evaluated by computed tomography (CT), spiral CT, magnetic resonance imaging (MRI), bone scans and/or other techniques considered clinically appropriate for the patient’s specific disease type using the RECIST criteria.
Pharmacokinetics
Plasma samples were collected from patients in parts 1 and 2 during the first week of cycle 1 (before cycle 1 day 1 (C1D1) dosing, after beginning C1D1 infusion (30, 60, 75, 90 and 120 min), 2, 4, 8 and 10–12 h after completion of C1D1 infusion, before cycle 1 day 2, before cycle 1 day 3 and at the end of cycle 1 to assess mipsagargin metabolism, inter-to-individual variability and possible correlation between clinical signs and mipsagargin exposure levels. The pharmacokinetic parameters included peak plasma concentration (Cmax), area under the plasma concentration–time curve (AUC), half-life (t1/2), clearance (CL) and volume of distribution in steady state (Vss). These PK parameters were determined in each subject by standard model independent method (WinNonlin Phoenix Version 6.2.1 (WinNonlin, 2011)). Plasma levels of mipsagargin were determined by liquid chromatography.
Statistical considerations
As this was a phase I study with primary objectives of establishing MTD, DLT, recommend phase II dose and evaluating pharmacokinetics, analysis of data was descriptive in nature. Data from all patients who received at least 1 infusion of G-202 were included in the safety analyses and determination of MTD and DLT. Descriptive analysis of antitumour activity, including rate of response (CR or PR) and disease control (CR or PR or SD), was summarised for those patients who received at least one cycle of G-202 and had at least one post-baseline disease assessment. Any patient with objective disease progression before the end of cycle 1 was included in the analysis of antitumour activity. Any evidence of CR required confirmation after a minimum 4-week interval.
Results
Patient population
From January 2010 through May 2013, 44 patients were enrolled and treated in this study. All patients had locally advanced or metastatic solid tumours, including 14 (32%) with colorectal, 9 (20%) with prostate and 6 (14%) with hepatocellular carcinoma (Table 2A). Overall, the median age of the treated patients was 64.5 years (range 48–83). The female/male ratio was 9 : 35 (20%:80%). The majority of patients were white (93%). At baseline, 25% of patients had ECOG PS of 0 and 75% had ECOG PS of 1. All patients had received prior surgery (64%), radiotherapy (52%) and/or systemic therapy (100%).
Dose escalation, MTD and RP2D
A total of 8 mipsagargin dose levels were evaluated in the dose escalation portion of the study (Table 2B). In this portion of the study, the same dose level was administered on days 1, 2 and 3 of each 28-day cycle. A protocol-defined MTD was not reached, but observations of Grade 2 infusion-related reaction (IRR; 2 patients) and Grade 2 creatinine elevation (1 patient) at 88 mg m−2 led to declaration of 66.8 mg m−2 as RP2D. One plausible explanation for the observations of IRR relates to the thapsigargin-induced inhibition of the SERCA pump that elevates cytosolic calcium levels in mast cells, resulting in degranulation and histamine release (Putney and Bird, 1993; Huber et al, 2000). Two protocol modifications were implemented in an effort to reduce the incidence of IRRs: the mipsagargin dose administered on day 1 was reduced to 40 mg m−2, with the RP2D of 66.8 mg m−2 administered on days 2 and 3, and a general premedication regimen with steroid, anti-histamine, anti-nausea, anti-pyretic and H2 blocker in accordance with institutional guidelines was recommended. In an effort to prevent elevations in serum creatinine, guidelines for prophylactic hydration by intravenous infusion of saline on each day of mipsagargin administration were provided. Thus, the RP2D was established as mipsagargin at 40 mg m−2 on day 1, followed by treatment at the RP2D (66.8 mg m−2) on days 2 and 3 of each 28-day cycle with routine premedications and prophylactic hydration on each day of infusion.
Safety
Mipsagargin was generally well tolerated in the population of patients with locally advanced or metastatic solid tumours. The most frequently reported AEs (occurring in >10% of patients) were nausea, fatigue and rash (Table 3A). The most common treatment-related AEs occurring in at least 5 (>10%) patients were fatigue, rash, nausea, IRR and pyrexia (Table 3B).
Overall, 29 (65.9%) patients experienced 83 (18.7%) Grade 3 or 4 AEs. The only AE of Grade ⩾3 occurring in at least 10% of patients was GGT elevation, experienced by 5 (11.4%) patients; however, only 1 occurrence of Grade 3 GGT elevation was judged as related to mipsagargin administration. All other AEs, including those related and unrelated to mipsagargin administration, of Grade ⩾3 occurred in >10% of patients.
All Grade 3 or Grade 4 AEs occurring in at least 3 (6%) patients, including those judged as related to mipsagargin administration and those judged as unrelated, are presented in Table 3C. Grade 3 or Grade 4 AEs judged as related to mipsagargin administration and occurring in at least 3 (6%) patients were thrombocytopenia and AST elevation, noted in Table 3D.
Among the 44 patients treated on the study and across the duration of the study, 17 (38.6%) experienced 34 serious adverse events (SAEs). The most frequent SAE was IRR that occurred in 4 (9.1%) patients and was typically observed on day 1 of the treatment cycle. With prophylactic premedications and a reduced dose level on day 1, as outlined above, only 1 of the 16 patients enrolled in the dose expansion cohort experienced an IRR, and it was Grade 1 in severity.
Several patients experienced renal observations during the study. Three (8.8%) patients experienced an AE with the preferred term of renal injury, including two of Grade 2 in severity that were attributed to study medication and one that was Grade 3 in severity and attributed to the combination of study medication and underlying disease. There were two SAEs with the preferred term of acute renal failure judged by the investigator as possibly or probably related to study drug. In each of these cases, the renal observations were represented as increased serum creatinine levels without other significant renal observations, such as acidosis, loss of urine output or need for haemodialysis. The renal changes generally were transient and reversed before the end of the 28-day treatment cycle; no patient with a renal change required haemodialysis or other long-term care for the AE.
One patient in the 66.8 mg m−2 dose group in part 1 experienced Grade 3 morbilliform rash, Grade 3 thrombocytopenia and Grade 2 renal injury and was discontinued from the study. The Grade 3 rash lasted for >5 days, constituted a DLT and led to a total of 6 patients enrolled in the 66.8 mg m−2 cohort. There were no additional observations of Grade 3 rash or other DLTs. Among the 16 patients in the expansion group treated at the RP2D (40 mg m−2on day 1 and 66.8 mg m−2 on days 2 and 3), 1 patient experienced Grade 3 renal injury that resulted in discontinuation of study drug. No other patients experience SAEs or AEs that resulted in discontinuation of study drug.
Serum chemistry, haematology and urinalysis laboratory values largely showed no notable changes over the study; notable changes in individual patients were reported as AEs. There were no notable changes in vital signs or ECG data over the study. There were no Grade 5 AEs and no deaths because of AEs.
Pharmacokinetics
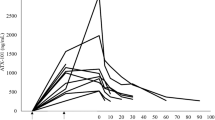
Mipsagargin was measured in blood collected before C1D1 dosing, after beginning C1D1 infusion (30, 60, 75, 90 and 120 min), 2, 4, 8 and 10–12 h after completion of C1D1 infusion, before C1D2 and before C1D3. Preclinical studies in mice, rats and monkeys demonstrated that levels of the cleavage product 12ADT-Asp were undetectable in plasma at any time after dosing; thus, levels of the cleavage product were not assessed in this clinical study. Results of pharmacokinetic analysis of the parent molecule mipsagargin are presented in Figure 2 and suggest a biexponential model with time-invariant kinetics. A biexponential function (without weighting) was considered to best represent the plasma concentration–time profiles of mipsagargin following 3 daily intravenous 1 h infusions. In general, the model adequately fitted the individual plasma concentration–time profiles of mipsagargin at each dose level and adequately predicted plasma concentrations of mipsagargin at 24 h after the second daily infusion, consistent with time-invariant kinetics of mipsagargin (i.e., plasma concentrations of mipsagargin after repeated dosing appear to be predictable based on linear kinetic theory). The model also indicated that approximate steady state would be reached after the third infusion.

Pharmacokinetics of mipsagargin in plasma after single and multiple intravenous infusions. Samples were collected at multiple timepoints after the first infusion of mipsagargin, 24 h after the second infusion of mipsagargin and 25 days after the third infusion of mipsagargin. The 66.8 mg m−2 dose is presented as 67 mg m−2 and the part 2 dosing regimen, 40 mg m−2 on day 1 and 66.8 mg m−2 on days 2 and 3, is presented as 40 mg m−2 with the dotted line.
Other calculated PK parameters are presented in Table 4. The geometric mean terminal (elimination) half-life (t1/2β) was 21.0 h (range 4.3–69.3). Greater than 90% of mipsagargin was eliminated during the secondary phase (i.e., associated with t1/2β). Geometric mean systemic clearance (CL) of mipsagargin was 199 ml h−1 m−2, and this is >1% of hepatic blood flow of patients (i.e., clearance was very low relative to hepatic blood flow).
The geometric mean apparent volume of distribution at steady state (Vss) of mipsagargin was 4995 ml m−2, not appreciably different from blood volume, suggesting that mipsagargin was largely confined to plasma and did not undergo extensive tissue distribution. In general, CL and Vss were reliably estimated at all dose levels, and the interpatient variability of systemic exposure to mipsagargin, characterised by CL and Vss, was low, with CVs of 45.4 and 39.2%, respectively.
Systemic exposure (AUC) to mipsagargin increased approximately dose proportionally; for a doubling in dose, AUC was predicted to increase, on average, 2.02-fold (95% CL: 1.86–2.19). The unadjusted confidence interval included unity, suggesting no statistical evidence for nonlinear kinetics of mipsagargin.
Response to treatment
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n=42; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months.
Discussion
The goal of this phase 1 first-in-man study was to evaluate the safety of mipsagargin and validate the TG prodrug strategy across a wide range of advanced solid tumours and identify specific tumour type(s) in which this strategic targeting of TG cytotoxicity would most likely provide clinical benefit. Prodrug therapy that targets a molecule critical for cellular homeostasis but only in PSMA-expressing cells, particularly the tumour vasculature, represents a new class of cancer therapeutics and a novel strategy that warrants clinical investigation.
Despite the advanced stage of disease in the heavily pretreated patients enrolled onto the study, a protocol-defined MTD was not reached. The phase II dosing regimen was derived on the basis of IRRs and creatinine elevations observed in the dose escalation portion of the study. The IRR to mipsagargin may be multifactorial. One possible explanation relates to thapsigargin-mediated inhibition of the SERCA pump and resultant elevation of cytosolic calcium levels in mast cells, resulting in degranulation and histamine release (Putney and Bird, 1993; Huber et al, 2000). Another plausible explanation is related to the excipients used for preparation of mipsagargin for infusion, polysorbate and propylene glycol. An association between polysorbate and incidence of IRRs, including in the oncology and vaccine setting, has been noted in the literature (Coors et al, 2005). The IRR, when present, generally occurred on day 1 of infusion and was not apparent on days 2 and 3. In an effort to reduce the occurrence and severity of IRRs, the mipsagargin dosing regimen was modified in the expansion cohort, so that a lower dose, 40 mg m−2, was administered on day 1 and the recommended phase II dose of 66.8 mg m−2 on days 2 and 3 of each 28-day cycle, along with standard premedications at the investigator’s discretion (steroid, anti-histamine, anti-nausea, anti-pyretic, H2 blocker). The specific choice of medication (dexamethasone or prednisone, for example) and dose of each premedication was at the investigator’s discretion and was allowed to be in accordance with each institution’s standard premedication guidelines. This modification appeared quite effective, with only one observation of IRR in the expansion cohort; this was a Grade 1 event that resolved and did not recur in subsequent cycles.
The elevation in creatinine was not unexpected based on evidence suggesting that cells of the proximal tubules in benign kidney express PSMA (Chang et al, 1999a). During the course of the study, it was observed anecdotally that among patients receiving prophylactic hydration as a component of their routine care, the incidence of creatinine elevation was infrequent. Thus, prophylactic hydration by intravenous infusion of 250–500 ml of saline on each day of mipsagargin infusion was incorporated into the treatment regimen. Thereafter, the incidence of creatinine elevation in patients receiving prophylactic hydration was greatly minimised, with only one observation of Grade 2 creatinine elevation noted: this single event was observed in one patient in the eleventh cycle of treatment; the creatine elevation resolved within 24 h and did not recur in cycle 12.
With the safety data are considered together, mipsagargin was manageable and with an acceptable safety profile in this population of patients with advanced disease. The MTD is 66.8 mg m−2 administered intravenously as a 1-h infusion on days 1, 2 and 3 every 4 weeks; a lower dose level, 40 mg m−2, is recommended for day 1, with standard premedications and prophylactic hydration.
Although no objective responses were observed in this study, some heavily pretreated patients demonstrated prolonged disease stabilisation. Despite the heterogeneity of the population enrolled into this study, a proportion of patients achieved a median PFS of 129 days, suggesting that a sub-group of patients or particular tumour types may preferentially derive benefit greater from PSMA-targeting approaches. This hypothesis is substantiated by observations in the expansion cohort, wherein whereby 5 patients with advanced HCC, refractory to sorafenib therapy, had prolonged disease stabilisation with an average PFS of 166 days. Given the typical median PFS of ∼2–3 months in the subgroup of patients with HCC who have progressed on prior sorafenib (Finn et al, 2012; Yau et al, 2012; Llovet et al, 2012), the suggested clinical benefit and the minimal AEs reported in this patient group are encouraging, although these data should be interpreted with caution because of the limited patient numbers and the nonrandomised study design. Given that the vascularity of HCC is frequently increased (Llovet et al, 2008; Yang and Poon, 2008) and PSMA expression is elevated in the vasculature of HCC (Furuya et al, 1996; Chang et al, 1999b; Haffner et al, 2009), it is feasible that mipsagargin, a PSMA-activated thapsigargin-based prodrug, might provide particular clinical benefit in this indication.
Recent clinical reports substantiate the logic of therapeutic approaches based on targeting PSMA-expressing tumours, namely castrate-resistant advanced prostate cancers. MLN2704 is an immunoconjugate designed to deliver the anti-microtubule agent maytansinoid-1 directly to prostate-specific membrane antigen-expressing cells via the PSMA-targeted monoclonal antibody MLN59. Clinical efficacy was observed, with two (22%) of the nine patients treated at 264 or 343 mg m−2 having a >50% decrease in PSA after treatment, accompanied by measurable tumour regression (Galsky et al, 2008). Similarly, in a clinical study of a single infusion of radiolabelled anti-PSMA monoclonal antibody J591 (lutetium-177; (177)Lu), 10.6% of patients experienced ⩾50% decline in PSA, 36.2% experienced ⩾30% decline and 59.6% experienced any PSA decline following a single infusion (Tagawa et al, 2013). The treatment was found to be well tolerated, with reversible myelosuppression as the most common AE. Finally, a PSMA antibody drug conjugate (ADC) consisting of a fully humanised anti-PSMA IgG1 monoclonal antibody conjugated to monomethyl auristatin E (MMAE), a microtubule-disrupting agent, via a protease-cleavable linker has entered clinical studies (Ma et al, 2006; Doronina et al, 2003). In a phase I clinical study with this PSMA ADC, antitumour activity was observed in approximately half of the patients treated with doses of 1.8 mg kg−1 or higher (Petrylak et al, 2013). In a phase II study in patients refractory to abiraterone and docetaxel therapies, treatment with 2.3 mg kg−1 PSMA ADC resulted in ⩾50% reductions in the number of circulating tumour cells in 74% of patients and ⩾30% reductions in serum PSA levels in 36% of patients (Petrylak et al, 2014). It is important to recognise that mipsagargin differs from these other PSMA-targeted approaches in two main ways. First, mipsagargin is designed to take advantage of the enzymatic activity of PSMA and is designed as a targeted therapy to deliver many more cytotoxic molecules to the tumour per single antigen (amplification). Secondly, the cytotoxic moiety of mipsagargin is expected to be released extracellularly and able to be taken up by cells within a limited diffusion radius (bystander effect). This may aid in destruction of tumours with heterogeneous expression of PSMA in different cells. Taken altogether, these early clinical studies are encouraging, and support the notion that a subset of cancer patients are likely to be more sensitive to PSMA-targeting agents. It is clear that additional studies will be required to better understand tumour responsiveness; these studies will likely include evaluation of the correlation between levels of intratumoural PSMA expression and clinical activity, as well as activity in other tumour types.
Mipsagargin exhibited a favourable PK profile in this study, and exposure appeared to be linear through all doses. Reversible renal toxicities were observed at doses of 66.8 mg m−1 that had an associated Cmax of ∼28 000 ng ml−1 and an AUC of ∼410 000 ng h−1 ml−1. These pharmacokinetic parameters are comparable to the Cmax of ∼20 500 ng ml−1 and AUC of ∼218 000 ng h−1 ml−1 observed in monkeys administered a 1 mg kg−1 dose where mild, reversible renal toxicities were also observed. These AUC levels are similar to the AUC of ∼470 000 ng h−1 ml−1 associated with a 5.6 mg kg−1 dose in nude mice under conditions where antitumour efficacy was observed. Preclinical studies in mice, rats and monkeys demonstrated that virtually undetectable levels of the ultimate PSMA cleavage product 12ADT-Asp were detectable in plasma at any time after dosing, and hence levels of this cleavage product were not assessed in this clinical study. These data suggest that preclinical models of efficacy and toxicity are supportive that target plasma levels were achieved in this study.
In conclusion, mipsagargin is a TG-based prodrug that is designed to be activated in PSMA-expressing epithelial cells, as well as tumour vasculature ECs. There are two particularly novel aspects of mipsagargin that distinguish it from other targeted anticancer agents in development. The first is incorporation of a synthetic masking peptide that is hydrolyzed by PSMA, allowing the cytotoxic analogue of TG, 12ADT, to be targeted specifically to tumours and the surrounding tumour vasculature. The second aspect that differentiates mipsagargin from other targeted agents is that the target, the SERCA pump, is a cellular molecule that is required for homeostasis. This is in contrast to many other targeted agents whose target is a cancer cell-specific molecule, allowing development of resistance by selection of cancer cells with increased, decreased or absent expression of the target or expression of an aberrant or alternate form of the target.
Mipsagargin is a novel agent with a distinct mechanism of action, acceptable tolerability and a favourable PK profile. Observations of disease stabilisation in patients with advanced disease suggest antitumour activity and support further exploration in the phase II setting. Observations of disease stabilisation in patients with hepatocellular carcinoma, in particular, suggest that mipsagargin may represent a therapeutic option for this patient population, and a phase II clinical trial is underway to evaluate the effect of mipsagargin on time to progression in patients with advanced, sorafenib-refractory hepatocellular carcinoma. Furthermore, results from this phase I study suggest the opportunity to explore PSMA-targeting prodrug therapies in a subset of tumours that have increased tumour vascularity and/or PSMA expression.
References
Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC (2003) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608–8618.
Chang SS, O’Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB (1999a) Prostate specific membrane antigen is produced in tumor-associated neovasculature. Clin Cancer Res 5: 2674–2681.
Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB (1999b) Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res 59: 3192–3198.
Coors EA, Seybold H, Merk HF, Mahler V (2005) Polysorbate 80 in medical products and nonimmunologic anaphylactoid reactions. Ann Allergy Asthma Immunol 95: 593–599.
Deniaud A, Sharaf el dein O, Malillier E, Poncet D, Kroemer G, Lemaire C, Brenner C (2008) Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27: 285–299.
Denmeade SR, Isaacs JT (1998) Enzymatic activation of prodrugs by prostate-specific antigen: targeted therapy for metastatic prostate cancer. Cancer J Sci Am 4 (Suppl 1): S15–S21.
Denmeade SR, Isaacs JT (2005) The SERCA pump as a therapeutic target: Making a ‘smart bomb’ for prostate cancer. Cancer Biol Ther 4: 14–22.
Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, Christensen SB, Isaacs JT (2003) Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst 95: 990–1000.
Denmeade SR, Lin XS, Tombal B, Isaacs JT (1999) Inhibition of caspase activity does not prevent the signaling phase of apoptosis in prostate cancer cells. Prostate 39: 269–279.
Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, Olesen C, Gurel B, DeMarzo AM, Wilding G, Carducci MA, Dionne CA, Moller JV, Nissen P, Christensen SB, Isaacs JT (2012) Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Trans Med 4: 1–12.
Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD (2003) Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 21: 778–784.
Finn RS, Kang YK, Mulcahy M, Polite BN, Lim HY, Walters I, Baudelet C, Manekas D, Park JW (2012) Phase II open-label study of brivanib as second-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res 18: 2090–2098.
Furuya Y, Krajewski S, Epstein JI, Reed JC, Isaacs JT (1996) Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clin Cancer Res 2: 389–398.
Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT (1994) The role of calcium, pH, and cell proliferation in the programmed (apoptotic) death of androgen-independent prostatic cancer cells induced by thapsigargin. Cancer Res 54: 6167–6175.
Galsky MD, Eisenberger M, Moore-Cooper S, Kelly WK, Slovin SF, DeLaCruz A, Lee Y, Webb IJ, Scher HI (2008) Phase I trial of the prostate-specific membrane antigen-directed immunoconjugate MLN2704 in patients with progressive metastatic castration-resistant prostate cancer. J Clin Oncol 26: 2147–2154.
Haffner MC, Kronberger IE, Ross JS, Sheehan CE, Zitt M, Mühlmann G, Ofner D, Zelger B, Ensinger C, Yang XJ, Geley S, Margreiter R, Bander NH (2009) Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum Pathol 40: 1754–1761.
Huber M, Hughes MR, Krystal G (2000) Thapsigargin-induced degranulation of mast cells is dependent on transient activation of phosphatidylinositiol-3 kinase. J Immunol 165: 124–133.
Jakobsen CM, Denmeade SR, Isaacs JT, Gady A, Olsen CE, Christensen SB (2001) Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J Med Chem 44: 4696–4703.
Lin XS, Denmeade SR, Cisek L, Isaacs JT (1997) Mechanism and role of growth arrest in programmed (apoptotic) death of prostatic cancer cells induced by thapsigargin. Prostate 33: 201–207.
Liu H, Moy P, Kim S, Xia Y, Rajasekaran A, Navarro V, Knudsen B, Bander NH (1997) Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res 57: 3629–3634.
Llovet JM, Decaens T, Raoul JL, Boucher E, Kudo M, Chang C, Kang YK, Assenat E, Lim HY, Boige V, Mathurin P, Fartoux L, Lin DY, Bruix J, Poon RT, Sherman M, Blanc JF, Finn R, Tak WY, Chao Y, Ezzeddine R, Liu D, Walters I, Park JW (2012) Brivanib versus placebo in patients with advanced hepatocellular carcinoma (HCC) who failed or were intolerant to sorafenib: results from the phase 3 BRISK-PS Study. ILC 2012 Abstract 1398.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, Cosme de Oliveira A, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359: 378–390.
Ma D, Hopf CE, Malewicz AD, Donovan GP, Senter PD, Goeckeler WF, Maddon PJ, Olson WC (2006) Potent antitumor activity of an auristatin-conjugated, fully human monoclonal antibody to prostate-specific membrane antigen. Clin Cancer Res 12: 2591–2596.
Petrylak DP, Kantoff PW, Mega AE, Vogelzang NJ, Stephenson J, Fleming MT, Stambler N, Petrini M, Blattman S, Israel RJ (2013) Prostate-specific membrane antigen antibody drug conjugate (PSMA ADC): a phase I trial in metastatic castration-resistant prostate cancer (mCRPC) previously treated with a taxane. J Clin Oncol 31 (Suppl 6): Abstract 119.
Petrylak DP, Smith DC, Appleman LJ, Fleming MT, Hussain A, Dreicer R, Sartor AO, Shore N, Vogelzang NJ, Youssoufian H, DiPippo VA, Stambler N, Huang K, Israel RJ (2014) A phase II trial of prostate-specific membrane antigen antibody drug conjugate (PSMA ADC) in taxane-refractory metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 32 (Suppl): Abstract 5023.
Putney JW Jr, Bird GS (1993) The signal for capacitative calcium entry. Cell 75: 199–201.
Randriamampita C, Tsien RY (1993) Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature 364: 809–814.
Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM (2002) Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem 277: 21836–21842.
Rasmussen U, Christensen SB, Sandberg F (1978) Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm Suec 15: 133–140.
Samplaski MK, Heston W, Elson P, Magi-Galluzzi C, Hansel DE (2011) Folate hydrolase (prostate specific antigen) 1 expression in bladder cancer subtypes and associated tumor neovasculature. Mod Pathol 24: 1521–1529.
Silver DA, Pellicer I, Fair WR, Heston WDW, Cordon-Cardo C (1997) Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 3: 81–85.
Singh P, Mhaka AM, Christensen SB, Gray JJ, Denmeade SR, Isaacs JT (2005) Applying linear interaction energy method for rational design of noncompetitive allosteric inhibitors of the sarco- and endoplasmic reticulum calcium-ATPase. J Med Chem 48: 3005–3014.
Søhoel H, Jensen AM, Møller JV, Nissen P, Denmeade SR, Isaacs JT, Olsen CE, Christensen SB (2006) Natural products as starting materials for development of second generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg Med Chem 14: 2810–2815.
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13: 184–190.
Tagawa ST, Milowsky MI, Morris M, Vallabhajosula S, Christos P, Akhtar NH, Osborne J, Goldsmith SJ, Larson S, Taskar NP, Scher HI, Bander NH, Nanus DM (2013) Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin Cancer Res 19: 5182–5191.
Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP (1990) Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA 87: 2466–2470.
Tombal B, Denmeade SR, Gillis JM, Isaacs JT (2002) A supramicromolar elevation of intracellular free calcium ([Ca2+]i) is consistently required to induce the execution phase of apoptosis. Cell Death Differ 9: 561–573.
Tombal B, Denmeade SR, Isaacs JT (1999) Assessment and validation of a microinjection method for kinetic analysis of [Ca2+]i in individual cells undergoing apoptosis. Cell Calcium 25: 19–28.
Tombal B, Weeraratna AT, Denmeade SR, Isaacs JT (2000) Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate 43: 303–317.
Toyoshima C, Nomura H, Sugita Y (2003) Crystal structures of Ca2+-ATPase in various physiological states. Ann NY Acad Sci 986: 1–8.
WinNonlin (2011) Phoenix Version 6.2.1. Pharsight Corporation Inc.: Mountain View, CA.
Winther AM, Liu H, Sonntag Y, Olesen C, Le Maire M, Soehoe Hl, Olsen CE, Christensen SB, Nissen P, Møller JV (2010) Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J Biol Chem 285: 28883–28892.
Yang ZF, Poon RTP (2008) Vascular changes in hepatocellular carcinoma. Anat Rec 291: 721–734.
Yau T, Wong H, Chan P, Yao TJ, Pang R, Cheung TT, Fan ST, Poon RT (2012) Phase II study of bevacizumab and erlotonib in the treatment of advanced hepatocellular patients with sorafenib-refractory disease. Invest New Drugs 30: 2384–2390.
Acknowledgements
We thank the patients, their families and the clinical teams participating in this study. GenSpera, Inc. provided assistance with preparation of this manuscript. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
M Kurman is a paid consultant to GenSpera, Inc. The other authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Mahalingam, D., Wilding, G., Denmeade, S. et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br J Cancer 114, 986–994 (2016). https://doi.org/10.1038/bjc.2016.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.72
Keywords
This article is cited by
-
Calcium signalling pathways in prostate cancer initiation and progression
Nature Reviews Urology (2023)
-
The unfolded protein response (UPR) pathway: the unsung hero in breast cancer management
Apoptosis (2023)
-
PERK signaling through C/EBPδ contributes to ER stress-induced expression of immunomodulatory and tumor promoting chemokines by cancer cells
Cell Death & Disease (2021)
-
Multi-level inhibition of coronavirus replication by chemical ER stress
Nature Communications (2021)
-
Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components
Cell Communication and Signaling (2020)
