
Abstract
Background:
Although there are reports that metronomic cyclophosphamide (CTX) can be immune stimulating, the impact of its combination with anti-CTLA-4 immunotherapy for the treatment of cancer remains to be evaluated.
Methods:
Murine EMT-6/P breast cancer, or its cisplatin or CTX-resistant variants, or CT-26 colon, were implanted into Balb/c mice. Established tumours were monitored for relative growth following treatment with anti-CTLA-4 antibody alone or in combination with; (a) metronomic CTX (ldCTX; 20 mg kg−1 day−1), b) bolus (150 mg kg−1) plus ldCTX, or (c) sequential treatment with gemcitabine (160 mg kg−1 every 3 days).
Results:
EMT-6/P tumours responded to anti-CTLA-4 therapy, but this response was less effective when combined with bolus plus ldCTX. Anti-CTLA-4 could be effectively combined with either ldCTX (without a bolus), or with regimens of either sequential or concomitant gemcitabine, including in orthotopic EMT-6 tumours, and independently of the schedule of drug administration. Tumour responses were confirmed with CT-26 tumours but were less pronounced in drug-resistant EMT-6/CTX or EMT-6/DDP tumour models than in the parent tumour. A number of tumour bearing mice developed spontaneous metastases under continuous therapy. The majority of cured mice rejected tumour re-challenges.
Conclusions:
Metronomic CTX can be combined with anti-CTLA-4 therapy, but this therapy is impaired by concomitant bolus CTX. Sequential therapy of anti-CTLA-4 followed by gemcitabine is effective in chemotherapy-naive tumours, although tumour relapses can occur, in some cases accompanied by the development of spontaneous metastases.
Similar content being viewed by others

Main
In 2010, 14 years after the report of CTLA-4 blockade causing tumour responses in preclinical models (Mokyr et al, 1998), by blocking the immune suppressive functions of the CTLA-4 protein, the anti-CTLA-4 antibody ipilimumab was approved by the FDA for the treatment of non-resectable or metastatic melanoma (Hodi et al, 2010). This approval was a pivotal event for cancer immunotherapy (Pardoll, 2012; Li et al, 2013), a field now enriched by additional targets such as PD-1, PD-L1, and LAG-3 (Pardoll, 2012; Li et al, 2013; Postow et al, 2015). Despite these successes, there remain several hurdles to be overcome in the quest for optimal anti-CTLA-4-based therapy regimens, including minimising the likelihood of the development of autoimmune toxicity (Maker et al, 2006; Li et al, 2013; Gangadhar and Vonderheide, 2014), or devising means to overcome the evolution of drug resistance to therapy (Tumeh et al, 2014). Currently, there is a growing interest to improve anti-CTLA-4 therapy by exploiting the immunostimulating properties of some conventional chemotherapeutics (Ghiringhelli et al, 2007; Galluzzi et al, 2012).
In this study, we tested whether continuous low-dose (metronomic) chemotherapy, in this case cyclophosphamide (CTX), which has been reported to act in part by boosting the immune system (Ghiringhelli et al, 2007; Chen et al, 2010; Pasquier et al, 2010; Andre et al, 2014), could be effectively combined with CTLA-4 antibody therapy for the treatment of breast cancer in a preclinical model. Surprisingly, we found that our previously designed protocol (Shaked et al, 2005), consisting of bolus (high-dose) CTX injection combined with oral low-dose CTX (ldCTX), actually hindered the anti-tumour efficacy of anti-CTLA-4 therapy. Conversely, we noted that metronomic CTX (without an upfront bolus) can enhance anti-CTLA-4 therapy. Furthermore, even more impressive tumour responses were obtained using a sequential regimen of CTLA-4 blockade followed by a previously described (Francia et al, 2012) metronomic gemcitabine chemotherapy (160 mg kg−1, every 3 days), irrespective of whether it was evaluated on the parent EMT-6/P tumour or on variants selected for resistance to cisplatin or to CTX. We also noted that acquired drug resistance (at least in a subset of mice) was observed with all therapies evaluated in this study, as was the emergence of spontaneous metastases. Our results contribute to our understanding of the preclinical benefits of chemotherapy regimens in combination with CTLA-4 blockade (Grosso and Jure-Kunkel, 2013; Jure-Kunkel et al, 2013; Lesterhuis et al, 2013). They also serve as cautionary notes in that some regimens (e.g. high-dose CTX) may hinder the beneficial anti-tumour effects of CTLA-4 blockade-based therapies, and that the use of chemotherapy naive tumours can sometimes produce overly simplistic tumour response results in preclinical studies.
Materials and methods
Drug preparation
Gemcitabine hydrochloride was purchased from Selleck Chemicals (Houston, TX, USA) and made up in sterile phosphate-buffered saline (PBS) immediately before i.p. administration. CTX was purchased from Sigma-Aldrich (St Louis, MO, USA) and made up in PBS before i.p. injection or before its addition to the mice’s drinking water. Metronomic ldCTX was administered at an estimated 20 mg kg−1 day−1 as previously described (Man et al, 2002). Some regimens (termed B+ldCTX) included an upfront bolus dose of CTX, administered on day 1 as a 150 mg kg−1 i.p. injection of CTX (Shaked et al, 2005; Francia et al, 2012).
Anti-CTLA-4 antibody preparation
Anti-mouse CD152 (CTLA-4), FG purified clone 9H10, purchased from Ebioscience (San Diego, CA, USA), was diluted in PBS immediately before i.p. injection. Mice were administered 100 μg of the antibody on day 1 of treatment, followed by a 35 μg injection on day 6.
Cell lines
Murine EMT-6/P mammary carcinoma cells (ER+/PR+/HER2+, B. Teicher–personal communication, and as previously reported (Chatterjee et al, 1995; Teicher, 1997), and the CTX-resistant EMT-6/CTX and cisplatin-resistant EMT-6/DDP variants, were a gift from Beverly Teicher, and they were grown in RPMI supplemented with 10% fetal bovine serum and 2 mM L-glutamine. Cells were grown in a humidified incubator at 37°C and 5% CO2.
In vivo tumour growth assessment
Six-week-old female Balb/c mice were purchased from Harlan (Indianapolis, IN, USA). Mice were allowed to acclimatise for 2 weeks before implantation of tumour cells. To prepare cells for injection, subconfluent plates were harvested with 1% trypsin-EDTA, and cells were then washed and resuspended in RPMI at 2 million cells per ml. Two hundred thousand EMT-6 cells were injected subcutaneously into the flank of the mice (for CT-26 cells, 1 million cells per mouse were implanted). Mice were monitored twice weekly for fluctuations in body weight, and for tumour growth, as measured by Vernier calipers, and tumour volume was calculated by the formula (length × width2)/2. Institutional guidelines were followed to determine when the experimental end points were reached. Results were also plotted as event-free survival (Kaplan–Meier analysis) over time, where duration of event-free survival is defined as time to primary tumour progression beyond 1200 mm3 or >15% weight loss, as per our previous study (du Manoir et al, 2006). Primary tumour fragments, or established lung metastases, were isolated from selected killed mice, and used to derive cell cultures as previously described (Francia et al, 2008, 2009). The orthotopic implant of EMT-6 and EMT-6DDP cells was carried out as previously described (Francia et al, 2008, 2009); 100 000 cells in 50 μl were implanted in the inguinal mammary fat pad of mice. All in vivo procedures and experiments were performed with the approval of the UTEP IACUC (IACUC reference #: A-201201-1).
Immunohistochemical analysis
Paraffin-embedded EMT-6 tumour sections were cut to 5 μ thickness and stained for anti-CD31 (Abcam 28364) used at a dilution of 1:400, using an antigen retrieval of citrate buffer pH6. Secondary antibody was goat anti-rabbit at a dilution of 1:200, using DAB for detection of positive staining, and counter stained with hematoxylin for contrast.
Statistical analysis
The analysis of variance among groups (ANOVA), followed by the Student-Newman–Keuls test, was used to assess the statistical differences of data in vivo. Tumour therapy results are reported as mean±s.d. Survival curves were plotted by the method of Kaplan and Meier and were tested for survival differences using the log-rank test. The level of significance was set at P<0.05. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA).
Results
Anti-CTLA-4 therapy combined with bolus plus ldCTX
To evaluate whether metronomic CTX can be effectively combined with anti-CTLA-4 treatment, we tested a combination regimen on subcutaneously implanted EMT-6/P tumours (Figures 1A and B). For the chemotherapy component, we sought to use a B+ldCTX protocol consisting of a bolus CTX (given i.p. on day 1) plus ldCTX (20 mg kg−1day−1, p.o.). Our choice was guided by our previous study (Shaked et al, 2005) in which the B+ldCTX protocol was shown to more effectively inhibit tumour growth than the sole ldCTX in the EMT-6/P tumour, as well as in other tumour models. Mice (n=32) bearing EMT-6/P tumours were treated with saline (control), B+ld CTX, anti-CTLA-4 antibody, or with B+ldCTX plus anti-CTLA-4 antibody. Figure 1B shows the resulting impact of the therapies on tumour growth. Thus, control treated tumours grew rapidly, the B+ldCTX treatment slowed down tumour growth, whereas anti-CTLA-4 antibody treatment caused tumour regressions over a 20 day period – followed by tumour relapses in the subsequent 15 days. Surprisingly, the B+ldCTX plus anti-CTLA-4 combination therapy did not produce tumour regressions and, furthermore, it produced a tumour growth rate that was only marginally slower than was observed with B+ldCTX alone. Thus, B+ldCTX significantly hinders the efficacy of anti-CTLA-4 therapy in the EMT-6/P tumour model. This was an unexpected finding, as we had recently reported that B+ldCTX could be effectively combined with an anti-VEGFR2 antibody (Francia et al, 2008), or with metronomic oral gemcitabine (LY2334737) (Francia et al, 2009). We also noted that by day 22 the CTLA-4 antibody monotherapy resulted in complete tumour regression in two mice, one of which then began to show tumour regrowth a few days later. All anti-CTLA-4 treated tumours shrank after the therapy began, although tumour relapses were eventually observed in 6 out of 7 mice in this group. Therefore, anti-CTLA-4 therapy is effective in the EMT-6/P tumour model, but its therapeutic efficacy is significantly hampered by concurrent B+ldCTX treatment.

Impact of bolus plus ldCTX combined with anti-CTLA-4 therapy on the growth of EMT-6/P tumours. (A) Schematic of evaluation of anti-CTLA-4 therapies with metronomic chemotherapy. EMT-6/P breast tumours, or CTX resistant (EMT-6/CTX) or DDP resistant (EMT-6/DDP) tumours were treated with anti-CTLA-4 antibody (administered on days 1 and 6). Chemotherapy regimens included low-dose metronomic CTX (ldCTX), bolus plus ldCTX, or gemcitabine. Confirmatory studies were carried out with the murine CT-26 colon tumour. (B) Murine EMT-6/P cells were implanted s.c. in female Balb/c mice. Therapies began when tumours were 50 mm3; the mice received control (n=8) saline (i.p.), anti-CTLA-4 (n=7), bolus plus ldCTX (n=10), or the combination (n=7) of anti-CTLA-4 together with bolus plus ldCTX. *P<0.05 vs control, #P<0.05 vs CTLA-4 (mean values±s.d.). (C) Mouse weights, as a measure of toxicity of the different treatments. (D) Impact of the different therapies as assessed by analysis of event-free survival (Kaplan–Meier analysis), where duration of event-free survival is defined as time to primary tumour progression beyond 1200 mm3 or >15% weight loss. Significant event-free survival was observed with anti-CTLA-4 therapy, but this benefit was reduced by the addition of bolus+low-dose CTX. The sole survivor, by day 46, in the anti-CTLA-4 therapy group was still alive and tumour-free at day 400 after tumour cell injection. *P<0.05 was taken as statistical indication of difference vs controls and between treated groups.
To assess the relative toxicity of the therapies, we monitored body weights of the mice in the course of the experiment (as per our previous studies (du Manoir et al, 2006; Francia et al, 2012)). Figure 1C shows that the treatments that included a B+ldCTX component produced a short-term weight loss (as previously reported (Shaked et al, 2005)), followed by a gain in weight by the treated mice. We also plotted the tumour responses as a Kaplan–Meier plot (Figure 1D), which shows that time to 50% event-free survival of CTLA-4 antibody treatment was 38 days, a significant increase compared with 22 days for the control group (P=0.0011 CTLA-4 vs control). B+ldCTX had no significant impact on survival. Figure 1D also shows that one anti-CTLA-4 antibody treated mouse, which had been bearing a palpable tumour in the first 2 weeks of this experiment, showed a tumour regression and remained tumour-free for the whole follow-up period. This mouse was still tumour-free 400 days later.
Anti-CTLA-4 therapy combined with ldCTX, or with sequential gemcitabine therapy
We next decided to test whether we could incorporate other chemotherapy regimens, either in combination with or subsequent to the anti-CTLA-4 administration. We reasoned that as high-dose CTX can be immunosuppressive (Emadi et al, 2009), the bolus CTX dose might impair the immune stimulating impact of the CTLA-4 blockade – thus negating its therapeutic benefit. Therefore, we either had to separate time of the dosing of chemotherapy from that of the anti-CTLA-4 antibody, or omit the bolus CTX component. We chose to evaluate two additional strategies. One was to combine anti-CTLA-4 administration with ldCTX (i.e. without a bolus). This choice was based on reports that (in contrast to the high-dose CTX regimens) ldCTX can stimulate the immune system (Ghiringhelli et al, 2007; Pasquier et al, 2010; Andre et al, 2014). Furthermore, in our previous studies, ldCTX effectively inhibited tumour growth without producing any obvious toxicity (Man et al, 2002; Francia et al, 2009). The second strategy involved the incorporation of a metronomic regimen of 160 mg kg−1 gemcitabine given every 3 days, as we recently observed that it can produce remarkable tumour responses in a preclinical breast cancer model (Francia et al, 2012). However, to avoid the possibility that gemcitabine might impair the therapeutic effect of the CTLA-4 blockade, we decided to administer the two drugs sequentially (i.e. after tumours began to regress following the anti-CTLA-4 treatment). To evaluate these alternative therapies, we implanted EMT-6/P tumours into 43 mice, which were subsequently divided into seven groups. Therapies began when all mice had established tumours and when the average tumour volume was ∼50 mm3. We noted no or minimal impact of tumour growth, compared with controls, in the groups treated with ldCTX (Figure 2A). We also noted that anti-CTLA-4 therapy led to a significant (initial) tumour regression in the first 2 weeks after therapy started, followed by tumour relapses. The combination of anti-CTLA-4 plus ldCTX (both co-administered from day 6 onwards) produced a greater inhibition of tumour growth that was observed with the anti-CTLA-4 monotherapy, although the results did not reach a statistical significance. The group treated with a single dose of gemcitabine unexpectedly showed weight loss in the days following treatment, and therefore this treatment was interrupted; the treatment caused initial inhibition of tumour growth, followed by tumour regrowth (Figure 2A), and a recovery of mouse body weight (Figure 2B). One group (treated with anti-CTLA-4 then Gem) initially received anti-CTLA-4 antibody, which again produced a tumour regression that lasted 2 weeks, followed by a tumour relapse (Figure 2A). For this group, as soon as tumours started to relapse (i.e. around day 21), the second-line therapy of gemcitabine was administered. In this group, we did not observe any weight loss after gemcitabine injection, and therefore gemcitabine treatment was continued. At the onset of the second-line gemcitabine therapy on day 21, all mice in this group had visible tumours and an average tumour volume of 145 mm3. Gemcitabine administration caused tumours to regress again, and by day 36 only two out of the six mice had any palpable tumours. These two mice eventually showed tumour re-growths, whilst under continuous gemcitabine therapy (Figure 2C). The remaining four mice showed significant (P<0.05) and complete tumour regression and are still tumour-free over 400 days later.

Effective combination of chemotherapy with anti-CTLA-4 therapy for the inhibition of the growth of EMT-6/P tumours. (A) Murine EMT-6/P cells were implanted s.c. in female Balb/c mice. Therapies began when tumours were 50 mm3; the mice received control saline (i.p.; n=7), anti-CTLA-4 (n=6), bolus plus ldCTX (n=7), a single dose of gemcitabine (160 mg kg−1; n=5), ldCTX (ldCTX; n=6), or the combination of anti-CTLA-4 plus ldCTX (n=6). One additional group received anti-CTLA-4 therapy as a first line treatment and then (when the tumours began to relapse around day 21) a second-line therapy consisting of gemcitabine (160 mg kg−1 every 3 days, i.p.; n=6), starting on day 21. *P<0.05 vs control, #P<0.05 vs CTLA-4 then gem (mean values±s.d.). (B) Mouse weights, as a measure of toxicity of the different treatments. (C) Kaplan–Meier plot of event-free survival, where duration of event-free survival is defined as time to primary tumour progression beyond 1200 mm3 or >15% weight loss. *P<0.05 was taken as statistical indication of difference vs controls and between treated groups. Significant event-free survival was observed with anti-CTLA-4 therapy, and this benefit could be improved by the combination of anti-CTLA-4 therapy plus metronomic CTX, or by the sequential regimen using a first line of anti-CTLA-4 followed by gemcitabine chemotherapy on relapsing tumours.
With regard to the relative toxicity of the tested therapies, other than the single gemcitabine treatment group as noted above, no significant changes in mouse weight were observed compared with controls (Figure 2B). As noted in the subsequent studies (detailed below), we infrequently observed toxicity following gemcitabine injection, and this was typically resolved by allowing the mice a break from the treatment, as in our previous study (Tang et al, 2010). No toxicity was observed in the CTLA-4 then gemcitabine group after the gemcitabine treatment started, and treatment continued for another 30 days without producing any obvious toxicity.
We also performed a Kaplan–Meier analysis (Figure 2C), which showed the observed event-free survival in four out of six mice in the CTLA-4 then gemcitabine group. We also noted long term event-free survival (>400 days) in one out of six mice for both the CTLA-4 monotherapy group and for the CTLA-4 plus ldCTX group.
With regards to the CTLA-4 then gemcitabine treated group, we note that the two mice with visible tumours after day 36 eventually showed tumour relapses, and that one of these mice developed advanced lung metastases (while under gemcitabine therapy), and was sacrificed on day 107 (Supplementary Figure 1 and Supplementary Table 1). These results show that although the anti-CTLA-4 then gemcitabine therapy is highly effective, tumour drug resistance eventually can develop in a subset of mice, and that metastatic disease (to the lungs) can develop, under therapy, in this tumour model.
Anti-CTLA-4-based therapies in EMT-6 tumours resistant to CTX
To evaluate the impact of selected CTLA-4-based therapies on drug-resistant tumours, we implanted 50 mice with the EMT-6/CTX tumour, a population previously selected for in vivo resistance to CTX (Teicher et al, 1990). We noted a similar relative response to the different therapies as we had noted with the parental EMT-6/P tumour, although therapeutic benefit was reduced in the EMT-6/CTX drug-resistant model (Figure 3A). The initial administration of gemcitabine, either on its own or following CTLA-4 therapy, did not produce any immediate toxicity, and was continued. Only after more than five cycles of gemcitabine did we see some drop in mouse weights (particularly with gemcitabine alone), but this was readily resolved by adopting our previously reported strategy (Tang et al, 2010) of giving the mice short breaks in therapy (as shown by the arrows in Figures 3A and B). Gemcitabine monotherapy initially produced a significant (P<0.05) impact on tumour growth, but eventually all mice developed drug resistance (while under continuous therapy) as shown in Figure 3C. Similarly, CTLA-4 therapy followed by gemcitabine treatment resulted in some tumours initially regressing, although in a number of mice drug resistance later developed under continuous gemcitabine therapy. 40% of mice in the CTLA-4 followed by gemcitabine therapy showed cures, whereas others developed drug resistance by day 80. The mice that did not develop resistance by day 80 were alive past day 200 – with no sign of tumour growth after cessation of therapy. Thus although in drug-resistant tumours (i.e. EMT-6-CTX) the CTLA-4-based therapies are less effective and drug resistance can readily emerge, we still noted a number of ‘cures’ with the CTLA-4-based therapies. Furthermore, for those tumours that showed the development of drug resistance, CTLA-4 followed by gemcitabine therapy was the most effective regimen in delaying tumour regrowth although not statistically significant if compared with gemcitabine alone (with the exception of the volume measurement at day 50). Instead, Figure 3C shows that CTLA-4 then gemcitabine treatment produced a significantly longer survival compared with controls (P=0.0002) and other treatments such as B+ldCTX (P=0.0003) or gemcitabine alone (P=0.0236). In contrast, metronomic CTX plus anti-CTLA-4 did not produce a significantly different response when compared with anti-CTLA-4 therapy alone.

Combination of chemotherapy with anti-CTLA-4 therapy for the inhibition of the growth of EMT-6/CTX tumours, including gemcitabine administration (as indicated by the green and orange arrows). (A) Murine EMT-6/CTX cells were implanted s.c. in female Balb/c mice. Therapies began when tumours were 50 mm3; the mice received control saline (i.p.; n=8), anti-CTLA-4 (n=7), gemcitabine (160 mg kg−1 every 3 days, i.p.; n=5), bolus plus ldCTX (n=8), metronomic CTX (n=7), or the combination of anti-CTLA-4 plus metronomic CTX (n=8). One additional group received anti-CTLA-4 therapy as a first line treatment and then a second-line therapy consisting of gemcitabine (160 mg kg−1 every 3 days, i.p.; n=7). *P<0.05 vs control (on days 13, 15, and 17, P<0.05 for gemcitabine, CTLA-4+ldCTX and B+ldCTX; whereas for CTLA-4 and for CTLA-4 then gem, P<0.05 on days 15 and 17), #P<0.05 vs CTLA-4 then gem (mean values±s.d.). (B) Mouse weights, as a measure of toxicity of the different treatments (θ indicates significant toxicity caused by gemcitabine treatment). (C) Kaplan–Meier plot of event-free survival, where duration of event-free survival is defined as time to primary tumour progression beyond 1200 mm3 or >15% weight loss. *P<0.05 was taken as statistical indication of difference vs controls and between treated groups. Significant event-free survival was observed with anti-CTLA-4 therapy, and this benefit could be improved by the sequential regimen using a first line of anti-CTLA-4 followed by gemcitabine chemotherapy on relapsing tumours. No significant difference was observed between anti-CTLA-4 therapy and combination of anti-CTLA-4 therapy plus metronomic CTX.
Anti-CTLA-4-based therapies in EMT-6 tumours resistant to cisplatin
Our results with the EMT-6/CTX model suggested that although metronomic CTX can be combined with CTLA-4 blockade, this regimen is not effective in tumours resistant to CTX. To further explore how drug-resistant tumours respond to CTLA-4-based therapies, we evaluated their impact on the EMT-6/DDP model, a variant selected for resistance to cisplatin treatment in vivo (Teicher et al, 1990). Figure 4A shows that in this drug-resistant tumour the CTLA-4 plus ldCTX had a greater anti-tumour effect than CTLA-4, although the difference was not statistically significant. The gemcitabine monotherapy was initially very effective, and did not produce toxicity, although all the mice eventually had tumours that became resistant to this therapy. The greatest anti-tumour effect was obtained with CTLA-4 followed by gemcitabine, and in this case we also did not see significant toxicity with the gemcitabine therapy (Figures 4A and B). Moreover, also in terms of survival this combination determined a significant (P=0.0018) benefit if compared with controls or CTLA-4 alone (P=0.0010) (Figure 4C). In this aggressive EMT-6/DDP model, we did not observe any CTLA-4 blockade-induced tumour regression, but only a growth delay. Consequently the second-line gemcitabine therapy started on day 19, when the data showed that tumours were no longer growth delayed. These results confirm the effectiveness of combining some chemotherapy regimens with anti-CTLA-4 therapy, but they also indicate that the use of chemotherapy naive tumours (e.g. EMT-6/P) may produce overly optimistic results on the therapeutic benefit of some combination therapies.

Combination of chemotherapy with anti-CTLA-4 therapy for the inhibition of the growth of EMT-6/DDP tumours. (A) Murine EMT-6/DDP cells were implanted s.c. in female Balb/c mice. Therapies began when tumours were 50 mm3; the mice received control saline (i.p.; n=7), anti-CTLA-4 (n=7), gemcitabine (160 mg kg−1 every 3 days, i.p.; n=6), ldCTX (ldCTX; n=7), or the combination of anti-CTLA-4 plus ldCTX (n=7). One additional group received anti-CTLA-4 therapy as a first line treatment and then a second-line therapy consisting of gemcitabine (160 mg kg−1 every 3 days, i.p.; n=7). *P<0.05 vs control (mean values±s.d.). (B) Mouse weights, as a measure of toxicity of the different treatments. (C) Kaplan–Meier plot of event-free survival, where duration of event-free survival is defined as time to primary tumour progression beyond 1200 mm3 or >15% weight loss. *P<0.05 was taken as statistical indication of difference vs controls and between treated groups. Event-free survival was observed with anti-CTLA-4 therapy, and this benefit could be improved or by the sequential regimen using a first line of anti-CTLA-4 followed by gemcitabine chemotherapy on relapsing tumours (note that, for this group, two green+signs indicate two mice with large tumours killed around day 60, revealing a subgroup of mice with tumours that were very responsive to therapy).
CTLA-4 therapies on orthotopically implanted tumours
Our results raised a number of questions: (1) Are the tumour responses also observable in orthotopic tumours?, (2) can the results be replicated in a tumour model other than the EMT-6, and (3) is metronomic chemotherapy more effective when given in conjunction with anti-CTLA-4, or should it be given sequentially? To answer the first question, we implanted EMT-6/P and EMT-6/DDP in the inguinal mammary fat pad, as previously described (Francia et al, 2008, 2009), and then evaluated our most effective therapy (i.e. anti-CTLA-4 with sequential gemcitabine). Anti-CTLA-4 produced a tumour growth delay in both models (Figures 5A and B), and the sequential addition of gemcitabine produced an initial tumour regression. Owing to the rapid growth of these orthotopic tumours, it was difficult to compare gemcitabine alone with anti-CTLA followed by gemcitabine, as the latter sequential treatment started with larger tumours (i.e. 400 mm3) following the end of CTLA-4 administration (i.e. around day 16 in both Figures 5A and B). Nonetheless, in spite of this challenge, the response obtained with the EMT-6/P and EMT-6/DDP models was consistent with our results with these tumours when grown subcutaneously (Figures 3 and 5). Thus, EMT-6/P respond to CTLA-4 with sequential gemcitabine therapy, but the drug-resistant EMT-6/DDP are less responsive to this regimen.

Anti-CTLA-4 therapy plus gemcitabine inhibits orthotopic EMT-6 tumours. (A) Murine EMT-6/P cells were implanted into the mammary fat pad of female Balb/c mice. (Top) The mice received control (n=4) saline (i.p.), anti-CTLA-4 (n=5), gemcitabine (n=5), or anti-CTLA-4 therapy as a first line treatment followed by second-line therapy consisting of gemcitabine (160 mg kg−1 every 3 days, i.p.; n=5). (Bottom) Mouse weights, as a measure of toxicity of the different treatments. (B) (Top) Murine EMT-6/DDP cells were implanted (n=10 mice per group) into the mammary fat pad of female Balb/c and treated with the same therapies described above. *P<0.05 vs control, #P<0.05 vs CTLA-4 then gem (mean values±s.d.). (Bottom) Mouse weights, as a measure of toxicity of the different treatments.
Evaluation of therapies in CT-26 tumours
Our preliminary studies with the murine CT-26 colon tumour (Supplementary Figures 2 and 3) showed that it responded to CTLA-4 therapies, and to gemcitabine. We therefore implanted CT-26 cells s.c. in Balb/c mice and evaluated our most effective combination therapy from our experiments with the EMT-6/P tumour (Figure 2). As shown in Figure 6, whereas CTLA-4 or gemcitabine monotherapies inhibited CT-26 tumour growth, the administration of CTLA-4 followed by metronomic gemcitabine led to tumour regression.

Impact of anti-CTLA-4 therapies on CT-26 colon tumours. (A) Murine CT-26 cells were implanted s.c. into female Balb/c mice. The mice received control saline (i.p.; n=7), anti-CTLA-4 (n=7), gemcitabine (n=8), or anti-CTLA-4 therapy as a first line treatment followed by second-line therapy consisting of gemcitabine (160 mg kg−1 every 3 days, i.p.; n=8). The anti-CTLA-4 treatment and the gemcitabine treatment inhibited tumour growth, whereas anti-CTLA-4 followed by gemcitabine led to tumour regression. *P<0.05 vs control, #P<0.05 vs CTLA-4 then gem (mean values±s.d.). (B) Mouse weights, as a measure of toxicity of the different treatments.
Evaluation of combination vs sequential treatment
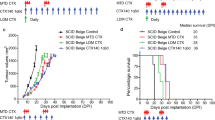
To test whether gemcitabine is more effective than CTX as a drug partner for CTLA-4 therapies, and whether sequential chemotherapy is better than combination therapy, we implanted the highly responsive EMT-6/P tumour and evaluated different CTLA-4-based therapies. As shown in Figure 7, anti-CTLA-4 is more effective when it is combined with gemcitabine – given either sequentially, or concomitantly – than when it is combined with metronomic CTX (Figure 7). Therefore, our data suggest that the drug partner for CTLA-4 (i.e. gemcitabine) is more critical for effective anti-tumour response than is the schedule by which the drugs are given.

Evaluation of different anti-CTLA-4 combination therapies on EMT-6/P tumour growth. (A) Murine EMT-6/P cells were implanted s.c. into female Balb/c mice (n=7 per group). The mice received control saline (i.p.), anti-CTLA-4, or anti-CTLA-4 followed by second-line therapy consisting of gemcitabine (160 mg kg−1 every 3 days, i.p.; CTLA-4 then gem), or anti-CTLA-4 therapy given concomitantly to gemcitabine therapy (CTLA-4+gem). In addition, one group received anti-CTLA-4 followed by second-line therapy consisting of CTX (20 mg kg−1 day−1, p.o.; CTLA-4 then CTX) or anti-CTLA-4 therapy with concomitant CTX therapy (CTLA-4+CTX). The results show that gemcitabine is a more effective therapeutic partner for anti-CTLA-4 than CTX, irrespective of whether the administration of gemcitabine is sequential or concomitant to the anti-CTLA-4. *P<0.05 vs control, #P<0.05 vs CTLA-4 then gem (mean values±s.d.). (B) Mouse weights, as a measure of toxicity of the different treatments.
Intratumoral CD31 staining and assessment of immune memory
To evaluate the impact of the different therapies on intratumoral blood vessel distribution (as a relative measure of angiogenic activity within treated tumours), we reassessed the data in Figures 1 and 2, and noted that maximal therapeutic response was noted 9–12 days after treatment started. This, taking note of the data in Figure 2, we implanted EMT-6 tumours s.c., and then administered the different therapies (using 5–7 mice per group, following the same regimens shown in Figure 2) when tumours reached an average size of 200 mm3. Thereafter tumours were measured daily, and the mice were sacrificed and the tumours excised after 7–12 days of therapy. At this termination point, tumours were 400–500 mm3 in size. Excised tumours were paraffin embedded and then evaluated for CD31 staining (Figure 8A). Analysis of the results (Figure 8B) showed that, as expected, ldCTX caused a relative reduction in intratumoral CD31 staining compared with controls – but no significant differences in staining were observed in tumours that received CTLA-4-based therapies.

Analysis of CD31-positive staining in EMT-6 tumour treated with anti-CTLA-4 therapies. (A)EMT-6/P tumours were implanted s.c. into female Balb/c mice. Tumour bearing mice were treated with (i.e. a repeat of the experiment shown in Figure 2) saline control, CTLA-4, metronomic CTX (ld CTX), gemcitabine (Gem), or CTLA-4 plus ld CTX, or CTLA-4 with sequential Gemcitabine. The experiment was terminated as the tumours were starting to respond to the different therapies, as assessed by caliper measurements. Tumours were resected, fixed, and then paraffin embedded, and 5μ sections were then stained (brown) for CD31 (black bar=100 μ). (B) Image analysis of × 20 pictures of CD31 immunohistochemistry of tumour tissues from control- and drug-treated groups of mice. The quantification of the images was performed by the image analysis software ImageJ (Image Processing and Analysis in Java; Wayne Rasband, Research Services Branch, National Institute of Mental Health, Bethesda, MD, USA) and the results are shown as the ratio between the CD31-positive area and the total area of the image. A one-way ANOVA analysis followed by Bartlett’s post test was applied to the results to demonstrate significant differences among the treatment groups. The statistical analysis of the data was performed with GraphPad Prism v.5.0. Blue lines, mean±s.d.; *P<0.05 vs control group.
In a number of our experiments (shown in Figures 2, 3, 4, 5), a few mice were cured by anti-CTLA-4 or combination therapies, and a number of mice survived for more than 500 days (some of the mice died of old age, after 400 or more days, without evidence of tumour regrowth). These results provided us with a very small pool of animals that had been cured of the implanted tumour, and these were used in tumour re-challenge experiments, where each mouse was given a second s.c. injection of originally implanted tumour cell line. Our results (Supplementary Figures 4–6 and Supplementary Table 2) show that in 11/14 cases, the mice rejected the re-implanted tumour (injected between 60 and 500 days after the first tumour implant), suggesting that CTLA-4 combination therapies can produce tumour responses that are accompanied by the establishment of immune memory. These observations are consistent with previously published data on anti-CTLA-4 therapies (Jure-Kunkel et al, 2013).
Discussion
A number of immunotherapy approaches have in recent years translated into significant increased survival in patients with cancers such as melanoma. For example, targeting CTLA-4 using ipilimumab is used for the treatment of non-resectable metastatic melanoma, and clinical trials are ongoing to test its use for the treatment of other malignancies, including lung cancer, prostate cancer, and breast cancer (www.clinicaltrials.gov). Furthermore, a number of active clinical trials are evaluating different combinations of chemotherapy and ipilimumab in melanomas and other types of cancer (Postow et al, 2015).
We previously reported extensively on experimental therapeutic studies of metronomic chemotherapy (Man et al, 2002; Emmenegger et al, 2007, 2011; Francia et al, 2009, 2012; Tang et al, 2010; Bocci et al, 2012; Hackl et al, 2013; Chow et al, 2014), including the use of metronomic CTX chemotherapy (Kerbel and Kamen, 2004) with an upfront bolus CTX dose (Shaked et al, 2005), and the use of sequential chemotherapy regimens, as well as second-line therapies (du Manoir et al, 2006; Chow et al, 2014). The proposed mechanisms of action for metronomic chemotherapy are many (Bocci et al, 2003; Kerbel and Kamen, 2004; Pasquier et al, 2010; Bocci et al, 2012; Francia et al, 2012), and they include inhibition of angiogenesis and inhibition of cancer stem cell growth. They also include activation of the immune system (Ghiringhelli et al, 2007; Pasquier et al, 2010; Andre et al, 2014). With regards to the latter, this has been documented for CTX (and for gemcitabine (Lesterhuis et al, 2013)), and it remains to be determined the extent to which metronomic dosing of other clinically used chemotherapy drugs can also activate the immune system.
As ipilimumab therapy is directed at immune activation (via inhibition of suppressor T-cells), and as combinations of ipilimumab with chemotherapy are being evaluated clinically, we sought to investigate whether the immune activation activity of CTLA-4 blocking could be augmented by the addition of metronomic CTX. Such combinations could provide data relevant to current clinical trials, such as the recently reported phase III trials of metronomic chemotherapy (Simkens et al, 2015; Colleoni et al, 2016). For example, treatment with CTLA-4 might be followed by a metronomic maintenance treatment, given the clinical low toxicity profile of metronomic chemotherapy (Kerbel and Kamen, 2004; Pasquier et al, 2010), as we have also reported (Lien et al, 2013; Orlandi et al, 2013; Derosa et al, 2014). We sought to use what, according to our previous studies, was the most effective metronomic-type CTX regimen; a protocol (Shaked et al, 2005) involving an upfront bolus (B) CTX dose, immediately followed by a metronomic CTX (ldCTX) regimen of adding CTX to the mice’s drinking water (Man et al, 2002). To our surprise, this B+ldCTX approach actually caused a less effective tumour response than the anti-CTLA-4 monotherapy alone. Therefore, our results serve as a cautionary note against the use of a bolus plus metronomic CTX component in therapies involving a CTLA-4 blockade.
We previously reported that bolus plus metronomic CTX could be improved by the addition of a targeting agent such as the anti-VEGFR2 antibody DC101 (Francia et al, 2008), or by the addition of metronomic LY2334737, an oral gemcitabine pro-drug (Francia et al, 2012). However, we had hitherto not yet observed that the bolus plus metronomic CTX could hinder the anti-tumour efficacy of a targeted therapy, or of other anti-tumour strategies. One possible interpretation of our results is that the bolus (high-dose) CTX, in our B+ldCTX regimen, is immunosuppressive thus blunting the therapeutic effect of anti-CTLA-4. We consequently sought to test two alternative strategies. One was to omit the bolus upfront CTX dose, and administer anti-CTLA-4 together with metronomic CTX. The second was to adopt gemcitabine chemotherapy (160 mg kg−1 every 3 days) as we recently reported that it produces notable responses in a LM2-4 preclinical breast cancer model (Francia et al, 2012). However, to avoid the possibility that gemcitabine administration would impair the CTLA-4 blocking strategy, we chose to separate its administration from that of the CTLA-4 antibody. Thus, we either combined a CTLA-4 with a sequential gemcitabine therapy, or we co-administered CTLA-4 antibody together with metronomic CTX. The results we obtained suggest that both strategies can improve a CTLA-4 monotherapy regimen, with the sequential gemcitabine therapy generating the more potent anti-tumour responses in the EMT-6/P model. Similar results were obtained with the CTX-resistant EMT-6/CTX and cisplatin-resistant EMT-6/DDP models, although in the drug-resistant models the benefits of these therapies were less evident and drug resistance to gemcitabine readily arose. The reduced sensitivity of the EMT-6/DDP tumour to CTLA-4 with sequential gemcitabine therapy, compared with the response seen with the parent EMT-6/P tumour, was confirmed in orthotopically implanted tumours. In addition, the effectiveness of the same sequential therapy was confirmed in CT-26 tumours. Furthermore, our data show that the effectiveness of CTLA-4 with sequential gemcitabine does not impact the relative intratumoral CD31 staining in EMT-6/P tumours, and we subsequently found that gemcitabine is equally effective irrespective of whether it is given sequentially or concomitantly with anti-CLTA-4 therapy. The intratumoral CD31-staining data we obtained are consistent with our previous study (Francia et al, 2012) showing that metronomic gemcitabine (and metronomic oral gemcitabine pro-drug) can inhibit tumour growth without impacting systemic angiogenesis. Overall, there are five aspects of this work that deserve to be highlighted: (1) bolus plus low-dose CTX can impair the antitumor efficacy of anti-CTLA-4 therapy. (2) Metronomic CTX, or metronomic gemcitabine, can effectively be combined with anti-CTLA-4 therapy and, (3) such combination therapies are also active against drug-resistant tumours (e.g. EMT-6/CTX and EMT-6/DDP). (4) The efficacy of chemotherapy plus anti-CTLA-4 is not dependent on the schedule of drug administration, and (5) produces anti-tumour effect in the absence of significant changes in intratumoral blood vessel distribution. Our results also suggest that alternative anti-tumour mechanisms are involved, including (as we previously suggested (Francia et al, 2012)), the direct targeting of tumour cells by the frequent (i.e. every 3 days) gemcitabine administration. Future studies will have to determine whether anti-CTLA-4 therapy impairs the induction of thromobospondin-1 by metronomic chemotherapy (Bocci et al, 2003), and determine if such combinations can activate the immune system. That could be assessed by testing whether such therapies result in an increase in intratumoral CD4 and CD8 staining, which would be consistent with studies showing activation of the immune system by metronomic chemotherapy (Ghiringhelli et al, 2007).
These results may be of interest to clinicians and translational researchers that are studying means of improving anti-CLTA-4 therapy, and they also caution that these therapies may be less effective in drug-resistant tumours. We did encounter a few cases of toxicity associated with gemcitabine administration, which may have been a consequence of tumour lysis syndrome, which has been reported by a few studies for this drug (Lin et al, 2007). However, in our experience, these problems were easily overcome by giving the mice short drug-free breaks, a procedure that is not infrequent with cancer patients, (Llovet et al, 2008), as we previously reported with metronomic sorafenib in a preclinical model (Tang et al, 2010).
We had expected that B+ldCTX would increase the efficacy of anti-CTLA-4 therapy – but our results proved otherwise. Nonetheless, we also observed that some chemotherapy regimens can be effectively combined with anti-CTLA-4 therapy. Our results are in agreement with studies by other groups; such as those by Mokyr et al (1998), who showed that low-dose melphalan can be effectively combined with anti-CTLA-4 therapy, and of Lesterhuis et al (2013), who recently reported that anti-CTLA-4 can be co-administered with gemcitabine to produce significant anti-tumour responses. Similarly, Jure-Kunkel et al (2013) recently showed the effective combination of anti-CTLA-4 plus chemotherapy, involving the injection of the anti-CTLA-4 antibody one day after the administration of chemotherapy (including gemcitabine). We had not initially considered such a regimen, because of our disappointing results with the B+ldCTX. In contrast to the aforementioned studies, one evident difference in our work is the inclusion of tumour variants selected in vivo for resistance to alkylating agents (Teicher et al, 1990), which we have previously used to study mechanisms of tumour drug resistance (Francia et al, 2004). These variants can be used to model the clinical situation where patients are eligible for immunotherapy following tumour relapses under standard chemotherapy regimens. In such cases, the response to immunotherapy might differ from that of chemotherapy-naive tumours. In that regard, the EMT-6/CTX and EMT-6/DDP variants produced less pronounced therapeutic benefits than the EMT-6/P tumour, a result that highlights how the sole use of chemotherapy naive tumours may exaggerate the potential preclinical benefit of a therapy. That is analogous to our observation that preclinical primary tumour models may in some cases exaggerate the impact of a therapy on the more clinically relevant metastatic disease (Francia et al, 2011). In that respect, a number of mice in this study eventually succumbed to spontaneous metastases. Overall, results obtained in this study show that although chemotherapy can augment the impact of anti-CTLA-4 therapy, caution is necessary in the design of such combinations, as some may be counterproductive.
Change history
31 January 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andre N, Carre M, Pasquier E (2014) Metronomics: towards personalized chemotherapy? Nat Rev Clin Oncol 11: 413–431.
Bocci G, Fioravanti A, Orlandi P, Di Desidero T, Natale G, Fanelli G, Viacava P, Naccarato AG, Francia G, Danesi R (2012) Metronomic ceramide analogs inhibit angiogenesis in pancreatic cancer through up-regulation of caveolin-1 and thrombospondin-1 and down-regulation of cyclin D1. Neoplasia 14: 833–845.
Bocci G, Francia G, Man S, Lawler J, Kerbel RS (2003) Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci USA 100: 12917–12922.
Chatterjee D, Liu C. J.-T., Northey D, Teicher BA (1995) Molecular characterization of the in vivo alkylating agent resistant murine EMT-6 mammary carcinoma tumors. Cancer Chemother Pharmacol 35: 423–431.
Chen CA, Ho CM, Chang MC, Sun WZ, Chen YL, Chiang YC, Syu MH, Hsieh CY, Cheng WF (2010) Metronomic chemotherapy enhances antitumor effects of cancer vaccine by depleting regulatory T lymphocytes and inhibiting tumor angiogenesis. Mol Ther 18: 1233–1243.
Chow A, Wong A, Francia G, Man S, Kerbel RS, Emmenegger U (2014) Preclinical analysis of resistance and cross-resistance to low-dose metronomic chemotherapy. Invest New Drugs 32: 47–59.
Colleoni M, Gray KP, Gelber S, Lang I, Thurlimann B, Gianni L, Abdi EA, Gomez HL, Linderholm BK, Puglisi F, Tondini C, Kralidis E, Eniu A, Cagossi K, Rauch D, Chirgwin J, Gelber RD, Regan MM, Coates AS, Price KN, Viale G, Goldhirsch A (2016) Low-dose oral cyclophosphamide and methotrexate maintenance for hormone receptor-negative early breast cancer: International Breast Cancer Study Group Trial 22-00. J Clin Oncol 34: 3400–3408.
Derosa L, Galli L, Orlandi P, Fioravanti A, di Desidero T, Fontana A, Antonuzzo A, Biasco E, Farnesi A, Marconcini R, Francia G, Danesi R, Falcone A, Bocci G (2014) Docetaxel plus oral metronomic cyclophosphamide: a phase II study with pharmacodynamic and pharmacogenetic analyses in castration-resistant prostate cancer patients. Cancer 120: 3923–3931.
du Manoir JM, Francia G, Man S, Mossoba M, Medin JA, Viloria-Petit A, Hicklin DJ, Emmenegger U, Kerbel RS (2006) Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res 12: 904–916.
Emadi A, Jones RJ, Brodsky RA (2009) Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol 6: 638–647.
Emmenegger U, Francia G, Chow A, Shaked Y, Kouri A, Man S, Kerbel RS (2011) Tumors that acquire resistance to low-dose metronomic cyclophosphamide retain sensitivity to maximum tolerated dose cyclophosphamide. Neoplasia 13: 40–48.
Emmenegger U, Shaked Y, Man S, Bocci G, Spasojevic I, Francia G, Kouri A, Coke R, Cruz-Munoz W, Ludeman SM, Colvin OM, Kerbel RS (2007) Pharmacodynamic and pharmacokinetic study of chronic low-dose metronomic cyclophosphamide therapy in mice. Mol Cancer Ther 6: 2280–2289.
Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS (2011) Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer 11: 135–141.
Francia G, Emmenegger U, Lee CR, Shaked Y, Folkins C, Mossoba M, Medin JA, Man S, Zhu Z, Witte L, Kerbel RS (2008) Long-term progression and therapeutic response of visceral metastatic disease non-invasively monitored in mouse urine using beta-human choriogonadotropin secreting tumor cell lines. Mol Cancer Ther 7: 3452–3459.
Francia G, Man S, Lee CJ, Lee CR, Xu P, Mossoba ME, Emmenegger U, Medin JA, Kerbel RS (2009) Comparative impact of trastuzumab and cyclophosphamide on HER-2-positive human breast cancer xenografts. Clin Cancer Res 15: 6358–6366.
Francia G, Man S, Teicher B, Grasso L, Kerbel RS (2004) Gene expression analysis of tumor spheroids reveals a role for suppressed DNA mismatch repair in multicellular resistance to alkylating agents. Mol Cell Biol 24: 6837–6849.
Francia G, Shaked Y, Hashimoto K, Sun J, Yin M, Cesta C, Xu P, Man S, Hackl C, Stewart J, Uhlik M, Dantzig AH, Foster FS, Kerbel RS (2012) Low-dose metronomic oral dosing of a prodrug of gemcitabine (LY2334737) causes antitumor effects in the absence of inhibition of systemic vasculogenesis. Mol Cancer Ther 11: 680–689.
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G (2012) The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov 11: 215–233.
Gangadhar TC, Vonderheide RH (2014) Mitigating the toxic effects of anticancer immunotherapy. Nat Rev Clin Oncol 11: 91–99.
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B (2007) Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 56: 641–648.
Grosso JF, Jure-Kunkel MN (2013) CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun 13: 5.
Hackl C, Man S, Francia G, Milsom C, Xu P, Kerbel RS (2013) Metronomic oral topotecan prolongs survival and reduces liver metastasis in improved preclinical orthotopic and adjuvant therapy colon cancer models. Gut 62: 259–271.
Hodi FS, O’Day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, Van Den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–723.
Jure-Kunkel M, Masters G, Girit E, Dito G, Lee F, Hunt JT, Humphrey R (2013) Synergy between chemotherapeutic agents and CTLA-4 blockade in preclinical tumor models. Cancer Immunol Immunother 62: 1533–1545.
Kerbel RS, Kamen BA (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4: 423–436.
Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, Harken JA, Robinson BW, Lake RA (2013) Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One 8: e61895.
Li Z, Chen L, Rubinstein MP (2013) Cancer immunotherapy: are we there yet? Exp Hematol Oncol 2: 33.
Lien K, Georgsdottir S, Sivanathan L, Chan K, Emmenegger U (2013) Low-dose metronomic chemotherapy: a systematic literature analysis. Eur J Cancer 49: 3387–3395.
Lin CJ, Lim KH, Cheng YC, Chen HH, Wu CJ (2007) Tumor lysis syndrome after treatment with gemcitabine for metastatic transitional cell carcinoma. Med Oncol 24: 455–457.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359: 378–390.
Maker AV, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Hughes M, Yellin MJ, Haworth LR, Levy C, Allen T, Mavroukakis SA, Attia P, Rosenberg SA (2006) Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother 29: 455–463.
Man S, Bocci G, Francia G, Green SK, Jothy S, Hanahan D, Bohlen P, Hicklin DJ, Bergers G, Kerbel RS (2002) Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res 62: 2731–2735.
Mokyr MB, Kalinichenko T, Gorelik L, Bluestone JA (1998) Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemotherapy-treated tumor-bearing mice. Cancer Res 58: 5301–5304.
Orlandi P, Fontana A, Fioravanti A, di Desidero T, Galli L, Derosa L, Canu B, Marconcini R, Biasco E, Solini A, Francia G, Danesi R, Falcone A, Bocci G (2013) VEGF-A polymorphisms predict progression-free survival among advanced castration-resistant prostate cancer patients treated with metronomic cyclophosphamide. Br J Cancer 109: 957–964.
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12: 252–264.
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7: 455–465.
Postow MA, Callahan MK, Wolchok JD (2015) Immune checkpoint blockade in cancer therapy. J Clin Oncol 33: 1974–1982.
Shaked Y, Emmenegger U, Francia G, Chen L, Lee CR, Man S, Paraghamian A, Ben-David Y, Kerbel RS (2005) Low-dose metronomic combined with intermittent bolus-dose cyclophosphamide is an effective long-term chemotherapy treatment strategy. Cancer Res 65: 7045–7051.
Simkens LH, Van Tinteren H, May A, Ten Tije AJ, Creemers GJ, Loosveld OJ, de Jongh FE, Erdkamp FL, Erjavec Z, Van Der Torren AM, Tol J, Braun HJ, Nieboer P, Van Der Hoeven JJ, Haasjes JG, Jansen RL, Wals J, Cats A, Derleyn VA, Honkoop AH, Mol L, Punt CJ, Koopman M (2015) Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): a phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 385: 1843–1852.
Tang TC, Man S, Xu P, Francia G, Hashimoto K, Emmenegger U, Kerbel RS (2010) Development of a resistance-like phenotype to sorafenib by human hepatocellular carcinoma cells is reversible and can be delayed by metronomic UFT chemotherapy. Neoplasia 12: 928–940.
Teicher BA (1997) Preclinical models for high-dose therapy. In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval, Teicher BA (ed). Humana Press: Totowa, NJ, USA.
Teicher BA, Herman TS, Holden SA, Wang YY, Pfeffer MR, Crawford JW, Frei E 3rd (1990) Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science 247: 1457–1461.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515: 568–571.
Acknowledgements
We thank Cynthia M. Rodriguez and Irving Miramontes for their technical assistance. GF was supported by an NIH-NCI SC2CA211029 grant, a BBRC Pilot grant, and by the National Institute on Minority Health and Health Disparities of the NIH (via RTRN CEA and RTRN Pilots grants) under Award Number U54MD008149, as well as RISE (to KP and NL) and MARC (to AG) Scholarship Awards.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Parra, K., Valenzuela, P., Lerma, N. et al. Impact of CTLA-4 blockade in conjunction with metronomic chemotherapy on preclinical breast cancer growth. Br J Cancer 116, 324–334 (2017). https://doi.org/10.1038/bjc.2016.429
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.429
Keywords
This article is cited by
-
Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier
Cancer Cell International (2022)
-
AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway
Molecular Cancer (2021)
-
Pharmacodynamic biomarkers in metronomic chemotherapy: multiplex cytokine measurements in gastrointestinal cancer patients
Clinical and Experimental Medicine (2021)
-
Immunostimulatory and anti-tumor metronomic cyclophosphamide regimens assessed in primary orthotopic and metastatic murine breast cancer
npj Breast Cancer (2020)
-
Prognostic significance of tumor poliovirus receptor and CTLA4 expression in patients with surgically resected non-small-cell lung cancer
Journal of Cancer Research and Clinical Oncology (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}