
Abstract
Background:
Evidences have shown that the RAS signalling pathway plays an important role in colorectal cancer (CRC). Moreover, RAS-GTPase-activating proteins (RASGAPs) as RAS signalling terminators are associated with tumourigenicity and tumour progression. In this study, we used bioinformatics analysis to predict and study important miRNAs that could target RAS p21 GTPase-activating protein 1 (RASA1), an important member of RASGAPs.
Methods:
The levels of RASA1 and miR-223 were analysed by real-time PCR, western blotting or in situ immunofluorescence analyses. The functional effects of miR-223 and the effects of miR-223-targeted inhibitors were examined in vivo using established assays.
Results:
Upregulation of miR-223 was detected in CRC tissues (P<0.01) and was involved in downregulation of RASA1 in CRC tissues. Furthermore, the direct inhibition of RASA1 translation by miR-223 and the activation of miR-223 by CCAAT/enhancer binding protein-β (C/EBP-β) were evaluated in CRC cells. An in vivo xenograft model of CRC suggested that the upregulation of miR-223 could promote tumour growth and that the inhibition of miR-223 might prevent solid tumour growth.
Conclusions:
These results identify that C/EBP-β-activated miR-223 contributes to tumour growth by targeting RASA1 in CRC and miR-223-targeted inhibitors may have clinical promise for CRC treatment via suppression of miR-223.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is one of the most common malignant tumours worldwide (Jemal et al, 2011). Although new and precise molecular mechanisms of CRC are being discovered, it is generally accepted that the development of CRC involves a multistep process involving the accumulation of both genetic and epigenetic changes (Wong et al, 2007; Grady and Carethers, 2008). Evidences indicate that the RAS signalling pathway plays an important role in CRC development, progression and resistance to treatment (Haigis et al, 2008; Jimeno et al, 2009). RAS can serve as a molecular microswitch of the RAS signalling pathway to control cellular functions, whose activity is regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) (Bos et al, 2007). Moreover, RAS-GTPase-activating proteins (RASGAPs) as RAS signalling terminators have been reported to be associated with tumourigenicity and tumour progression (Kolfschoten et al, 2005; Ohta et al, 2009; McLaughlin et al, 2013). As an important member of RASGAPs, RAS p21 GTPase-activating protein 1 (RASA1) contains two SH2, SH3, PH and CaLB/C2 domains in N-terminal part. These protein domains allow various functions, such as anti-/pro-apoptosis, proliferation and cell migration, depending on their distinct partners in angiogenesis and cancer (Pamonsinlapatham et al, 2009). Lapinski et al (2012) revealed that RASA1 could maintain the lymphatic vasculature in a quiescent functional state through its ability to inhibit RAS signal transduction induced by growth factor receptors such as VEGFR-3 in mice. RASA1 gene mutations are associated with capillary malformation–arteriovenous malformations and lymphatic abnormalities in patients (Boon et al, 2005; Burrows et al, 2013). Nevertheless, the role of RASA1 in CRC is rarely reported.
MicroRNAs (miRNAs) have raised considerable attention for their important roles in numerous cellular and developmental processes (Bushati and Cohen, 2007). In particular, miRNAs are key players in human tumours, acting as oncogenes and tumour suppressors (Calin and Croce, 2006). Emerging evidences suggest that a variety of miRNAs are aberrantly expressed in the serum, plasma and tissues of CRC patients, indicating that miRNAs may have vital roles in CRC (Schetter et al, 2008; Ng et al, 2009; Wang and Gu, 2012). The biological functions of these aberrantly expressed miRNAs involving the RAS signalling pathway in CRC have not been extensively studied (Akao et al, 2006; Chen et al, 2009; Tsang and Kwok, 2009). Anand et al (2010) reported that miR-132 served as an angiogenic microswitch predominantly through targeting RASA1, leading to neovascularisation in an orthotopic xenograft mouse model of human breast carcinoma. Sun et al (2013) revealed that miR-31 functioned as an oncogenic miRNA to activate the RAS/MAPK pathway by repressing RASA1 in CRC. However, it is unknown whether other miRNAs could target RASA1 in CRC.
The CCAAT/enhancer binding protein-β (C/EBP-β) is a transcription factor that can either activate or repress transcription (Descombes and Schibler, 1991). Evidences suggest that it plays a critical role in various malignancies, including glioma, pancreatic tumour and skin tumourigenesis (Homma et al, 2006; Sterneck et al, 2006; Shimizu et al, 2007). The C/EBP-β was found to overexpress and induce Cox-2 gene expression in gastric carcinogenesis (Regalo et al, 2006). In addition, Fukao et al (2007) discovered that there was a C/EBP-β-binding element in pri-miR-223 promoter. Recent studies showed that C/EBP-β could respectively regulate let-7i following microbial infection and miR-145 in cancer cells (O'Hara et al, 2010; Sachdeva et al, 2012). Although Rask et al (2000) reported that C/EBP-β markedly increased in all CRCs compared with normal colon mucosa, it is not clear whether C/EBP-β could regulate miRNAs in CRC.
In this study, we used bioinformatics analysis to predict and find six important miRNAs that could target RASA1 by binding the 3′-UTR of RASA1. The results of in situ immunofluorescence analysis and western blotting analysis highlighted that miR-223 and RASA1 demonstrated an inverse correlation in CRC patient tissues. In addition, in vitro studies on the direct inhibition of RASA1 by miR-223, the activation mechanism of miR-223 by C/EBP-β in CRC and the potential role of miR-223 to promote colorectal cell proliferation were experimentally investigated. The influence of miR-223 on CRC was further studied in an immunodeficient mouse xenograft tumour model by over- or down-expression of miR-223.
Materials and methods
Clinical samples, cell lines and chemical reagents
Paired CRC and adjacent, nontumour tissue (NAT) samples were obtained from patients who underwent radical resection at Jinling Hospital (Nanjing, Jiangsu, China) from 2011 to 2013. Ethical approval was obtained from the local ethics committee. A total of 24 patients were randomly chosen and numbered for this study. The information of patients is shown in Table 1 and Supplementary Table 1. Caco-2 cells and HT-29 cells were cultured as described previously (Sun et al, 2013). All chemical reagents were purchased from Sigma-Aldrich (St Louis, MO, USA).
Assay to detect mutant K-RAS
Paraffin-embedded tumour sections were deparaffinised and air dried, and DNA was isolated using proteinase K and a DNeasy minispin column (Qiagen, Valencia, CA, USA). Then, K-RAS point mutations in codon 12 and codon 13, two hot spots that include >95% of mutations in this gene, were detected as previously reported (Pinto et al, 2011).
RNA extraction and quantitative real-time PCR (qRT–PCR) assay
RNA was isolated from tissues and cells and made into complementary DNA using methods previously described (Sun et al, 2013). The qRT–PCR for miRNAs and mRNA was performed according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA) using the specific primers listed in Supplementary Table 2. The qRT–PCR data for miRNA and mRNA are expressed relative to U6 snRNA or β-actin mRNA, respectively.
Histology and in situ immunofluorescence staining assays
The CRC and NAT samples were fixed in 4% (wt/vol) paraformaldehyde. After paraffin embedding, 5-μm sections were cut and stained with haematoxylin and eosin (H&E; Lerner, New Haven, CT, USA) for histopathological examination. For miRNA in situ hybridisation, a 5′-Cy5-conjugated miR-223 probe (Exiqon, Vedbaek, Denmark) was used as previously described (de Planell-Saguer et al, 2010)
Protein extraction and analysis
Western blotting was carried out as previously described (Sun et al, 2013). Antibodies included: RASA1 (1 : 200 dilution, Santa Cruz, Santa Cruz, CA, USA), RAS-GTP (1 : 200 dilution, Thermo Scientific, Hudson, NH, USA), total RAS (1 : 1000 dilution, BD Biosciences, San Jose, CA, USA), p-MEK1/2, total MEK1/2, p-ERK1/2 or total ERK1/2 (1 : 1000 dilution, Cell Signaling Technologies, Danvers, MA, USA). We performed the normalisation by blotting the same membranes with an antibody against GAPDH (1 : 10 000 dilution, KangChen Bio-tech, Shanghai, China).
Cell transfection/infection assays
Caco-2 or HT-29 cells were transfected with precursor oligonucleotides (pre-miR-223), antisense oligonucleotides (anti-miR-223) (Ambion, Austin, TX, USA) or their corresponding controls (pre-ncRNA or anti-ncRNA) using the Lipofectamine 2000 transfection reagent (Life Technologies) according to the manufacturer’s instructions. To obtain HT-29 cells stably expressing miR-223 or ncRNA, HT-29 cells were infected with recombinant lentiviruses respectively carrying pre-miR-223 precursor (pre-miR-223-LV) or pre-non-coding sequence (pre-NC-LV) (GenePharma, Shanghai, China) in the presence of 8 mg ml−1 polybrene (GenePharma) for 12 h, and the medium was then replaced with fresh medium including 10 μg ml−1 puromycin. The cells stably expressing miR-223 were sorted by a FACS (fluorescent-activated cell sorter, Becton Dickinson, Mountain View, CA, USA) based on the expression of GFP carried by pGLV3 plasmid. The sorted cells were taken as the overexpression model for the following experiments.
Plasmid constructs and Luciferase reporter assay
The RASA1 luciferase reporter plasmid was constructed as previously described (Sun et al, 2013). This construct was co-transfected with pre/anti-miR-223 into Caco-2 cells. The binding-site mutant luciferase plasmid (binding site: AACTGAC replaced by TTGACTG) was also transfected as a control.
The full-length human C/EBP-β expression plasmid (pcDNA3.1-C/EBP-β) was obtained from GeneCopoeia (Rockville, MD, USA). The C/EBP-β-responsive sequence in the pri-miR-223 promoter (Fukao et al, 2007) was cloned into the MluI/BgII sites in the pGL3-promoter vector (E1761; Promega, San Luis Obispo, CA, USA) with a luciferase gene, creating pGL3 miR-223−198. The PGL3 miR-223−198 was then co-transfected with pcDNA3.1-C/EBP-β or C/EBP-β siRNA into Caco-2 cells, and the cells were lysed to measure the luciferase activity 24 h later. A plasmid encoding β-gal was co-transfected and used for normalisation.
Cell proliferation assay
The cell proliferation assay was performed as previously described (Sun et al, 2013). Briefly, the number of Caco-2 cells with over- or down-expression of miR-223 was determined using WST-8 staining with a Cell Counting Kit-8 (CCK-8, Dojindo, Tokyo, Japan) according to the manufacturer’s instructions. Moreover, the growth of Caco-2 cells at 48 h post transfection in the presence or absence of 10 μ M U0126 (MEK1/2 inhibitor) was also assessed using the proliferation index KI-67 and PCNA.
The growth of human colorectal tumour in an immunodeficient mouse xenograft model
The 4-week-old, thymic BALB/c male, nude (nu/nu) mice were obtained from Model Animal Research Center of Nanjing University (Nanjing, Jiangsu, China) and maintained under pathogen-limited conditions. All animals received care according to Chinese legal requirements and the animals were equally divided into two groups (seven mice per group). The HT-29 cells that stably overexpressed miR-223 (pre-miR-223-LV) or ncRNA (pre-ncRNA-LV) were subcutaneously injected into their left flanks with the viable cell number of 1 × 107. After subcutaneous (s.c.) implantation of cells, the animals were observed daily for tumour growth as described previously (Sun et al, 2013).
Therapeutic effect of a miR-223 inhibitor on an immunodeficient mouse xenograft model
The miR-223 AMOs with full phosphorothioate linkage were designed and synthesised by SBS Genetech Co., Ltd (Shanghai, China), and the most efficient one (5′-GGGGTATTTGACAAACTGAC-3′) was selected. Polyetherimide (PEI, 25 kDa)/miR-223 AMO complexes were prepared as already reported (Huang et al, 2014).
Xenografts were initiated by s.c. injection of 1 × 107 HT-29 cells into the right flanks of nude mice. After 3 weeks, the tumours were aseptically dissected and mechanically minced; and 1 mm × 1 mm diameter tumour pieces were transplanted subcutaneously into the nude mice using a trocar needle. When tumours had grown to a volume of ∼100 mm3 on day 10 after tumour transplantation, the mice were randomised into specific treatment group, control treatment group and nontreatment group (7 mice per group). The 10 nmol PEI/miR-223 AMO complexes, PEI/scramble DNA complexes or PEI alone were injected directly into the tumours every 3 days beginning on day 10 after tumour transplantation. Tumour volumes were monitored every 3 days as indicated in the figures and, upon termination of the experiment, mice were killed and tumours removed. Pieces of each tumour were immediately snap frozen for RNA preparation.
Statistical analysis
All experiments were conducted in triplicate and repeated at least three independent times. Data are expressed as the mean±s.e.m. The difference among groups was determined using ANOVA analysis, and comparison between two groups was analysed using Student’s t-test and GraphPad Prism software version 4.0 (GraphPad Software, Inc., San Diego, CA, USA). A value of P<0.05 was considered statistically significant.
Results
Patient characteristics and detection of K-RAS mutations by direct sequencing
Table 1 summarises patient demographic data and CRC characteristics. The cohort in the study consists of 24 CRC samples, including stage T1, T2, T3 and T4 patients. The average patient age is 56.5 years (range, 29–72) and most patients (79.2%) have well or moderately differentiated cancers. Among the 24 patients, 10 patients harbour a somatic K-RAS mutation (41.7%) and of the total K-RAS mutations, 80.0% and 20.0% were observed in codon 12 and codon 13, respectively, that is consistent with the previous report (Samowitz et al, 2000) (for patient information, refer to Supplementary Table 1).
MiR-223 is upregulated in human CRC
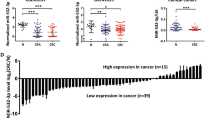
We performed bioinformatics analysis to search for RASA1-targeted miRNAs. Targetscan, Miranda, PICTAR5 and PITA were independently used to predict the miRNAs. Combination of these bioinformatics prediction methods revealed that six important miRNAs could target RASA1, including miR-24, miR-132, miR-223, miR-182, miR-31 and miR-335. Therefore, we performed qRT–PCR assay on 24 pairs of CRC tissues and NAT to quantify the expressions of these six miRNAs. As shown in Figure 1A, qRT–PCR analysis revealed that the levels of miR-223 and miR-31 in CRC samples significantly increased (P<0.01); the levels of miR-335 and miR-182 in CRC samples were enhanced (P<0.05); but no significant changes were observed in the levels of miR-24 and miR-132 between CRC and NAT samples (P>0.05). Because miR-31 was found to act as an oncogenic miRNA in CRC in our previous report (Sun et al, 2013), we hypothesised that miR-223, 65 times higher than miR-31 in NAT samples as shown in Figure 1B, might play a significant role in CRC. Interestingly, miR-223 was upregulated in 24 CRC samples whereas 10 CRC samples were K-RAS mutated tumours, suggesting that miR-223 could be a broader mechanism for CRC than for K-RAS mutated tumours. The action of miR-223 in CRC was thereby investigated in the following.

Expression analysis of six RASA1-targeted miRNAs in paired CRC and NAT samples. (A) The levels of six miRNAs tested using qRT–PCR assay and shown as scatter plots. (B) The levels of six miRNAs tested using qRT–PCR assay and shown as histograms. Values are expressed as the means±s.e. *P<0.05. **P<0.01. NS (P>0.05)=no significant change.
Identification of RASA1 as a target of miR-223
The correlation between miR-223 and RASA1 was further examined by using in situ immunofluorescence staining, qRT–PCR and western blotting assays in patient samples from sample 1 to sample 12 in Supplementary Table 1. As shown in Figure 2A, following the examination of H&E staining for sample 1, the immunofluorescence staining demonstrated that miR-223 was upregulated whereas the RASA1 protein was downregulated in CRC sample 1 compared with NAT sample 1, suggesting an inverse correlation between miR-223 and the RASA1 in sample 1. The similar results of the immunofluorescence staining were observed in other 11 samples (figures not shown). Consistently, the overall level of miR-223 was increased by 6.85-fold in CRC samples compared with NAT samples in qRT–PCR analysis (Figure 2B), whereas the overall level of RASA1 in CRC samples was 64.9% lower than that in NAT samples (Figure 2C).

Inverse correlations between RASA1 and miR-223 in paired CRC and NAT samples. (A) Representative photos of three individual experiments of H&E staining and in situ immunofluorescence staining for miR-223 and RASA1 for sample 1 (red, miR-223; green, RASA1; blue, DAPI nuclear staining). Pictures were imaged at × 40 magnification on a Nikon confocal microscope. Scale bar, 25 μm. (B) The level of miR-223 in CRC and NAT samples tested using qRT–PCR. The data shown are the means±s.e. of three individual experiments. (C) The level of RASA1 protein in CRC and NAT samples, as demonstrated using western blotting. The data shown are representative of three individual western blotting analyses. **P<0.01. A full colour version of this figure is available at the British Journal of Cancer journal online.
It was not clear whether miR-223 downregulated RASA1 by binding to the 3′-UTR of the RASA1 mRNA; therefore, additional experiments were carried out in CRC cells. As Figure 3A shows, when compared with mock experiments using the luciferase reporter assay, overexpression of miR-223 exerted a repressive effect with ∼53.9% reduction in reporter activity. Meanwhile, inhibition of miR-223 resulted in an 18.6% increase in reporter activity compared with the control, and mutation of the miR-223-binding site abolished the above effects. Next, we used western blotting to examine the RASA1 protein level after over- or down-expression of miR-223. As Figure 3B shows, compared with the control, overexpression of miR-223 resulted in a significant decrease in RASA1, whereas downexpression of miR-223 led to an increase in RASA1. However, in contrast with the variation of RASA1 protein, the level of RASA1 mRNA was not repressed by over- or down-expression of miR-223, as shown in Figure 3C. These results demonstrated that miR-223 regulated RASA1 expression only via a translational repression mechanism, rather than by affecting its mRNA stability. Taken together, these results proved that miR-223 might negatively regulate RASA1 in CRC.

Identification of miR-223-targeting RASA1 and activation mechanism of miR-223 in CRC cells. (A) The relative luciferase activity after transfection with pre/anti-miR-223 or the corresponding control in Caco-2 cells. The data shown are the means±s.e. of three individual experiments. (B) Western blotting analysis of RASA1 in whole-cell lysates from CRC cells transfected with pre/anti-miR-223 or the corresponding control. Left panel: Caco-2 cells; right panel: HT-29 cells. The data shown are representative of three individual western blotting analyses. (C) The qRT–PCR analysis of RASA1 mRNA in total RNA from CRC cells transfected with pre/anti-miR-223 or the corresponding control. Left panel: Caco-2 cells; right panel: HT-29 cells. The data shown are the means±s.e. of three individual experiments. (D) The qRT–PCR analysis of C/EBP-β mRNA in total RNA from CRC and NAT samples. The data shown are the means±s.e. of three individual experiments. (E) A schematic description of the luciferase reporter construct: pGL3 miR-223−198. The conserved genomic region in pri-miR-223 promoter for C/EBP-β binding is also shown. (F) Effect of C/EBP-β on the promoter of miR-223. Caco-2 cells were co-transfected with pGL3 miR-223−198 and pcDNA 3.1-C/EBP-β or C/EBP-β siRNA. After 24 h, the reporter activity was measured using luciferase assay. The data shown are the means±s.e. of three individual experiments. (G) The qRT–PCR analysis of miR-223 in total RNA from Caco-2 cells transfected with pcDNA 3.1-C/EBP-β or C/EBP-β siRNA at 48 h post transfection. The data shown are the means±s.e. of three individual experiments. (H) Western blotting analysis of RASA1 in whole-cell lysates from Caco-2 cells transfected with pcDNA 3.1-C/EBP-β or C/EBP-β siRNA at 48 h post transfection. The data shown are representative of three individual western blotting analyses. **P<0.01. NS=no significant change.
C/EBP-β activates miR-223 expression in CRC
In CRC, the expression of miR-223 was upregulated. However, the reason for this is not clear yet. Therefore, we searched for factors that might be responsible for the regulation of miR-223 expression in CRC. As shown in Figure 3D, we discovered that the C/EBP-β mRNAs were significantly upregulated in 24 CRC samples, indicating a possible positive correlation between miR-223 and C/EBP-β. To further study whether miR-223 could be modulated by C/EBP-β, we generated a miR-223 promoter reporter plasmid carrying luciferase: pGL3 miR-223−198 (Figure 3E). As shown in Figure 3F, the overexpression of C/EBP-β resulted in a 4.8-fold increase in pGL3 miR-223−198 luciferase reporter activity over the control, whereas the inhibition of C/EBP-β with C/EBP-β siRNA resulted in a 62.4% reduction in luciferase activity compared with the control. However, pGL3 basic vector abolished these effects. Consistent with these results, qRT–PCR assay detected an increase of miR-223 level in the cells transfected with pcDNA3.1-C/EBP-β and a significant reduction of miR-223 level in the cells transfected with C/EBP-β siRNA (Figure 3G). In addition, the level of RASA1 protein also obviously changed after over- or down-expression of C/EBP-β (Figure 3H). These results demonstrated that C/EBP-β might be a positive regulator of miR-223, thereby resulting in a significant reduction of RASA1.
MiR-223 regulates the RAS/MAPK pathway and influences cell proliferation in CRC cells
To study the effect of miR-223 targeting RASA1, we detected the expression of RAS-GTP and total RAS, p-MEK1/2 and total MEK1/2 and p-ERK1/2 and total ERK1/2 using western blotting assay after transfection with pre/anti-miR-223 or the corresponding control. As shown in Figure 4A and B, miR-223 had no obvious effect on the expressions of total RAS, total MEK1/2 or total ERK1/2; however, it strengthened the ability of RAS to bind to GTP and enhanced phosphorylation of the MEK1/2 and ERK1/2 proteins. Overexpression of miR-223 led to a 2.39-fold increase in p-ERK1/2, and downexpression of miR-223 decreased p-ERK1/2 expression by 70.4%. In addition, as shown in Figure 4B, the expression of p-ERK1/2 was not significantly increased or reduced compared with the control after Caco-2 cells were transfected with pre-miR-223 or anti-miR-223 in the presence of U0126, a MEK1/2 inhibitor. In a word, overexpression of miR-223 significantly suppressed RASA1, increased the level of RAS-GTP and activated the MAPK signalling pathway.

The miR-223 regulates the RAS/MAPK pathway and influences CRC cell proliferation. (A) Whole-cell lysates from Caco-2 cells transfected for 48 h with pre/anti-miR-223 or the corresponding control were subjected to western blotting analysis of RAS-GTP, total RAS, p-MEK1/2, total MEK1/2 and GAPDH (as a loading control). The data shown are representative of three individual western blotting analyses. (B, upper panel) Whole-cell lysates from Caco-2 cells transfected for 48 h with pre/anti-miR-223 or the corresponding control were subjected to western blotting analysis of p-ERK1/2, total ERK1/2 and GAPDH (as a loading control) without 10 μ M U0126. (B, lower panel) Whole-cell lysates from Caco-2 cells transfected for 48 h with pre/anti-miR-223 or the corresponding control were subjected to western blotting analysis of p-ERK1/2, total ERK1/2 and GAPDH (as a loading control) with 10 μ M U0126. The data shown are representative of three individual western blotting analyses. (C) Cell proliferation of Caco-2 transfected with pre/anti-miR-223 or the corresponding control was measured using CCK-8. The data shown are the means±s.e. of three individual experiments. (D) Total RNA from Caco-2 cells transfected for 48 h with pre/anti-miR-223 or the corresponding control were subjected to qRT–PCR analysis of KI-67 and PCNA without or with 10 μ M U0126. The data shown are the means±s.e. of three individual experiments. **P<0.01. NS=no significant change.
Cell proliferation was further monitored and analysed using the CCK-8 kit. Statistical analysis showed that overexpression of miR-223 significantly increased the proliferation of Caco-2 cells, whereas downexpression of miR-223 obviously inhibited the proliferation of Caco-2 cells (Figure 4C). The qRT–PCR results showed that mRNA levels of KI-67 and PCNA were increased when miR-223 was overexpressed, whereas downexpression of miR-223 reduced mRNA levels of KI-67 and PCNA. However, when Caco-2 cells were treated with U0126 after transfection with pre/anti-miR-223 or the corresponding control, neither overexpression of miR-223 nor downexpression of miR-223 influenced mRNA levels of KI-67 and PCNA (Figure 4D). Based on the results above, we summarised that overexpression of miR-223 promoted CRC cell proliferation by activating the RAS/MAPK signalling pathway.
The effect of miR-223 on colorectal tumour growth in vivo
Taking into account that miR-223 was upregulated in 24 CRC samples and in order to examine the effect of miR-223 on tumour growth in vivo, we successfully constructed HT-29 cells stably overexpressing miR-223. The sorted HT-29 cells were observed based on the expression of GFP as shown in Figure 5A, indicating that the cells were successfully infected by pre-miR-223-LV or pre-ncRNA-LV. As shown in Figure 5B, the level of miR-223 in HT-29 cells infected with pre-miR-223-LV was 15.2-fold of the control that resulted in a significant decrease of cellular RASA1 and suggested that the sorted pre-miR-223-LV-infected HT-29 cells could be used as stably overexpressing miR-223 cells in the immunodeficient mouse xenograft tumour model. As shown in Figure 5C, the tumour volumes from pre-miR-223-LV group grew more rapidly than those from pre-ncRNA-LV group, and the excised tumours from pre-miR-223-LV group were 51.2% heavier than those from pre-ncRNA-LV group on day 34 after implantation (Figure 5D), suggesting that overexpression of miR-223 greatly stimulated the growth of HT-29-inducing tumours in vivo. Representative photographs of mice bearing the tumours and the corresponding tumours from each group are shown in Figure 5E and F.

Effect of miR-223 on the growth of colorectal tumour in vivo. (A) Representative photos of HT-29 cells infected with pre-miR-223-LV and pre-ncRNA-LV sorted by a FACS based on the GFP expression. Pictures were imaged at × 10 magnification on a Nikon confocal microscope. Scale bar, 100 μm. (B, upper panel) The qRT–PCR analysis of miR-223 from HT-29 cells stably expressing miR-223 or ncRNA. The data shown are the means±s.e. of three individual experiments. (B, lower panel) Western blotting analysis of RASA1 from HT-29 cells stably expressing miR-223 (pre-miR-223-LV group) or ncRNA (pre-ncRNA-LV group). The data shown are representative of three individual western blotting analyses. (C) The volumes of the xenograft tumours in nude mice derived from s.c. implantation of HT-29 cells stably expressing miR-223 (pre-miR-223-LV group) or ncRNA (pre-ncRNA-LV group). (D) The weights of the xenograft tumours in nude mice 34 days after s.c. implantation of HT-29 cells stably expressing miR-223 (pre-miR-223-LV group) or ncRNA (pre-ncRNA-LV group). (E) A representative photograph of mice bearing tumours 34 days post implantation. (F) A corresponding, representative photograph of excised tumours 34 days post implantation. Values are expressed as the means±s.e. **P<0.01.
Therapeutic effect of a miR-223 inhibitor in vivo
In order to attempt to analyse miR-223 as a therapeutic target in CRC, we designed a miR-223 AMO to inhibit miR-223 expression in the nude mouse xenograft model of human CRC. As shown in Figure 6A and B, when HT-29 cells were transfected with miR-223 AMO, the level of miR-223 declined by 67.3% and the level of RASA1 was enhanced by ∼41.5% compared with the scramble DNA. To assess the antitumour activity of miR-223 AMO in vivo, nude mice with s.c xenograft tumours of ∼100 mm3 were treated by intratumoural administration of PEI/miR-223 AMO complexes, PEI/scramble DNA complexes or PEI alone. As shown in Figure 6C, the mice treated with PEI/miR-223 AMO complexes showed the obviously decreased tumour volumes whereas the tumour volumes of mice, among PEI/scramble DNA complexes, PEI alone and control (nontreatment) groups, were not significantly different. Similarly, the tumour weights of PEI/miR-223 AMO complexes-treated mice were 47.2% less than those from other three groups (Figure 6D). Representative photographs of mice bearing the tumours and the corresponding tumours from each group are displayed in Figure 6E and F. In addition, qRT–PCR results showed that the levels of miR-223, KI-67 and PCNA in tumours had a significant decline after the xenografts were treated with PEI/miR-223 AMO complexes, as shown in Figure 6G and H, suggesting miR-223 AMO could be a potentially effective drug against human CRC.

Antitumour activity of miR-223 AMO in vivo. (A) The qRT–PCR analysis of miR-223 from HT-29 cells transfected with miR-223 AMO or scramble DNA for 48 h. The data shown are the means±s.e. of three individual experiments. (B) Western blotting analysis of RASA1 from HT-29 cells transfected with miR-223 AMO or scramble DNA for 48 h. The data shown are representative of three individual western blotting analyses. (C) The volumes of the xenograft tumours in nude mice from s.c. implantation of HT-29 cells injected with 10 nmol PEI/miR-223 AMO complexes, PEI/scramble DNA complexes, PEI alone or control at indicated time. (D) The weights of xenograft tumours in nude mice 31 days after s.c. implantation of HT-29 cells. (E) A representative photograph of mice bearing tumours 31 days after s.c. implantation of HT-29 cells. (F) Corresponding tumours dissected from mice 31 days after s.c. implantation of HT-29 cells. (G) The qRT–PCR analysis of miR-223 from xenograft tumours in nude mice. The data shown are the means±s.e. of three individual experiments. (H) The qRT–PCR analysis of KI-67 and PCNA from xenograft tumours in nude mice. The data shown are the means±s.e. of three individual experiments. **P<0.01.
Discussion
Because the tumour suppressor RASA1 and its downstream RAS signalling pathway play crucial roles in controlling cell growth, RASA1-targeted miRNA profiling analyses are very important for a better understanding of the miRNA regulation mechanism through the RAS pathway in CRC. In the present study, we used bioinformatics analyses to find six important miRNAs of miR-24, miR-132, miR-182, miR-335, miR-223 and miR-31 that could target RASA1 depending on the ‘seed region’ of the RASA1 3′-UTR, but these ‘seed regions’ for six miRNAs recognition were diverse, suggesting that these miRNAs may play similar or differing roles in CRC. Among the six miRNAs, the significantly upregulated expressions of miR-223 and miR-31 were found in CRC and miR-223 showed very high abundance relative to that of miR-31 in NAT samples. It was also reported that miR-223 was upregulated in larger series of CRC patients than 24 patients (Schetter et al, 2008; Zhang et al, 2014). Especially, Zhang et al (2014) reported that 48 CRC patients with stage T3 or stage T4 presented an even higher expression of miR-223 compared with 42 CRC patients with low-grade stages, indicating that upregulation of miR-223 may play a key role in CRC. The results of in situ immunofluorescence analysis and western blotting assay further highlighted the importance of miR-223 in the CRC transformation process. Moreover, emerging evidence shows that miR-223 is a plasma or serum biomarker in gastric cancer and nasopharyngeal carcinoma (Zeng et al, 2012; Li et al, 2012a), but so far the biological roles of miR-223 in solid tumours are complicated because it may function as an oncogene or as a tumour suppressor, probably associated with the varying pathogeneses of different types of cancers (Wong et al, 2008; Li et al, 2011a, 2011b, 2012b; Kurashige et al, 2012). Therefore, it is very necessary to investigate the role of miR-223 targeting RASA1 in CRC for a clearer view of its dynamic behaviour.
In the present study, we proved that miR-223 might stimulate CRC cell proliferation by targeting RASA1 and, therefore, activating the RAS/MAPK signalling pathway. These results were further supported by the findings that overexpression of miR-223 could promote tumour growth in nude mice. Obviously, miR-223 functions as an oncogenic miRNA by targeting RASA1 in CRC. The other targets of miR-223 have been reported to be FBXW7 in oesophageal squamous cell carcinoma and EPB41L3 in gastric cancer when miR-223 acts as an oncogenic miRNA in solid tumours (Li et al, 2011b; Kurashige et al, 2012). FBXW7 is another tumour suppressor and thereby negatively regulates oncoproteins, such as cyclin E, c-Myc, c-Jun and Notch in CRC (Akhoondi et al, 2007; Iwatsuki et al, 2010). FBXW7 was also predicted to affect colorectal cell proliferation probably mainly through the WNT and TGF-β pathways rather than the RAS/MAPK pathway (Cancer Genome Atlas Network, 2012). However, our results demonstrated that the use of a MEK1/2 inhibitor distinctly inhibited miR-223-induced cell proliferation, implying the importance of the RAS/MAPK pathway in CRC. As a possible target of miR-223 in CRC, EPB4IL3 expression was not detected in human CRC cells, although EPB4IL3 could maintain normal cellular proliferation and adhesion, thereby preventing malignant transformation of intestinal epithelial cells (Ohno et al, 2004). Taken together, upregulation of miR-223 promotes tumour growth in CRC predominantly by targeting RASA1 and activating the RAS/MAPK pathway.
Some transcription factors have been reported to regulate expression of miR-223. For example, C/EBP-α and PU.1 can upregulate the level of miR-223 during granulopoiesis (Fazi et al, 2005; Fukao et al, 2007). The metastasis-promoting transcription factor Twist can drive miR-223 transcription in gastric cancer (Li et al, 2011b). Cell-cycle regulator E2F1 can inhibit miR-223 transcription in acute myeloid leukemia (Pulikkan et al, 2010). Among miR-223-regulating transcription factors, C/EBP-β is paid great attention because the increased expression of C/EBP-β is correlated with invasiveness of human CRC (Rask et al, 2000). Moreover, the CRC cells display reduced tumourigenic potential when transplanted into the C/EBP-β-deficient animals, suggesting that C/EBP-β is involved in colorectal tumourigenesis (Staiger et al, 2009). Our findings in this study further indicate that C/EBP-β serves as a positive regulator for miR-223 expression and has a critical role in RASA1-mediating colorectal tumour growth. Moreover, Xi et al (2010) found that C/EBP-β could mediate the activation of miR-31 in normal human respiratory epithelia and lung cancer cells. The miR-31 was demonstrated to directly target RASA1 in CRC in our previous report. Therefore, we speculate that miR-31 may also be regulated by C/EBP-β and play the same role as miR-223 in CRC. Whether miR-223 and miR-31 work synergistically in CRC and whether the combination with miR-223 and miR-31 inhibitors result in superior reduction of RASA1 would be further studied in the future.
As deregulated cell proliferation is a significant mechanism for neoplastic progression, we speculate that therapies targeting miR-223 might be used to prevent or treat CRC in the future. Especially, miR-223 will implicate as an alternative target for tumour inhibition in CRC patients with a K-RAS mutation because upregulated miR-223 is found in almost all CRC patients with both wild-type K-RAS and K-RAS mutations. Additional findings on miRNA regulation of the gene expression networks of cancers imply the prospect of more therapies. Kota et al (2009) found that miR-26a was downregulated in a murine liver cancer model and that treatment with miR-26a overexpressing adenovirus through the tail vein inhibited the growth of hepatocellular carcinoma. In addition, the first cancer-targeted miRNA drug, a liposome-based miR-34 mimic, entered phase I clinical trials in patients with advanced hepatocellular carcinoma (Ling et al, 2013). In this study, our results revealed that intratumoural injection of PEI/miR-223 AMO complexes significantly inhibited the growth of colorectal tumours in nude mice because of a significant reduction of miR-223 level, suggesting an important role of miR-223 in CRC and providing a potential therapeutic tool that targets miR-223 for CRC treatment. Therefore, other types of delivery for miR-223 AMO, such as intraperitoneal delivery, can be further investigated for the clinical CRC therapy.
In conclusion, we studied six important miRNAs that could target RASA1 in CRC patients and identified that miR-223 was significantly upregulated and its expression was inversely correlated with RASA1 protein expression. Our findings further revealed that miR-223 activated by C/EBP-β could stimulate colorectal cell proliferation and tumour growth by targeting RASA1 and thereby activating the RAS/MAPK pathway in vitro and in vivo. The miR-223 AMO significantly inhibited the growth of colorectal tumours in nude mice, providing a promising approach that may be valuable for CRC treatment.
Change history
28 April 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Akao Y, Nakagawa Y, Naoe T (2006) Let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull 29 (5): 903–906.
Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C (2007) FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 67 (19): 9006–9012.
Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN, Lapinski PE, King PD, Weis SM, Cheresh DA (2010) MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med 16 (8): 909–U109.
Boon LM, Mulliken JB, Vikkula M (2005) RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev 15 (3): 265–269.
Bos JL, Rehmann H, Wittinghofer A (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129 (5): 865–877.
Burrows PE, Gonzalez-Garay ML, Rasmussen JC, Aldrich MB, Guilliod R, Maus EA, Fife CE, Kwon S, Lapinski PE, King PD, Sevick-Muraca EM (2013) Lymphatic abnormalities are associated with RASA1 gene mutations in mouse and man. Proc Natl Acad Sci USA 110 (21): 8621–8626.
Bushati N, Cohen SM (2007) MicroRNA functions. Annu Rev Cell Dev Biol 23: 175–205.
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6 (11): 857–866.
Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487 (7407): 330–337.
Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, Cai X, Wang K, Wang G, Ba Y, Zhu L, Wang J, Yang R, Zhang Y, Ren Z, Zen K, Zhang J, Zhang CY (2009) Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 28 (10): 1385–1392.
de Planell-Saguer M, Rodicio MC, Mourelatos Z (2010) Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nat Protoc 5 (6): 1061–1073.
Descombes P, Schibler U (1991) A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67 (3): 569–579.
Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I (2005) A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBP alpha regulates human granulopoiesis. Cell 123 (5): 819–831.
Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, Kawamura A, Nakamura K, Takeuchi T, Tanabe M (2007) An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 129 (3): 617–631.
Grady WM, Carethers JM (2008) Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135 (4): 1079–1099.
Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T (2008) Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet 40 (5): 600–608.
Homma J, Yamanaka R, Yajima N, Tsuchiya N, Genkai N, Sano M, Tanaka R (2006) Increased expression of CCAAT/enhancer binding protein beta correlates with prognosis in glioma patients. Oncol Rep 15 (3): 595–601.
Huang Z, Shi T, Zhou Q, Shi S, Zhao R, Shi H, Dong L, Zhang C, Zeng K, Chen J, Zhang J (2014) MiR-141 regulates colonic leukocytic trafficking by targeting CXCL12 beta during murine colitis and human Crohn’s disease. Gut 63 (8): 1247–1257.
Iwatsuki M, Mimori K, Ishii H, Yokobori T, Takatsuno Y, Sato T, Toh H, Onoyama I, Nakayama KI, Baba H, Mori M (2010) Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: clinical significance. Int J Cancer 126 (8): 1828–1837.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61 (2): 69–90.
Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, Eckhardt SG (2009) KRAS mutations and sensitivity to epidermal growth factor receptor inhibitors in colorectal cancer: practical application of patient selection. J Clin Oncol 27 (7): 1130–1136.
Kolfschoten IGM, van Leeuwen B, Berns K, Mullenders J, Beijersbergen RL, Bernards R, Voorhoeve PM, Agami R (2005) A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell 121 (6): 849–858.
Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT (2009) Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137 (6): 1005–1017.
Kurashige J, Watanabe M, Iwatsuki M, Kinoshita K, Saito S, Hiyoshi Y, Kamohara H, Baba Y, Mimori K, Baba H (2012) Overexpression of microRNA-223 regulates the ubiquitin ligase FBXW7 in oesophageal squamous cell carcinoma. Br J Cancer 106 (1): 182–188.
Lapinski PE, Kwon S, Lubeck BA, Wilkinson JE, Srinivasan RS, Sevick-Muraca E, King PD (2012) RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J Clin Invest 122 (2): 733–747.
Li BS, Zhao YL, Guo G, Li W, Zhu ED, Luo X, Mao XH, Zou QM, Yu PW, Zuo QF, Li N, Tang B, Liu KY, Xiao B (2012a) Plasma microRNAs, miR-223, miR-21 and miR-218, as novel potential biomarkers for gastric cancer detection. PLoS One 7 (7): e41629.
Li G, Cai M, Fu D, Chen K, Sun M, Cai Z, Cheng B (2012b) Heat shock protein 90B1 plays an oncogenic role and is a target of microRNA-223 in human osteosarcoma. Cell Physiol Biochem 30 (6): 1481–1490.
Li S, Li Z, Guo F, Qin X, Liu B, Lei Z, Song Z, Sun L, Zhang HT, You J, Zhou Q (2011a) MiR-223 regulates migration and invasion by targeting Artemin in human esophageal carcinoma. J Biomed Sci 18: 24.
Li X, Zhang Y, Zhang H, Liu X, Gong T, Li M, Sun L, Ji G, Shi Y, Han Z, Han S, Nie Y, Chen X, Zhao Q, Ding J, Wu K, Daiming F (2011b) MiRNA-223 promotes gastric cancer invasion and metastasis by targeting tumor suppressor EPB41L3. Mol Cancer Res 9 (7): 824–833.
Ling H, Fabbri M, Calin GA (2013) MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov 12 (11): 847–865.
McLaughlin SK, Olsen SN, Dake B, De Raedt T, Lim E, Bronson RT, Beroukhim R, Polyak K, Brown M, Kuperwasser C, Cichowski K (2013) The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer cell 24 (3): 365–378.
Ng EKO, Chong WWS, Jin H, Lam EKY, Shin VY, Yu J, Poon TCW, Ng SSM, Sung JJY (2009) Differential expression of microRNAs in plasma of patients with colorectal cancer: a potential marker for colorectal cancer screening. Gut 58 (10): 1375–1381.
Ohno N, Terada N, Murata S, Yamakawa H, Newsham IF, Katoh R, Ohara O, Ohno S (2004) Immunolocalization of protein 4.1B/DAL-1 during neoplastic transformation of mouse and human intestinal epithelium. Histochem Cell Biol 122 (6): 579–586.
Ohta M, Seto M, Ijichi H, Miyabayashi K, Kudo Y, Mohri D, Asaoka Y, Tada M, Tanaka Y, Ikenoue T, Kanai F, Kawabe T, Omata M (2009) Decreased expression of the RAS-GTPase activating protein RASAL1 is associated with colorectal tumor progression. Gastroenterology 136 (1): 206–216.
O’Hara SP, Splinter PL, Gajdos GB, Trussoni CE, Fernandez-Zapico ME, Chen XM, LaRusso NF (2010) NF kappaB p50-CCAAT/enhancer-binding protein beta (C/EBP beta)-mediated transcriptional repression of microRNA let-7i following microbial infection. J Biol Chem 285 (1): 216–225.
Pamonsinlapatham P, Hadj-Slimane R, Lepelletier Y, Allain B, Toccafondi M, Garbay C, Raynaud F (2009) P120-Ras GTPase activating protein (RasGAP): a multi-interacting protein in downstream signaling. Biochimie 91 (3): 320–328.
Pinto P, Rocha P, Veiga I, Guedes J, Pinheiro M, Peixoto A, Pinto C, Fragoso M, Sanches E, Araujo A, Alves F, Coutinho C, Lopes P, Henrique R, Teixeira MR (2011) Comparison of methodologies for KRAS mutation detection in metastatic colorectal cancer. Cancer Genet 204 (8): 439–446.
Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Muller-Tidow C, Bohlander SK, Tenen DG, Behre G (2010) Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 115 (9): 1768–1778.
Rask K, Thorn M, Ponten F, Kraaz W, Sundfeldt K, Hedin L, Enerback S (2000) Increased expression of the transcription factors CCAAT-enhancer binding protein-beta (C/EB Beta) and C/EBzeta (CHOP) correlate with invasiveness of human colorectal cancer. Int J Cancer 86 (3): 337–343.
Regalo G, Canedo P, Suriano G, Resende C, Campos ML, Oliveira MJ, Figueiredo C, Rodrigues-Pereira P, Blin N, Seruca R, Carneiro F, Machado JC (2006) C/EBP beta is over-expressed in gastric carcinogenesis and is associated with COX-2 expression. J Pathol 210 (4): 398–404.
Sachdeva M, Liu Q, Cao J, Lu Z, Mo YY (2012) Negative regulation of miR-145 by C/EBP-beta through the Akt pathway in cancer cells. Nucleic Acids Res 40 (14): 6683–6692.
Samowitz WS, Curtin K, Schaffer D, Robertson M, Leppert M, Slattery ML (2000) Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev 9 (11): 1193–1197.
Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DLW, Au GKH, Liu CG, Calin GA, Croce CM, Harris CC (2008) MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 299 (4): 425–436.
Shimizu Y, Kishimoto T, Ohtsuka M, Kimura F, Shimizu H, Yoshidome H, Miyazaki M (2007) CCAAT/enhancer binding protein-beta promotes the survival of intravascular rat pancreatic tumor cells via antiapoptotic effects. Cancer Sci 98 (11): 1706–1713.
Staiger J, Lueben MJ, Berrigan D, Malik R, Perkins SN, Hursting SD, Johnson PF (2009) C/EBP beta regulates body composition, energy balance-related hormones and tumor growth. Carcinogenesis 30 (5): 832–840.
Sterneck E, Zhu S, Ramirez A, Jorcano JL, Smart RC (2006) Conditional ablation of C/EBP beta demonstrates its keratinocyte-specific requirement for cell survival and mouse skin tumorigenesis. Oncogene 25 (8): 1272–1276.
Sun DF, Yu F, Ma YT, Zhao R, Chen X, Zhu J, Zhang CY, Chen JN, Zhang JF (2013) MicroRNA-31 activates the RAS pathway and functions as an oncogenic microRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). J Biol Chem 288 (13): 9508–9518.
Tsang WP, Kwok TT (2009) The miR-18a*microRNA functions as a potential tumor suppressor by targeting on K-Ras. Carcinogenesis 30 (6): 953–959.
Wang LG, Gu J (2012) Serum microRNA-29a is a promising novel marker for early detection of colorectal liver metastasis. Cancer Epidemiol 36 (1): E61–E67.
Wong JJ, Hawkins NJ, Ward RL (2007) Colorectal cancer: a model for epigenetic tumorigenesis. Gut 56 (1): 140–148.
Wong QW, Lung RW, Law PT, Lai PB, Chan KY, To KF, Wong N (2008) MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology 135 (1): 257–269.
Xi S, Yang M, Tao Y, Xu H, Shan J, Inchauste S, Zhang M, Mercedes L, Hong JA, Rao M, Schrump DS (2010) Cigarette smoke induces C/EBP-beta-mediated activation of miR-31 in normal human respiratory epithelia and lung cancer cells. PLoS One 5 (10): e13764.
Zeng X, Xiang J, Wu M, Xiong W, Tang H, Deng M, Li X, Liao Q, Su B, Luo Z, Zhou Y, Zhou M, Zeng Z, Shen S, Shuai C, Li G, Fang J, Peng S (2012) Circulating miR-17, miR-20a, miR-29c, and miR-223 combined as non-invasive biomarkers in nasopharyngeal carcinoma. PLoS One 7 (10): e46367.
Zhang JF, Luo X, Li HM, Yue XP, Deng L, Cui YY, Lu YX (2014) MicroRNA-223 functions as an oncogene in human colorectal cancer cells. Oncol Rep 32 (1): 115–120.
Acknowledgements
We thank Dr Guangxin Zhou for his assistance in clinical tissue sample collection and pathological diagnosis. This work was supported by the National Science Fund for Distinguished Young Scholars (81025019), the National Basic Research Program of China (2012CB517603), the Program for New Century Excellent Talents in University (NCET-13-0272), the National Natural Science Foundation of China (31070722, 31271013, 31071232, 31170751, 31200695, 51173076, 91129712 and 81102489), the Key Project of the Chinese Ministry of Education (108059), the PhD Programs Foundation of the Ministry of Education of China (20130091110037) and Jiangsu Planned Projects for Postdoctoral Research Funds (1302009B).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Sun, D., Wang, C., Long, S. et al. C/EBP-β-activated microRNA-223 promotes tumour growth through targeting RASA1 in human colorectal cancer. Br J Cancer 112, 1491–1500 (2015). https://doi.org/10.1038/bjc.2015.107
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.107
Keywords
This article is cited by
-
Modulation of microRNAs and claudin-7 in Caco-2 cell line treated with Blastocystis sp., subtype 3 soluble total antigen
BMC Microbiology (2022)
-
CBFβ promotes colorectal cancer progression through transcriptionally activating OPN, FAM129A, and UPP1 in a RUNX2-dependent manner
Cell Death & Differentiation (2021)
-
microRNAs as therapeutic targets in intestinal diseases
ExRNA (2019)
-
Therapeutic prospects of microRNAs in cancer treatment through nanotechnology
Drug Delivery and Translational Research (2018)
-
MicroRNA-199a-5p promotes tumour growth by dual-targeting PIAS3 and p27 in human osteosarcoma
Scientific Reports (2017)
