
Abstract
Background:
Polycomb group genes (PcGs) are epigenetic effectors implicated in most cancer hallmarks. The mutational status of all PcGs has never been systematically assessed in solid tumours.
Methods:
We conducted a multi-step analysis on publically available databases and patient samples to identify somatic aberrations of PcGs.
Results:
Data from more than 1000 cancer patients show for the first time that the PcG member PHC3 is amplified in three epithelial neoplasms (rate: 8–35%). This aberration predicts poorer prognosis in lung and uterine carcinomas (P<0.01). Gene amplification correlates with mRNA overexpression (P<0.01), suggesting a functional role of this aberration.
Conclusion:
PHC3 amplification may emerge as a biomarker and potential therapeutic target in a relevant fraction of epithelial tumours.
Similar content being viewed by others


Main
Epigenetic aberrations have been widely reported in human neoplasms, and preclinical studies have established conclusive links between epigenetics and most cancer hallmarks (Baylin and Jones, 2011). Despite this, the only epigenetic drugs approved for clinical use are DNA-methyltransferase and histone-deacetylase inhibitors, whose employment is restricted to a few haematological malignancies (Piekarz and Bates, 2009). Even the clinical exploitation of epigenetic biomarkers as prognostic/predictive factors is currently limited to a few notable exceptions (Wick et al, 2009).
Polycomb group genes (PcGs) are epigenetic effectors organised into two main repressive complexes (PRC1, PRC2). The PRCs silence tumour-suppressor genes through histone modifications, thereby driving cancer cell proliferation, metastasis and drug resistance (Crea et al, 2012). Despite a plethora of preclinical studies demonstrating their crucial role in human neoplasms, PcGs still lack clinical value. Most molecular research has focused on the histone-methyltransferase EZH2 (PRC2 member), which acts as an oncogene in several neoplasms (Chase and Cross, 2011). To the contrary, the role of PRC1 in many solid tumours is still largely overlooked.
The recent identification of EZH2-specific inhibitors has been welcomed as a fundamental advancement in epigenetic therapy (Melnick, 2012). Those molecules are particularly effective on lymphoma cells harbouring EZH2-activating mutations, but not on cells with simple mRNA upregulation (McCabe et al, 2012). In addition, inhibition of the catalytic subunit of PRC1 induces cancer cell apoptosis (Wen et al, 2013), thereby paving the way for the development of PRC1-targeting drugs. As PcG mutations have been described almost exclusively in haematological malignancies (Chase and Cross, 2011), there is currently no genetic evidence to predict their efficacy in solid tumours. Most reports on PcG status in solid neoplasms are restricted to gene expression analysis of a few popular targets (EZH2, BMI1), overlooking the evidence that human PcGs are encoded by least 18 different genes (Christophersen and Helin, 2010), and not taking into account possible mutational events. For this reason, we explored publically available databases and patient data sets to investigate the mutational status and clinical significance of all PcGs in solid tumours.
Methods
We conducted a two-step analysis on publically available databases. First, we queried the cBio database for somatic aberrations (gene amplification, insertion, deletion, point mutation) in each PcG (listed in Christophersen and Helin (2010), Supplementary Table 1). This database includes mutational data from exome sequencing, copy number calls from GISTC 2.0, and for some studies, mRNA levels expressed as Z score from RNA seq (Cerami et al, 2012). The cBio portal provides access to more than 5000 samples from over 20 cancer studies. Herein we report results only for those genes showing a mutational rate higher than 5%. We restricted our analysis to studies available for publication (bladder, breast, ovarian, glioblastoma, lung, prostate, and uterine carcinomas). To confirm our findings in an independent data set, we selected the most frequently mutated gene (PHC3), and investigated its clinical relevance in both cBio and Oncomine (Rhodes et al, 2007) databases (more than 100 additional data sets, more than 10 000 patients screened), searching for correlations between gene expression/amplification and clinico/pathological characteristics in lung, uterine and ovarian neoplsams. To further reduce the false discovery rate, we considered significant results with P-value<0.01 and fold-change >2.0. Amplification and overexpression of PHC3 in lung cancer was assessed in an additional data set comprised of 169 lung adenocarcinoma (AC) and 92 lung SCC samples, of which 35 AC and 13 SCC tumours had patient-matched normal tissue (GSE31800) (Starczynowski et al, 2011).
Results
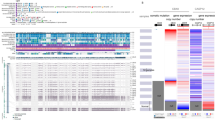
Our global mutational analysis is summarised in Figure 1A. We found no relevant deletions and only two missense mutations with a frequency higher than 5%. To the contrary, several loci were amplified in at least one investigated neoplasm. According to previous reports (Chase and Cross, 2011), EZH2 is not mutated in most common solid tumours, although we found an interesting rate of EZH2 amplification (5.6%) in ovarian serous carcinoma. This neoplasm shows relevant amplification of four different PcGs. Gene amplification was significantly correlated with higher mRNA levels (Supplementary Figure 1), thereby suggesting that these epigenetic effectors are crucial for its development.

Somatic aberrations of Polycomb genes in solid tumours. (A) Somatic aberration rate in different tumour types. All displayed percentages refer to genomic amplification, except the last two columns (EZH2_mut and PHC3_mut), which refer to missense mutation. The mutation rate was derived from the cBio portal studies. If not specified, cancer type is adenocarcinoma. SCC=squamous cell carcinoma. Patient sample size (tumours with complete information): bladder, 58; breast, 463; glioblastoma, 91; ovarian, 316; lung SCC, 178; prostate, 85; uterine corpus, 232. If two studies with partially overlapping patient sets were present in the database, we present the published rather than the ‘provisional’ data set. (B–D) Correlation between genomic amplification and mRNA levels in different neoplasms. Black and grey rectangles refer to the percentage of samples showing upregulated and non-upregulated Phc3 mRNA, respectively. Gene is considered upregulated when Z score is >1.0. **P<0.001, ***P<0.0001 (Fisher exact test).
The most striking result arising from our query was the high rate of PHC3 genetic amplifications. This aberration was found in lung (34.8%), ovarian (20.6%), and uterine ACs (7.7%). In order to explore the functional role of this genetic aberration, we computed the correlation between gene amplification and mRNA upregulation in those three neoplasms. Notably, gene amplification was significantly correlated with mRNA upregulation in each tumour type (Figure 1B–D).
Oncomine data confirmed that PHC3 is selectively upregulated in SCC, compared with other non-small-cell lung cancer subtypes (Figure 2A, P=1.32E−11, fold-change=2.137). Furthermore, PHC3 overexpression was found to be associated with shorter 3-year survival rate in lung cancer (Figure 2B, P=0.005, fold-change=2.267, Oncomine analysis). PHC3 mRNA was also upregulated in cervical, colorectal, gastric, and prostate ACs, compared with normal tissues (P<0.01, fold-change >2.0, Oncomine data not shown).

PHC3 gene expression and amplification in lung cancers. (A) Correlation between PhC3 mRNA level and lung cancer histotype (138 patients from ‘Lee Lung’ study). (B) Correlation between PhC3 mRNA level and 3-year survival in lung cancer (nine patients from ‘TCGA Lung’ study). (C) Correlation between genomic amplification and mRNA levels in PHC3 and eight additional genes located at 3q62.2 (4 next to the 5′ end and 4 next to the 3′ end of the PHC3 locus, displayed from 5′ to 3′). The mRNA is considered upregulated when Z score is ⩾1.0. Data are from Cbio lung SCC data set. (D–F) Data from matched normal and neoplastic lung tissues. AC=adenocarcinoma (35 samples). SqCC=lung squamous cell carcinoma (13 samples). Data from A and C: Oncomine (Compendia Bioscience, Ann Arbor, MI) was used for analysis and visualisation. **P<0.01 (A and B, Oncomine analysis; D–F, two-sided t-test).
PHC3 is located at 3q62.2, a genomic region that has been described as amplified in solid tumours (Lavigne et al, 2004; Linxweiler et al, 2012). For this reason, we investigated amplification and expression level of eight genes flanking the PHC3 locus (Figure 2C). The rate of genomic amplification was very similar for all eight genes (34–35% in lung cancer, data not shown). However, only two genes (PHC3 and PRKCI) showed mRNA upregulation in more than 50% of tumours with genomic amplification, suggesting that the genetic aberration has a functional role in those loci. The remaining six genes showed an mRNA upregulation rate lower than 35%, even in the presence of an amplified gene.
We confirmed the high frequency of PHC3 amplification and overexpression in lung SCC using an additional publically available data set of 169 AC and 92 SCC tumours. PHC3 was amplified in 71 out of 92 (77%) lung SCC samples and 36 out of 169 (21%) AC and overexpressed in 7 of 13 (54%) and 5 of 35 (14%) SCC and AC cases, respectively. Moreover, comparison of mRNA expression levels between AC and SCC revealed no difference in PHC3 mRNA levels in non-malignant tissue (Figure 2D, P=0.76), but significantly higher expression in SCC tumours relative to AC tumours (Figure 2E, P=0.002) and SCC tumours relative to matched non-malignant tissue (Figure 2F, P=0.0024).
A very interesting clinical correlation was identified in uterine carcinoma. In this neoplasm, PHC3 amplification also predicted shorter disease-free survival (Supplementary Figure 2). Notably, PHC3 amplification was detectable only in grade 3 neoplasms. Despite this, our analysis indicates that PhC3 amplification is an independent prognostic factor, even when contrasted with tumour grade (Table 1). To the contrary, PhC3 amplification was not significantly associated with prognosis in ovarian cancer (Supplementary Figure 3).
Discussion
Taken together, our analyses of data from three different data sets indicate that the PHC3 gene is amplified in three epithelial tumours, with percentages ranging from 8 to 35%. Gene amplification is highly correlated with mRNA overexpression and, at least for lung and uterine cancer, associated with poorer prognosis. PHC3 is rarely deleted and almost never downregulated in cancer vs normal tissue. Oncomine analysis revealed that cancer-specific PHC3 upregulation might be a common phenomenon in several epithelial neoplasms. To the best of our knowledge, PHC3 genomic amplification and mRNA upregulation have never been described before. However, PHC3 has been found to be downregulated in human sarcomas, where it acts as a tumour suppressor (Deshpande et al, 2007).
PHC3 is a member of PRC1, which catalyses histone H2A ubiquitination. PRC1 is composed of RING1B (catalytic subunit) and other proteins responsible for interactions with transcription factors, epigenetic regulators, and DNA sequences. PhC proteins bridge distant chromatin templates, thereby recruiting PRC1 to selected loci (Lavigne et al, 2004). Currently, three PhC isoforms are known, with different tissue distribution and partially overlapping functions (Deshpande et al, 2007). PRC1 has been implicated in tumorigenesis and progression of several cancer types. Tobacco smoke is able to activate PRC1 genes, thereby increasing the tumorigenicity of lung cancer cells (Hussain et al, 2009). Amplification of the chromosomal region harbouring the PHC3 locus (3q26) has been described in preneoplastic and invasive lung SCC, and its rate increases with disease progression (Pelosi et al, 2007). Future molecular studies should dissect the role of PHC3 in lung tumorigenicity.
As genomic amplification can occur at a multigenic scale, we investigated if PHC3 copy number gain was associated with similar alteration in nearby loci. Our results indicate that the PHC3-proximity region is frequently amplified in solid tumours. However, when we contrasted mRNA expression with genomic amplification, we observed that only PHC3 and PRKCI mRNA levels were frequently upregulated in tumours with copy number gain (Figure 2D). The remaining six genes did not show a correlation between copy number gain and increased gene expression. Interestingly, PRKCI (protein kinase C iota) has been described as a potential oncogene in lung cancer (Regala et al, 2009; Linxweiler et al, 2012). Our data indicate that copy number gain may have a functional role in the PHC3 locus too, and that PHC3 upregulation is functionally relevant for cancer cells. In conclusion, we identified for the first time that PHC3 copy number gain as a novel and possibly clinically significant genetic alteration in three common epithelial neoplasms (lung SCC, uterine carcinoma, and ovarian serous carcinoma). Future molecular studies should dissect the role of PHC3 in each specific context. However, our data indicate that PHC3 may emerge as a novel oncogene, and possibly a prognostic marker and a target for epigenetic therapy, in epithelial neoplasms.
Change history
17 September 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer 11 (10): 726–734.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2 (5): 401–404.
Chase A, Cross NC (2011) Aberrations of EZH2 in cancer. Clin Cancer Res 17 (9): 2613–2618.
Christophersen NS, Helin K (2010) Epigenetic control of embryonic stem cell fate. J Exp Med 207 (11): 2287–2295.
Crea F, Paolicchi E, Marquez VE, Danesi R (2012) Polycomb genes and cancer: time for clinical application? Crit Rev Oncol/Hematol 83 (2): 184–193.
Deshpande AM, Akunowicz JD, Reveles XT, Patel BB, Saria EA, Gorlick RG, Naylor SL, Leach RJ, Hansen MF (2007) PHC3, a component of the hPRC-H complex, associates with E2F6 during G0 and is lost in osteosarcoma tumors. Oncogene 26 (12): 1714–1722.
Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, Caragacianu D, Schrump DS (2009) Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res 69 (8): 3570–3578.
Lavigne M, Francis NJ, King IF, Kingston RE (2004) Propagation of silencing; recruitment and repression of naive chromatin in trans by polycomb repressed chromatin. Mol Cell 13 (3): 415–425.
Linxweiler M, Linxweiler J, Barth M, Benedix J, Jung V, Kim YJ, Bohle RM, Zimmermann R, Greiner M (2012) Sec62 bridges the gap from 3q amplification to molecular cell biology in non-small cell lung cancer. Am J Pathol 180 (2): 473–483.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Iii AD, Diaz E, Lafrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492 (7427): 108–112.
Melnick A (2012) Epigenetic therapy leaps ahead with specific targeting of EZH2. Cancer Cell 22 (5): 569–570.
Pelosi G, Del Curto B, Trubia M, Nicholson AG, Manzotti M, Veronesi G, Spaggiari L, Maisonneuve P, Pasini F, Terzi A, Iannucci A, Viale G (2007) 3q26 Amplification and polysomy of chromosome 3 in squamous cell lesions of the lung: a fluorescence in situ hybridization study. Clin Cancer Res 13 (7): 1995–2004.
Piekarz RL, Bates SE (2009) Epigenetic modifiers: basic understanding and clinical development. Clin Cancer Res 15 (12): 3918–3926.
Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP (2009) Atypical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res 69 (19): 7603–7611.
Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM (2007) Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9 (2): 166–180.
Starczynowski DT, Lockwood WW, Delehouzee S, Chari R, Wegrzyn J, Fuller M, Tsao MS, Lam S, Gazdar AF, Lam WL, Karsan A (2011) TRAF6 is an amplified oncogene bridging the RAS and NF-kappaB pathways in human lung cancer. J Clin Invest 121 (10): 4095–4105.
Wen W, Peng C, Kim MO, Ho Jeong C, Zhu F, Yao K, Zykova T, Ma W, Carper A, Langfald A, Bode AM, Dong Z (2013) Knockdown of RNF2 induces apoptosis by regulating MDM2 and p53 stability. Oncogene e-pub ahead of print 14 Jan 2013 doi:10.1038/onc.2012.605.
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, Sabel MC, Koeppen S, Ketter R, Meyermann R, Rapp M, Meisner C, Kortmann RD, Pietsch T, Wiestler OD, Ernemann U, Bamberg M, Reifenberger G, von Deimling A, Weller M (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27 (35): 5874–5880.
Acknowledgements
We thank Kelsie L Thu for helping with data analyses and for insightful discussions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Crea, F., Sun, L., Pikor, L. et al. Mutational analysis of Polycomb genes in solid tumours identifies PHC3 amplification as a possible cancer-driving genetic alteration. Br J Cancer 109, 1699–1702 (2013). https://doi.org/10.1038/bjc.2013.454
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.454
Keywords
This article is cited by
-
Hypoxia increases the biogenesis of IGF2BP3-bound circular RNAs
Molecular Biology Reports (2024)
-
Sulforaphane inhibits self-renewal of lung cancer stem cells through the modulation of sonic Hedgehog signaling pathway and polyhomeotic homolog 3
AMB Express (2021)
-
Neonatal tumours
Pediatric Surgery International (2013)
