
Abstract
Vascular dementia (VaD) is a progressive neurodegenerative disease with a high prevalence. Several studies have recently reported that VaD patients present cholinergic deficits in the brain and cerebrospinal fluid (CSF) that may be closely related to the pathophysiology of cognitive impairment. Moreover, cholinergic therapies have shown promising effects on cognitive improvement in VaD patients. The precise mechanisms of these cholinergic agents are currently not fully understood; however, accumulating evidence indicates that these drugs may act through the cholinergic anti-inflammatory pathway, in which the efferent vagus nerve signals suppress pro-inflammatory cytokine release and inhibit inflammation, although regulation of oxidative stress and energy metabolism, alleviation of apoptosis may also be involved. In this paper, we provide a brief overview of the cholinergic treatment strategy for VaD and its relevant mechanisms of anti-inflammation.
Similar content being viewed by others

Introduction
Vascular dementia (VaD), a disease with a high prevalence among the elderly, is the second most common cause of dementia, after Alzheimer's disease (AD)1. Moreover, the number of VaD patients will double by around 2020 as the population ages2. The increasing public costs have become a heavy burden on society and have attracted attention worldwide. VaD is regarded as a heterogeneous clinical entity; the disease varies in clinical-pathological phenotype and in pathophysiological mechanisms. However, cerebrovascular disease (CVD) resulting from vascular or circulatory pathologies is a common finding among VaD patients and is either the sole or primary cause of dementia3. Although multiple etiopathogeneses are involved in the VaD process, cerebral ischemia is the most common pathology, and accumulating evidence indicates that stroke has also become one of the leading causes of the high prevalence of VaD, which shows an exponential increase with age4.
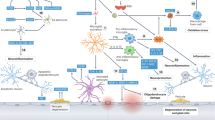
The precise mechanisms involved in VaD remain unclear, but accumulating evidence from various experimental cerebral ischemia models indicates that in addition to cholinergic deficiency, post-ischemic inflammation occurs in response to ischemic injury and contributes to delayed brain damage5, 6. More interestingly, cholinergic agents, including acetylcholinesterase (AChE) inhibitors, have shown considerable benefits in VaD therapy, and these effects were recently reported to be associated with the cholinergic anti-inflammatory pathway7, suggesting that modulation of this pathway may provide a useful therapeutic strategy to ameliorate VaD (Figure 1).

Pathways of ischemic vascular cognitive impairment/dementia and possible therapeutic approaches. Following cerebral ischemia, energy failure and subsequent events including inflammation, glutamate-mediated excitotoxicity, calcium overload, initiation of intracellular death pathways, oxidative stress, and structural and functional changes occur. Mediators of these events interact with each other and contribute to cellular damage, in which a cholinergic deficit is involved, and finally cause cognitive impairment or dementia. Current neuroprotective treatment options cover all of the molecular targets of the dementia cascades. Interestingly, protective effects of cholinergic agents, especially AChE inhibitors, involve multiple mechanisms (I48, 127, 128, 129; II130, 131, 132; III85, 98; IV127, 133; V90, 133, 134; references to literature regarding VI and VII can be found throughout this review). Abbreviations: ATP, adenosine triphosphate; CaM, calmodulin; Hup A, huperzine A; JAK/STAT, Janus kinase/signal transducer and activator of transcription; MMP, matrix metalloproteinase; NGF, nerve growth factor; NMDA, N-methyl-D-aspartic acid; PAF, platelet activating factor.
The aim of this review is to discuss research progress in the study of cholinergic deficiency in VaD, relevant therapeutic strategies and inflammation-related mechanisms, focusing particularly on ischemic vascular disease-caused dementia.
Cholinergic deficiency in VaD animal models and VaD patients
The pathology of vascular cognitive disorder shows focal, multifocal or diffuse vascular and/or ischemic lesions involving various brain areas and neuronal networks, with deafferentation of frontal and limbic cortical structures and interruption of basal ganglia, thalamus, white matter and subfrontal areas8, 9. Cholinergic dysfunction which resembles that observed in patients with AD occurs due to a dense network of cholinergic fibers exists in the injured area. As a matter of fact, numerous studies have documented the occurrence of cholinergic dysfunction in murine models of cerebrovascular injuries and in patients with VaD10, 11.
Many studies of cholinergic deficit in cerebral ischemia-related VaD models have shown persistent reductions in several cholinergic markers. First, in rats, bilateral common carotid artery occlusion (BCCAO), which leads to chronic cerebral hypoperfusion, has been shown to result in the loss of cholinergic neurons, as demonstrated by decreased choline acetyltransferase (ChAT) and AChE activities12, 13, as well as reduced mRNA expression of the m3 and m5 muscarinic acetylcholine (ACh) receptors14. Second, decreased ACh content and corresponding impairments in learning and memory were found in rats with 4-vessel occlusion15. Third, ChAT immunostaining was shown to be decreased in rats with transient occlusion of the middle cerebral artery (MCAO)16, 17, 18. Fourth, rats with multiple small embolizations have been shown to produce multiple infarctions and exhibit decreases in cholinergic markers19. Finally, there is a significant reduction in cholinergic markers, including ACh, in the neocortex, hippocampus and cerebrospinal fluid (CSF) in the spontaneously hypertensive stroke prone rat (SHspR), which is the best model for essential hypertension and stroke20, 21.
It has been suggested that patients with VaD also exhibit cholinergic deficits (reviewed in ref 22). Postmortem examinations have revealed significant reductions in ChAT activity in the hippocampus and temporal cortex of VaD patients23, 24. Tohgi et al and Wallin et al observed significantly reduced CSF ACh concentrations in patients with Binswanger or multi-infarct dementia (MID)25, 26. These results are consistent with the finding that the number of cholinergic neurons in the nucleus basalis of Meynert is reduced in MID27. Mesulam et al and Jessica et al demonstrated cholinergic denervation from pathway lesions in the absence of AD, in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a pure genetic form of VaD28, 29. Furthermore, one study reported a loss of cholinergic neurons in 40% of VaD patients, accompanied by reduced ACh activity in the cortex, hippocampus, striatum, and CSF30.
Cholinergic anti-inflammation and current cholinergic therapy for VaD
In view of the overlap of cholinergic pathology between VaD and AD, cholinergic agents have been proposed for relieving symptoms of VaD10. Although different mechanisms may be involved in the protective effects of cholinergic therapy in VaD and AD, increasing evidence shows that anti-inflammation accounts at least partially for the protective effects of cholinergic therapy31. Inflammatory injury is another significant characteristic of VaD pathology. Inflammatory events following cerebral ischemia include upregulation of inflammatory mediators such as intercellular adhesion molecule 1, selectins, tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), nitric oxide (NO), cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS); peripheral leukocyte recruitment; and resident glial cell (microglia and astrocyte) activation32. Once activated, immune cells may release a variety of toxic mediators, such as additional pre-inflammatory cytokines and reactive oxygen species, NO, and glutamate33. These substances may be harmful to neurons and may disrupt the blood-brain barrier (BBB). In turn, BBB disruption may induce secondary ischemic brain damage by potentiating inflammation34. In addition, the further recruitment of peripheral leukocytes may lead to microvascular obstruction, which in turn worsens local brain damage35. In fact, several studies have shown that inhibiting inflammatory cascades may protect against cerebral ischemic lesion36.
The concept of the “cholinergic anti-inflammation pathway” was proposed by Tracey and coworkers at the turn of the century37. Recent work has shown that ACh released from cholinergic axon terminals can interact with α7 nicotinic ACh receptors (nAChRs) on vicinal immune cells38, 39. The nicotinic receptors then translate the cholinergic signal into suppression of cytokine release37, 40. Therefore, cholinergic agents may ameliorate cerebral ischemia injury via their anti-inflammatory activity. Cholinergic agents include ACh precursors, which increase the synthesis of ACh; nicotinic or M1 muscarinic agonists, which directly stimulate cholinergic receptors or allosterically modulate nAChR41; and synaptic AChE inhibitors, which prevent the degradation of ACh. Recent preclinical and clinical evidence indicates that treatment with cholinergic agents has beneficial effects on dementia of vascular origin42, 43, 44, 45, 46, 47, 48.
Cholinergic precursors
Cholinergic precursor loading therapy was the first attempt to relieve cognitive impairment in dementia disorders. In the 1970's, Wecker and Schmidt observed that neurons incorporated more exogenous choline under conditions of reduced cholinergic synthesis and increased neuronal demand49, which suggested that the systemic administration of a choline precursor may antagonize biochemical disorders of the cholinergic system. Clinical trials have evaluated the effects of cholinergic precursors — including lecithin (also known as phosphatidylcholine), cytidine 5′-diphosphocholine (CDP-choline or citicoline) and choline alphoscerate — on dementia of vascular origin (reviewed in ref50).
Lecithin was the first cholinergic precursor used, however, it showed no clear clinical benefits with respect to symptoms of dementia disorders51. This is probably because lecithin provides choline for ACh synthesis only under conditions of stimulated neurotransmitter release52. CDP-choline, another phospholipid involved in choline biosynthetic pathways, promoted modest improvements in cognitive function in dementias of vascular origin. Specifically, CDP-choline administration improved global and neurological functions and recovered motor and cognitive performance in a subgroup of moderate to severe stroke cases44. A Cochrane meta-analysis also concluded that CDP-choline had a positive effect on memory and behavior over 20 days to one year, primarily in patients suffering from cognitive deficits associated with CVD53. The molecular mechanisms underlying CDP-choline-induced cognitive enhancement are still unclear, but one possibility is that CDP-choline counters the progression of ischemic damage by reducing the release of free fatty acids44. Choline alphoscerate, a semi-synthetic derivative of lecithin that does not carry the electrical charge of endogenous choline, can be incorporated into brain phospholipids within 24 h of absorption, resulting in a more rapid increase in free plasma choline than other uncharged choline precursors54. Preclinical studies have demonstrated that choline alphoscerate increases the release of ACh in the rat hippocampus and facilitates learning and memory in animal models of aging55. In addition, choline alphoscerate also significantly attenuates the extent of glial reaction in the hippocampus of SHspR56, suggesting that the compound may protect the brain from injury of vascular origin. Indeed, clinical studies have shown that choline alphoscerate can improve memory, attention and cognitive impairment in patients with mild to moderate degenerative dementia disorders, VaD, or acute cerebral vascular disease43, 57.
The above-mentioned clinical findings regarding the treatment of dementias of vascular origin with cholinergic precursors showed either no effect or modest symptomatic relief in memory and cognition. The reason for these divergent effects is unclear, although the effect may depend on the availability of the affected neurotransmitter55. However, further studies are necessary to demonstrate the molecular mechanisms of these drugs and how they function in VaD therapy.
Acetylcholine receptor agonists
Activation of cholinergic receptor is another way to stimulate cholinergic system. Several nicotinic agonists, such as nicotine and SIB-1553A ((+/−)4-[[2-(1-methyl-2-pyrrolidinyl)ethyl]thio]phenol hydrochloride), a compound that acts predominantly at β4 subunit-containing human nAChRs subtypes, were reported to be efficacious in improving cognitive performance in mice58. However, further reports of nicotine and SIB-1553A in VaD have not been documented. Nefiracetam (DM-9384), a pyrrolidone nootropic drug that potentiates the α4β2-type current by acting on neuronal nAChRs, has been shown to improve learning and memory in microsphere-embolized rats in which sustained cerebral ischemia and stroke symptoms occur59. However, the clinical trials on nefiracetam in the treatment of post-stroke VaD were halted for lack of efficacy in Phase III60. This failure may be attributable to the opposing effect of nefiracetam on rat and human nAChRs61. Another reason for the ineffectiveness of nicotinic agonists in VaD treatment may be due to desensitization of nAChRs, which leads to tolerance and loss of efficacy after long-term use62, 63.
Muscarinic AChRs are also implicated in learning and memory improvement. Pooled studies suggest that stimulation of M1 muscarinic AChRs may reverse cognitive deficits, although sufficient evidence on their effect in VaD is still lacking. Chotosan, a Kampo (traditional medicine of Japan) prescription medicine, consists of 10 medicinal herbs and Gypsum fibrosum and is used to treat chronic headache and hypertension. Pharmacological studies have reported that Chotosan prevented the occurrence of stroke, prolonged the life span of SHspR64, and ameliorated cognitive dysfunction in stroke patients65, 66. Recent data indicate that Chotosan improves chronic cerebral hypoperfusion-induced spatial learning deficit via stimulation of M1 muscarinic AChRs42. Muscarinic agonists have shown certain beneficial effects on learning and memory in a few cases; however, clinical utility was limited by poor bioavailability, short duration of action and excessive adverse events, such as syncope, nausea and vomiting67. Therefore, ACh receptor agonists do not appear to be promising for VaD therapy.
Acetylcholinesterase inhibitors
Cholinergic deficits have been found in AD patients, and AChE inhibitors are currently the most commonly prescribed treatment for mild to moderate AD68. Similarly, cholinergic deficits exist in VaD patients as well; therefore, AChE inhibitors may also provide benefit for these patients69. These AChE inhibitors include the active compound from the Chinese herb huperzine A and the US Food and Drug Administration-approved drugs donepezil, rivastigmine, and galantamine. Moreover, as mentioned above, the cholinergic system is probably affected in both AD and VaD and may underlie the cognitive deficits seen in such patients. Therefore, AChE inhibitors may represent a potential therapeutic option for impaired cognitive status in dementia of vascular origin. In fact, the effects of these AChE inhibitors have already been well evaluated in the treatment of VaD. Although the results were inconsistent, application of AChE inhibitors showed promising benefits in ameliorating the learning and memory impairments as well as other deficits found in VaD patients and VaD animal models6, 16, 70, 71, 72, 73.
Huperzine A
Huperzine A, an alkaloid isolated from the Chinese folk medicine huperzia serrata, is a reversible and selective inhibitor of AChE74. It has been widely used as an anti-AD drug in China75, 76 and has shown promising clinical effects with low toxicity. The therapeutic effects of huperzine A in VaD have been extensively evaluated and have consistently shown favorable outcomes. In a randomized, matched and double-blinded study early in 1991, huperzine A administration was shown to produce significant memory improvement in 56 patients with MID77. In a subsequent study with a self-controlled design, a 4-week treatment with huperzine A produced marked improvements in memory deficits78. In agreement with these studies, routine treatment-controlled trials also indicated that memory deficiency and recognition decline were greatly improved in VaD patients treated with huperzine A73, 76, 79, 80 . Moreover, huperzine A was shown to be more effective than pyritinol in the treatment of MID81. However, clinical trials with larger sample sizes and better-defined criteria will be needed to further understand the effect of huperzine A in VaD therapy.
In the past decade, great efforts have been made to clarify the clinical benefits of huperzine A. Consistent with the clinical data, huperzine A exhibits neuroprotective effects against ischemia-induced injury both in vitro and in vivo. Long-term treatment of huperzine A showed beneficial effects on learning deficits and brain neuronal damage in rats induced by permanent BCCAO, a chronic cerebral hypoperfusion model82. Similarly, in a gerbil model of transient global ischemia, huperzine A administration significantly reduced memory impairment and neuronal degeneration in the CA1 region of the hippocampus and partially restored hippocampal ChAT activity83. Moreover, huperzine A treatment showed significant protection from neuropathology damage and associated behavior in the hypoxic-ischemic neonatal rat model84, the transient cerebral ischemia and reperfusion mice model85, and the MCAO rat model48.
Although the precise mechanisms by which huperzine A produced the above-mentioned preclinical and clinical effects remain unclear, our previous study indicated that the benefits of huperzine A may depend at least partly on the anti-inflammatory property. In agreement with previous reports33, 86, 87, our study indicates that ischemia triggers a strong inflammatory response that involves activation of endogenous glial cells and overexpression of various proinflammatory factors, including TNF-α, IL-1β, NO, iNOS, and COX-2, and contributes to delayed brain damage. Interestingly, huperzine A decreased overexpression of proinflammatory factors in the ipsilateral cortex and striatum, and huperzine A suppressed activation of astrocytes and microglia in the ischemic penumbra48. Recently, we found that the beneficial effects of huperzine A on cerebral hypoperfusion-induced cognitive injury might also involve suppression of glial activation (unpublished data). In agreement with the in vivo data, huperzine A is also able to attenuate iNOS, COX-2, and NO overproduction, and it can increase cell survival in oxygen-glucose deprivation (OGD)-treated C6 rat glioma cells88. Nuclear factor-kappa B (NF-κB) is a principal mediator of the postischemic inflammatory response89. Further investigation showed that MCAO/OGD led to increased phosphorylation and degradation of IκB, as well as the nuclear translocation of p65, which indicated activation of NF-κB signaling. However, these phenomena could be dramatically inhibited by huperzine A treatment48, 88. Moreover, in mouse microglia BV-2 cells, huperzine A reduced hypoxia-induced TNF-α production by regulating the phosphorylation of p38 and JNK, two mitogen-activated protein kinases (MAPKs) (unpublished data). It is suggested that nAChR might be involved in these protective effects because these effects can be partially reversed by mecamylamine, a nAChR antagonist48, 88, and this finding is consistent with the “cholinergic anti-inflammation” hypothesis.
Although one function of huperzine A is AChE inhibition, this function is not sufficient as the mechanism by which huperzine A acts in VaD therapy because accumulating evidence indicates that huperzine A exerts multiple neuroprotective effects through several molecular sites that do not include inhibition on AChE activity. Previous reports have suggested that huperzine A can improve mitochondrial dysfunction90 and regulate anti-oxidative enzyme activities and apoptotic gene expressions91 in ischemic animal models. Whether these effects contribute to VaD therapy and reflect responsible cellular targets remain open questions.
Donepezil
Donepezil is the most widely prescribed drug for AD therapy, and it is also used for VaD treatment in New Zealand, India, the Philippines, Romania, South Korea, and Thailand (http://www.eisai.co.jp/enews/enews200609.html). The safety and efficacy of donepezil have been studied in the largest clinical trial of pure VaD to date92. Six-month treatment with donepezil was shown to significantly improve Clinical Dementia Rating (CDR) scores and activities of daily living in VaD patients (excluding AD and mixed dementia (VaD/AD) patients)93. In a later study, donepezil was further demonstrated to significantly improve cognitive function, global function, and activities of daily living versus placebo-treated subjects in a randomized, double-blind, placebo-controlled, 24-week clinical trial comprising 1219 patients with mild to moderate cognitive decline due to probable or possible VaD (according to the NINCDS/AIREN criteria and the Hachinski Ischemia Scale), although adverse effects were reported in the higher-dose group71, 92, 94, 95. Moreover, a recent randomized double-blind trial of donepezil in CADASIL showed a significant benefit on executive function and processing speed46, although this result was later questioned by Schneider45.
The beneficial effects and mechanisms involved in the protection by donepezil were further studied in animal models of VaD. Fujiki et al reported that pretreatment with a single oral dose of donepezil significantly attenuated cerebral infarction induced by permanent MCAO in rats96. Similarly, treatment with a single oral dose of donepezil immediately after mild traumatic brain injury also significantly attenuated neuronal death and cognitive impairment97. These neuroprotective effects of donepezil were probably related to the facilitation of nicotinic acetylcholinergic transmission. Several studies have found that donepezil can up-regulate the expression of nAChR and activate them, especially α4 and α7 receptor subtypes98, 99. The subsequent action of downstream signaling pathways, including the phosphatidylinositol 3-kinase-Akt signaling pathway and the MAPK pathway, makes neurons more sensitive to the protection by donepezil98, 100. Consistent with this finding, Fujiki and co-workers found that the reduction of cerebral infarct and traumatic brain injury after donepezil administration was prevented by coinjection with mecamylamine, indicating that protection of donepezil is mediated by nAChR activation96, 97. Moreover, another study showed that donepezil could markedly inhibit lipopolysaccharide-induced enhancement of AChE activity and suppress the elevated expression of IL-2 in several brain regions in mice101. Furthermore, using collected peripheral blood from AD patients, Reale et al found that donepezil modulated production of monocyte chemotactic protein-1, a positive regulator of Th2 differentiation, and IL-4, an anti-inflammatory factor102. Therefore, the anti-inflammation effect of donepezil may also occur, at least in part, through the cholinergic system, which may provide another explanation for the delayed VaD progression in patients treated with donepezil.
Rivastigmine
Rivastigmine, a dual inhibitor of AChE and butyryl-cholinesterase103, has also shown promise in VaD therapy. Open-label extension phase data from a preliminary study using a small number of patients with frontosubcortical VaD showed that 12-month rivastigmine treatment improved executive function and behavior compared with baseline and a control group receiving cardioaspirin. Furthermore, these beneficial effects were maintained for 22 months104, 105. Another clinical trial, with 16 subcortical VaD patients, showed similar results, indicating that rivastigmine may provide targeted treatment to brain areas that are particularly affected in this kind of patient population70. Moreover, a large, double-blind, randomized, placebo-controlled clinical trial showed that rivastigmine provided greater benefits in patients with AD and VaD than in patients with pure AD106. Most recently, clinical data showed the prospective result that long-term treatment with rivastigmine may produce significant improvements in all behavioral symptoms in subcortical VaD and MID, except delusions107.
In agreement with the clinical findings, rivastigmine also showed promising effects in ischemic animal models. Pretreatment with rivastigmine mitigated the abnormalities in the cerebral cholinergic system in the BCCAO-induced gerbil ischemic model108, 109. Post-ischemic administration of rivastigmine showed the same result in prevention of the decrease in cholinergic activity in head trauma rats110 together with reduced motor and neurological deficits and faster recovery. Moreover, the investigators also demonstrated that prevention of delayed neuron death and amelioration of accumulation of astrocytes in the hippocampal CA1 region contributed to the protective mechanisms of rivastigmine108, 111. These protective effects could be prevented by the simultaneous injection of mecamylamine, but not by scopolamine110, suggesting that the therapeutic effects on cerebrovascular type dementia of rivastigmine may be related to nAChR-mediated cholinergic enhancement. Moreover, in a mouse model of multiple sclerosis, rivastigmine administration ameliorated neurological dysfunction and memory deficits, and this treatment decreased reactivity of T cells and reduced the production of TNF-α and interferon-γ (INF-γ), which can also be abolished by α7 nACh receptor antagonist112. Rivastigmine also lowered lipopolysaccharide-induced over-expression of IL-2 in mice through inhibiting the enhanced activity of AChE101. The above-mentioned findings indicate that rivastigmine may influence central nervous system inflammation by up-regulating cholinergic function, which may contribute to its protective effects in VaD patients and animal models.
Galantamine
In addition to inhibiting AChE activity, galantamine also modulates central nicotinic receptors to enhance cholinergic transmission113. Galantamine was the cholinergic drug used in the first large-scale, randomized, controlled trial in patients with either probable VaD or mixed dementia (possible AD and VaD). In this trial, 6-month galantamine treatment showed convincing and clinically relevant benefits in cognition (ADAS-cog), global function (CIBIC-plus), functional abilities (DAD) and behavioral symptoms (NPI)72, 114. However, galantamine provided no significant benefits over placebo in patients with pure VaD. In the second larger trial, cognition and executive function were significant enhanced by galantamine compared to placebo, although fewer data were generated45, 115. Moreover, a post-ischemic single administration of galantamine also showed a beneficial effect on the recovery of learning ability in rats116, which suggests a direct effect of galantamine on the early pathologic changes of CNS damage. In addition, continuous administration of galantamine could protect pyramidal neurons in the hippocampal CA1 region of ischemic gerbils and lead to the recovery of spatial memory in a transient brain global ischemic model in gerbils117, 118. Furthermore, galantamine attenuated the release of cytokines from activated murine microglia119. These protective effects were mediated partly by nAChRs. As a potential nicotinic allosteric ligand, galantamine facilitates synaptic transmission in the mammalian central nervous system, which could be an important determinant of its therapeutic effect120, 121, 122. Indeed, galantamine was able to reverse the learning impairment induced by mecamylamine122, and the activation of nicotinic receptors makes anti-inflammation of galantamine possible. In addition to nAChR, central muscarinic receptors may also be involved in the anti-inflammatory process of galantamine123.
Other AChE inhibitors
Aside from the four AChE inhibitors described above, which have been investigated extensively in Europe, the United States and China, other AChE inhibitors may have potential protective effects against dementia of vascular origin. Methanesulfonyl fluoride (MSF), a highly selective CNS inhibitor of AChE, has recently been demonstrated to promote improvement in cognitive performance in patients with AD124, and it was also shown to attenuate simple learning and memory deficits in the MCAO rat model16. The seed extract of Cassia obtusifolia (COE), which has been found to inhibit AChE activity both in vitro and ex vivo, attenuated memory impairment induced by scopolamine or BCCAO in the passive-avoidance, Y-maze, and Morris water-maze tests in ICR mice47. Another agent that could increase ChAT activity and inhibit AChE activity is Z-Ligustilide (LIG). This drug is suggested to have significant neuroprotective effects in transient forebrain ischemia in mice125 and permanent focal cerebral ischemia in rats126 via antioxidant and antiapoptotic mechanisms. LIG has also been shown to alleviate cognitive deficits and prevent neuronal loss and astrocyte activation in the rat hippocampus due to chronic hypoperfusion6. Although these findings seem promising, further clinical studies will be needed to determine the efficacy of these AChE inhibitors in VaD therapy.
Conclusion
Although there is currently no cure for VaD owing to its multiple etiologies, evidence from a considerable number of studies and controlled clinical trials support the benefits of cholinergic agents — especially AChE inhibitors, including huperzine A, donepezil, galantamine and rivastigmine — in improving cognitive function, clinical global impression and activities of daily in living patients with probable or possible mild to moderate VaD. These findings provide new insight into the pharmacological application of cholinergic functional enhancement accompanied by the adoption of the “cholinergic anti-inflammation pathway”. Additionally, the non-cholinergic effects of these drugs, including anti-glutamate-mediated excitotoxicity, anti-oxidative stress, and anti-apoptosis, might also contribute to their clinical benefits. These findings show promise for the use of AChE inhibitors in VaD therapy, and will provide valuable clues for future VaD drug development.
However, certain AChE inhibitors do not always provide consistent results in probable VaD patients, and the data indicating efficacy in cognitive outcomes was derived from older patients likely to have concomitant AD pathology. This caveat supports an existing argument that the putative cholinergic deficit in VaD reflects the presence of concomitant AD pathology. Therefore, because of these controversial results, it will be necessary to establish specific clinical diagnostic criteria and rating scales for VaD and extend studies for longer periods in order to better evaluate the efficacy of these cholinergic agents in future clinical practice.
References
Kalaria RN, Maestre GE, Arizaga R, Friedland RP, Galasko D, Hall K, et al. Alzheimer's disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol 2008; 7: 812–26.
Sloane PD, Zimmerman S, Suchindran C, Reed P, Wang L, Boustani M, et al. The public health impact of Alzheimer's disease, 2000–2050: potential implication of treatment advances. Annu Rev Public Health 2002; 23: 213–31.
Sorrentino G, Migliaccio R, Bonavita V . Treatment of vascular dementia: the route of prevention. Eur Neurol 2008; 60: 217–23.
Bonita R . Epidemiology of stroke. Lancet 1992; 339: 342–4.
Kumaran D, Udayabanu M, Kumar M, Aneja R, Katyal A . Involvement of angiotensin converting enzyme in cerebral hypoperfusion induced anterograde memory impairment and cholinergic dysfunction in rats. Neuroscience 2008; 155: 626–39.
Kuang X, Du JR, Liu YX, Zhang GY, Peng HY . Postischemic administration of Z-Ligustilide ameliorates cognitive dysfunction and brain damage induced by permanent forebrain ischemia in rats. Pharmacol Biochem Behav 2008; 88: 213–21.
Pavlov VA, Tracey KJ . The cholinergic anti-inflammatory pathway. Brain Behav Immun 2005; 19: 493–9.
Jellinger KA . The pathology of ischemic-vascular dementia: An update. J Neurol Sci 2002; 203–204: 153–7.
Jellinger KA . The pathology of “vascular dementia”: a critical update. J Alzheimers Dis 2008; 14: 107–23.
Roman GC . Cholinergic dysfunction in vascular dementia. Curr Psychiatry Rep 2005; 7: 18–26.
Roman GC, Kalaria RN . Vascular determinants of cholinergic deficits in Alzheimer disease and vascular dementia. Neurobiol Aging 2006; 27: 1769–85.
Ni JW, Matsumoto K, Li HB, Murakami Y, Watanabe H . Neuronal damage and decrease of central acetylcholine level following permanent occlusion of bilateral common carotid arteries in rat. Brain Res 1995; 673: 290–6.
Tanaka K, Ogawa N, Asanuma M, Kondo Y, Nomura M . Relationship between cholinergic dysfunction and discrimination learning disabilities in Wistar rats following chronic cerebral hypoperfusion. Brain Res 1996; 729: 55–65.
Zhao Q, Murakami Y, Tohda M, Obi R, Shimada Y, Matsumoto K . Chotosan, a kampo formula, ameliorates chronic cerebral hypoperfusion-induced deficits in object recognition behaviors and central cholinergic systems in mice. J Pharmacol Sci 2007; 103: 360–73.
Zhang LL, Wang JZ, Liu SW, Chen ME . Changes of somatostatin and acetylcholine contents in vascular dementia rats. Acta Acad Med Milit Tert 2004; 26: 714–6. In Chinese (with English abstract).
Borlongan CV, Sumaya IC, Moss DE . Methanesulfonyl fluoride, an acetylcholinesterase inhibitor, attenuates simple learning and memory deficits in ischemic rats. Brain Res 2005; 1038: 50–8.
Francis A, Pulsinelli W . The response of GABAergic and cholinergic neurons to transient cerebral ischemia. Brain Res 1982; 243: 271–8.
Nyberg P, Waller S . Age-dependent vulnerability of brain choline acetyltransferase activity to transient cerebral ischemia in rats. Stroke 1989; 20: 495–500.
Naritomi H . Experimental basis of multi-infarct dementia: memory impairments in rodent models of ischemia. Alzheimer Dis Assoc Disord 1991; 5: 103–11.
Togashi H, Matsumoto M, Yoshioka M, Hirokami M, Minami M, Saito H . Neurochemical profiles in cerebrospinal fluid of stroke-prone spontaneously hypertensive rats. Neurosci Lett 1994; 166: 117–20.
Kimura S, Saito H, Minami M, Togashi H, Nakamura N, Nemoto M, et al. Pathogenesis of vascular dementia in stroke-prone spontaneously hypertensive rats. Toxicology 2000; 153: 167–78.
Kalaria R . Similarities between Alzheimer's disease and vascular dementia. J Neurol Sci 2002; 203–204: 29–34.
Waller SB, Ball MJ, Reynolds MA, London ED . Muscarinic binding and choline acetyltransferase in postmortem brains of demented patients. Can J Neurol Sci 1986; 13: 528–32.
Gottfries CG, Blennow K, Karlsson I, Wallin A . The neurochemistry of vascular dementia. Dementia 1994; 5: 163–7.
Tohgi H, Abe T, Kimura M, Saheki M, Takahashi S . Cerebrospinal fluid acetylcholine and choline in vascular dementia of Binswanger and multiple small infarct types as compared with Alzheimer-type dementia. J Neural Transm 1996; 103: 1211–20.
Wallin A, Sjogren M, Blennow K, Davidsson P . Decreased cerebrospinal fluid acetylcholinesterase in patients with subcortical ischemic vascular dementia. Dement Geriatr Cogn Disord 2003; 16: 200–7.
Mann DM, Yates PO, Marcyniuk B . The nucleus basalis of Meynert in multi-infarct (vascular) dementia. Acta Neuropathol 1986; 71: 332–7.
Keverne JS, Low WC, Ziabreva I, Court JA, Oakley AE, Kalaria RN . Cholinergic neuronal deficits CADASIL. Stroke 2007; 38: 188–91.
Mesulam M, Siddique T, Cohen B . Cholinergic denervation in a pure multi-infarct state: observations on CADASIL. Neurology 2003; 60: 1183–5.
Court J, Perry E, Kalaria R . Neurotransmitter control of the cerebral vasculature and abnormalities in vascular dementia. In: Erkinjuntti T, Gauthier S, editors. Vascular cognitive impairment. Martin Dunitz, London; 2002. p 167–85.
Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T . Anti-inflammatory properties of cholinergic up-regulation: a new role for acetylcholinesterase inhibitors. Neuropharmacology 2006; 50: 540–7.
Wang Q, Tang XN, Yenari MA . The inflammatory response in stroke. J Neuroimmunol 2007; 184: 53–68.
Barone FC, Feuerstein GZ . Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab 1999; 19: 819–34.
Liu R, Wen Y, Perez E, Wang X, Day AL, Simpkins JW, et al. 17beta-Estradiol attenuates blood-brain barrier disruption induced by cerebral ischemia-reperfusion injury in female rats. Brain Res 2005; 1060: 55–61.
Pan J, Konstas AA, Bateman B, Ortolano GA, Pile-Spellman J . Reperfusion injury following cerebral ischemia: pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007; 49: 93–102.
Mehta SL, Manhas N, Raghubir R . Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 2007; 54: 34–66.
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405: 458–62.
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421: 384–8.
Conejero-Goldberg C, Davies P, Ulloa L . Alpha7 nicotinic acetylcholine receptor: a link between inflammation and neurodegeneration. Neurosci Biobehav Rev 2008; 32: 693–706.
Pavlov VA, Tracey KJ . Controlling inflammation: the cholinergic anti-inflammatory pathway. Biochem Soc Trans 2006; 34: 1037–40.
Maelicke A . Allosteric modulation of nicotinic receptors as a treatment strategy for Alzheimer's disease. Dement Geriatr Cogn Disord 2000; 11 Suppl 1: 11–8.
Murakami Y, Zhao Q, Harada K, Tohda M, Watanabe H, Matsumoto K . Choto-san, a Kampo formula, improves chronic cerebral hypoperfusion-induced spatial learning deficit via stimulation of muscarinic M1 receptor. Pharmacol Biochem Behav 2005; 81: 616–25.
De Jesus Moreno Moreno M . Cognitive improvement in mild to moderate Alzheimer's dementia after treatment with the acetylcholine precursor choline alfoscerate: a multicenter, double-blind, randomized, placebo-controlled trial. Clin Ther 2003; 25: 178–93.
Adibhatla RM, Hatcher JF . Cytidine 5'-diphosphocholine (CDP-choline) in stroke and other CNS disorders. Neurochem Res 2005; 30: 15–23.
Kavirajan H, Schneider LS . Efficacy and adverse effects of cholinesterase inhibitors and memantine in vascular dementia: a meta-analysis of randomised controlled trials. Lancet Neurol 2007; 6: 782–92.
Dichgans M, Markus HS, Salloway S, Verkkoniemi A, Moline M, Wang Q, et al. Donepezil in patients with subcortical vascular cognitive impairment: a randomised double-blind trial CADASIL. Lancet Neurol 2008; 7: 310–8.
Kim DH, Yoon BH, Kim YW, Lee S, Shin BY, Jung JW, et al. The seed extract of Cassia obtusifolia ameliorates learning and memory impairments induced by scopolamine or transient cerebral hypoperfusion in mice. J Pharmacol Sci 2007; 105: 82–93.
Wang ZF, Wang J, Zhang HY, Tang XC . Huperzine A exhibits anti-inflammatory and neuroprotective effects in a rat model of transient focal cerebral ischemia. J Neurochem 2008; 106: 1594–603.
Wecker L, Schmidt DE . Central cholinergic function: relationship to choline administration. Life Sci 1979; 25: 375–84.
Parnetti L, Mignini F, Tomassoni D, Traini E, Amenta F . Cholinergic precursors in the treatment of cognitive impairment of vascular origin: ineffective approaches or need for re-evaluation? J Neurol Sci 2007; 257: 264–9.
Higgins JP, Flicker L . Lecithin for dementia and cognitive impairment. Cochrane Database Syst Rev 2003; 3: CD001015.
Domino EF, Mathews BN, Tait SK, Ortiz A . Effects of oral phosphatidylcholine on mouse brain choline and acetylcholine. Arch Int Pharmacodyn Ther 1983; 265: 49–54.
Fioravanti M, Yanagi M . Cytidinediphosphocholine (CDP-choline) for cognitive and behavioural disturbances associated with chronic cerebral disorders in the elderly. Cochrane Database Syst Rev 2005; 2: CD000269.
Abbiati G, Fossati T, Lachmann G, Bergamaschi M, Castiglioni C . Absorption, tissue distribution and excretion of radiolabelled compounds in rats after administration of 14CL-alpha-glycerylphosphorylcholine. Eur J Drug Metab Pharmacokinet 1993; 18: 173–80.
Amenta F, Franch F, Ricci A, Vega JA . Cholinergic neurotransmission in the hippocampus of aged rats: influence of L-alpha-glycerylphosphorylcholine treatment. Ann N Y Acad Sci 1993; 695: 311–3.
Tomassoni D, Avola R, Mignini F, Parnetti L, Amenta F . Effect of treatment with choline alphoscerate on hippocampus microanatomy and glial reaction in spontaneously hypertensive rats. Brain Res 2006; 1120: 183–90.
Parnetti L, Amenta F, Gallai V . Choline alphoscerate in cognitive decline and in acute cerebrovascular disease: an analysis of published clinical data. Mech Ageing Dev 2001; 122: 2041–55.
Bontempi B, Whelan KT, Risbrough VB, Lloyd GK, Menzaghi F . Cognitive enhancing properties and tolerability of cholinergic agents in mice: a comparative study of nicotine, donepezil, and SIB-1553A, a subtype-selective ligand for nicotinic acetylcholine receptors. Neuropsychopharmacology 2003; 28: 1235–46.
Fukatsu T, Miyake-Takagi K, Nagakura A, Omino K, Okuyama N, Ando T, et al. Effects of nefiracetam on spatial memory function and acetylcholine and GABA metabolism in microsphere-embolized rats. Eur J Pharmacol 2002; 453: 59–67.
Crespi F . Nefiracetam. Daiichi Seiyaku. Curr Opin Investig Drugs 2002; 3: 788–93.
Zhao X, Kuryatov A, Lindstrom JM, Yeh JZ, Narahashi T . Nootropic drug modulation of neuronal nicotinic acetylcholine receptors in rat cortical neurons. Mol Pharmacol 2001; 59: 674–83.
Wonnacott S, Marks MJ . Nicotine: not just for cigarettes anymore. Drug Discov Today 1999; 4: 490–492.
Maelicke A, Albuquerque E . New approach to drug therapy in Alzheimer's dementia. Drug Discov Today 1996; 1: 53–9.
Shimada Y, Yang Q, Yokoyama K, Goto H, Kasahara Y, Sekiya N, et al. Choto-san prevents occurrence of stroke and prolongs life span in stroke-prone spontaneously hypertensive rats. Am J Chin Med 2003; 31: 79–85.
Terasawa K, Shimada Y, Kita S . Choto-san in the treatment of vascular dementia: a double-blind, placebo-controlled study. Phytomedicine 1997; 4: 15–22.
Shimada Y, Terasawa K, Yamamoto T . A well-controlled study of Choto-san and placebo in the treatment of vascular dementia. J Tradit Med 1994; 11: 246–55.
Thal LJ, Forrest M, Loft H, Mengel H . Lu 25–109, a muscarinic agonist, fails to improve cognition in Alzheimer's disease. Lu 25–109 Study Group. Neurology 2000; 54: 421–6.
Birks J . Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev 2006; 1: CD005593.
Farlow MR . Use of antidementia agents in vascular dementia: beyond Alzheimer disease. Mayo Clin Proc 2006; 81: 1350–8.
Moretti R, Torre P, Antonello RM, Cazzato G, Griggio S, Bava A . An open-label pilot study comparing rivastigmine and low-dose aspirin for the treatment of symptoms specific to patients with subcorticl vascular dementia. Curr Ther Res 2002; 63: 443–58.
Black S, Roman GC, Geldmacher DS, Salloway S, Hecker J, Burns A, et al. Efficacy and tolerability of donepezil in vascular dementia: positive results of a 24-week, multicenter, international, randomized, placebo-controlled clinical trial. Stroke 2003; 34: 2323–30.
Erkinjuntti T, Kurz A, Small GW, Bullock R, Lilienfeld S, Damaraju CV . An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003; 25: 1765–82.
Yin FM, Du YY, Wang LE . Huperzine A interventions on vascular dementia. Modern Rehabilit 2001; 5: 74–5. In Chinese.
Wang YE, Yue DX, Tang XC . Anticholinesterase activity of huperzine A. Acta Pharmacol Sin 1986; 7: 110–3. In Chinese (with English abstract).
Zhang ZX, Wang XD, Chen QT, Shu L, Wang JZ, Shan GL . Clinical efficacy and safety of huperzine A in treatment of mild to moderate Alzheimer's disease, a placebo-controlled, doube-blind, randomized trial. Natl Med J China 2002; 82: 941–4. In Chinese (with English abstract).
Wang RP, Zhao ZK, Hu LL . Effect of huperzine A on cognitive function and quality of life in vascular dementia: 36 cases for 6 months follow up study. Chin J Clin Rehabilit 2004; 8: 3892. In Chinese.
Zhang RW, Tang XC, Han YY, Sang GW, Zhang YD, Ma YX, et al. Drug evaluation of huperzine A in the treatment of senile memory disorders. Acta Pharmacol Sin 1991; 12: 250–2. In Chinese (with English abstract).
Sun CY, Chen XY . A self-control study of huperzine A on memory deficits in patients with multiple infarctions. Henan Yi Yao Xin Xi 1998; 6: 31–2. In Chinese.
Wang YQ . Treatment of tablet huperzine A on 17 patients with vascular dementia. China Pharm 2000; 9: 32. In Chinese.
Zhong ZG, Liang KZ . Clinical observation of huperzine A on 29 patients with vascular dementia. J Hainan Med Coll 2004; 10: 251–2. In Chinese.
Chang JJ . Huperzine A vs pyritinol in treating dementia from mulltiple infarctions. New Drugs Clin Remedies 1997; 16: 333–4. In Chinese (with English abstract).
Wang LM, Han YF, Tang XC . Huperzine A improves cognitive deficits caused by chronic cerebral hypoperfusion in rats. Eur J Pharmacol 2000; 398: 65–72.
Zhou J, Zhang HY, Tang XC . Huperzine A attenuates cognitive deficits and hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci Lett 2001; 313: 137–40.
Wang LS, Zhou J, Shao XM, Tang XC . Huperzine A attenuates cognitive deficits and brain injury in neonatal rats after hypoxia-ischemia. Brain Res 2002; 949: 162–70.
Wang ZF, Tang LL, Yan H, Wang YJ, Tang XC . Effects of huperzine A on memory deficits and neurotrophic factors production after transient cerebral ischemia and reperfusion in mice. Pharmacol Biochem Behav 2006; 83: 603–11.
Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, et al. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res 1999; 45: 500–9.
Hedtjarn M, Mallard C, Hagberg H . Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J Cereb Blood Flow Metab 2004; 24: 1333–51.
Wang ZF, Tang XC . Huperzine A protects C6 rat glioma cells against oxygen-glucose deprivation-induced injury. FEBS Lett 2007; 581: 596–602.
Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, et al. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 2004; 35: 987–91.
Zheng CY, Zhang HY, Tang XC . Huperzine A attenuates mitochondrial dysfunction after middle cerebral artery occlusion in rats. J Neurosci Res 2008; 86: 2432–40.
Wang R, Yan H, Tang XC . Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol Sin 2006; 27: 1–26.
Pratt RD, Perdomo CA . Donepezil-treated patients with probable vascular dementia demonstrate cognitive benefits. Ann N Y Acad Sci 2002; 977: 513–22.
Mendez MF, Younesi FL, Perryman KM . Use of donepezil for vascular dementia: preliminary clinical experience. J Neuropsychiatry Clin Neurosci 1999; 11: 268–70.
Wilkinson D, Doody R, Helme R, Taubman K, Mintzer J, Kertesz A, et al. Donepezil in vascular dementia: a randomized, placebo-controlled study. Neurology 2003; 61: 479–86.
Malouf R, Birks J . Donepezil for vascular cognitive impairment. Cochrane Database Syst Rev 2004; 1: CD004395.
Fujiki M, Kobayashi H, Uchida S, Inoue R, Ishii K . Neuroprotective effect of donepezil, a nicotinic acetylcholine-receptor activator, on cerebral infarction in rats. Brain Res 2005; 1043: 236–41.
Fujiki M, Kubo T, Kamida T, Sugita K, Hikawa T, Abe T, et al. Neuroprotective and antiamnesic effect of donepezil, a nicotinic acetylcholine-receptor activator, on rats with concussive mild traumatic brain injury. J Clin Neurosci 2008; 15: 791–6.
Takada-Takatori Y, Kume T, Ohgi Y, Izumi Y, Niidome T, Fujii T, et al. Mechanism of neuroprotection by donepezil pretreatment in rat cortical neurons chronically treated with donepezil. J Neurosci Res 2008; 86: 3575–83.
Hiranita T, Nawata Y, Sakimura K, Anggadiredja K, Yamamoto T . Suppression of methamphetamine-seeking behavior by nicotinic agonists. Proc Natl Acad Sci U S A 2006; 103: 8523–7.
Takada-Takatori Y, Kume T, Ohgi Y, Fujii T, Niidome T, Sugimoto H, et al. Mechanisms of alpha7-nicotinic receptor up-regulation and sensitization to donepezil induced by chronic donepezil treatment. Eur J Pharmacol 2008; 590: 150–6.
Tyagi E, Agrawal R, Nath C, Shukla R . Effect of anti-dementia drugs on LPS induced neuroinflammation in mice. Life Sci 2007; 80: 1977–83.
Reale M, Iarlori C, Gambi F, Feliciani C, Isabella L, Gambi D . The acetylcholinesterase inhibitor, donepezil, regulates a Th2 bias in Alzheimer's disease patients. Neuropharmacology 2006; 50: 606–13.
Ogura H, Kosasa T, Kuriya Y, Yamanishi Y . Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find Exp Clin Pharmacol 2000; 22: 609–13.
Moretti R, Torre P, Antonello RM, Cazzato G . Rivastigmine in subcortical vascular dementia: a comparison trial on efficacy and tolerability for 12 months follow-up. Eur J Neurol 2001; 8: 361–2.
Moretti R, Torre P, Antonello RM, Cazzato G, Bava A . Rivastigmine in subcortical vascular dementia: an open 22-month study. J Neurol Sci 2002; 203–204: 141–6.
Kumar V, Anand R, Messina J, Hartman R, Veach J . An efficacy and safety analysis of Exelon in Alzheimer's disease patients with concurrent vascular risk factors. Eur J Neurol 2000; 7: 159–69.
Moretti R, Torre P, Antonello RM, Cazzato G, Pizzolato G . Different responses to rivastigmine in subcortical vascular dementia and multi-infarct dementia. Am J Alzheimers Dis Other Demen 2008; 23: 167–76.
Tanaka K, Ogawa N, Mizukawa K, Asanuma M, Kondo Y, Nishibayashi S, et al. Acetylcholinesterase inhibitor ENA-713 protects against ischemia-induced decrease in pre- and postsynaptic cholinergic indices in the gerbil brain following transient ischemia. Neurochem Res 1994; 19: 117–22.
Tanaka K, Ogawa N, Asanuma M, Hirata H, Kondo Y, Nakayama N, et al. Effects of the acetylcholinesterase inhibitor ENA-713 on ischemia-induced changes in acetylcholine and aromatic amine levels in the gerbil brain. Arch Int Pharmacodyn Ther 1993; 323: 85–96.
Chen Y, Shohami E, Bass R, Weinstock M . Cerebro-protective effects of ENA713, a novel acetylcholinesterase inhibitor, in closed head injury in the rat. Brain Res 1998; 784: 18–24.
Tanaka K, Mizukawa K, Ogawa N, Mori A . Post-ischemic administration of the acetylcholinesterase inhibitor ENA-713 prevents delayed neuronal death in the gerbil hippocampus. Neurochem Res 1995; 20: 663–7.
Nizri E, Irony-Tur-Sinai M, Faranesh N, Lavon I, Lavi E, Weinstock M, et al. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J Neuroimmunol 2008; 203: 12–22.
Woodruff-Pak DS, Lander C, Geerts H . Nicotinic cholinergic modulation: galantamine as a prototype. CNS Drug Rev 2002; 8: 405–26.
Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV . Efficacy of galantamine in probable vascular dementia and Alzheimer's disease combined with cerebrovascular disease: a randomised trial. Lancet 2002; 359: 1283–90.
Craig D, Birks J . Galantamine for vascular cognitive impairment. Cochrane Database Syst Rev 2006; CD004746.
Iliev AI, Traykov VB, Mantchev GT, Stoykov I, Prodanov D, Yakimova KS, et al. A post-ischaemic single administration of galanthamine, a cholinesterase inhibitor, improves learning ability in rats. J Pharm Pharmacol 2000; 52: 1151–6.
Ji X, Li C, Lu Y, Chen Y, Guo L . Post-ischemic continuous administration of galantamine attenuates cognitive deficits and hippocampal neurons loss after transient global ischemia in gerbils. Neurosci Lett 2007; 416: 92–5.
Lorrio S, Sobrado M, Arias E, Roda JM, Garcia AG, Lopez MG . Galantamine postischemia provides neuroprotection and memory recovery against transient global cerebral ischemia in gerbils. J Pharmacol Exp Ther 2007; 322: 591–9.
Giunta B, Ehrhart J, Townsend K, Sun N, Vendrame M, Shytle D, et al. Galantamine and nicotine have a synergistic effect on inhibition of microglial activation induced by HIV-1 gp120. Brain Res Bull 2004; 64: 165–70.
Santos MD, Alkondon M, Pereira EF, Aracava Y, Eisenberg HM, Maelicke A, et al. The nicotinic allosteric potentiating ligand galantamine facilitates synaptic transmission in the mammalian central nervous system. Mol Pharmacol 2002; 61: 1222–34.
Dajas-Bailador FA, Heimala K, Wonnacott S . The allosteric potentiation of nicotinic acetylcholine receptors by galantamine is transduced into cellular responses in neurons: Ca2+ signals and neurotransmitter release. Mol Pharmacol 2003; 64: 1217–26.
Woodruff-Pak DS, Vogel RW 3rd, Wenk GL . Mecamylamine interactions with galantamine and donepezil: effects on learning, acetylcholinesterase, and nicotinic acetylcholine receptors. Neuroscience 2003; 117: 439–47.
Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, et al. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun 2009; 23: 41–5.
Moss DE, Berlanga P, Hagan MM, Sandoval H, Ishida C . Methanesulfonyl fluoride (MSF): a double-blind, placebo-controlled study of safety and efficacy in the treatment of senile dementia of the Alzheimer type. Alzheimer Dis Assoc Disord 1999; 13: 20–5.
Kuang X, Yao Y, Du JR, Liu YX, Wang CY, Qian ZM . Neuroprotective role of Z-ligustilide against forebrain ischemic injury in ICR mice. Brain Res 2006; 1102: 145–53.
Peng HY, Du JR, Zhang GY, Kuang X, Liu YX, Qian ZM, et al. Neuroprotective effect of Z-ligustilide against permanent focal ischemic damage in rats. Biol Pharm Bull 2007; 30: 309–12.
Zhou J, Fu Y, Tang XC . Huperzine A and donepezil protect rat pheochromocytoma cells against oxygen-glucose deprivation. Neurosci Lett 2001; 306: 53–6.
Nobili F, Vitali P, Canfora M, Girtler N, De Leo C, Mariani G, et al. Effects of long-term donepezil therapy on rCBF of Alzheimer's patients. Clin Neurophysiol 2002; 113: 1241–8.
Lojkowska W, Ryglewicz D, Jedrzejczak T, Minc S, Jakubowska T, Jarosz H, et al. The effect of cholinesterase inhibitors on the regional blood flow in patients with Alzheimer's disease and vascular dementia. J Neurol Sci 2003; 216: 119–26.
Ved HS, Koenig ML, Dave JR, Doctor BP . Huperzine A, a potential therapeutic agent for dementia, reduces neuronal cell death caused by glutamate. Neuroreport 1997; 8: 963–8.
Li W, Pi R, Chan HH, Fu H, Lee NT, Tsang HW, Pu Y, et al. Novel dimeric acetylcholinesterase inhibitor bis7-tacrine, but not donepezil, prevents glutamate-induced neuronal apoptosis by blocking N-methyl-D-aspartate receptors. J Biol Chem 2005; 280: 18179–88.
Akasofu S, Kimura M, Kosasa T, Sawada K, Ogura H . Study of neuroprotection of donepezil, a therapy for Alzheimer's disease. Chem Biol Interact 2008; 175: 222–6.
Ezoulin MJ, Dong CZ, Liu Z, Li J, Chen HZ, Heymans F, et al. Study of PMS777, a new type of acetylcholinesterase inhibitor, in human HepG2 cells. Comparison with tacrine and galanthamine on oxidative stress and mitochondrial impairment. Toxicol In Vitro 2006; 20: 824–31.
Casademont J, Miro O, Rodriguez-Santiago B, Viedma P, Blesa R, Cardellach F . Cholinesterase inhibitor rivastigmine enhance the mitochondrial electron transport chain in lymphocytes of patients with Alzheimer's disease. J Neurol Sci 2003; 206: 23–6.
Acknowledgements
The works that referred to huperzine A were supported by the Ministry of Science and Technology of China (No 2004CB518907); the National Natural Science Foundation of China (No 30801402) and the Knowledge Innovation Program of the Chinese Academy of Sciences (No SIMM0709QN-2).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, J., Zhang, Hy. & Tang, Xc. Cholinergic deficiency involved in vascular dementia: possible mechanism and strategy of treatment. Acta Pharmacol Sin 30, 879–888 (2009). https://doi.org/10.1038/aps.2009.82
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.82
Keywords
This article is cited by
-
Daidzein Attenuates Ovariectomy-Induced Cognitive Deficits by Improving Cortical Endothelial Function in Rats
Revista Brasileira de Farmacognosia (2023)
-
Nootropic effect of Syzygium polyanthum (Wight) Walp leaf extract in chronic cerebral hypoperfusion rat model via cholinergic restoration: a potential therapeutic agent for dementia
Advances in Traditional Medicine (2023)
-
Fosgonimeton, a Novel Positive Modulator of the HGF/MET System, Promotes Neurotrophic and Procognitive Effects in Models of Dementia
Neurotherapeutics (2023)
-
Hexahydrocurcumin Attenuates Neuronal Injury and Modulates Synaptic Plasticity in Chronic Cerebral Hypoperfusion in Rats
Molecular Neurobiology (2023)
-
Ameliorative effect of Clerodendrum volubile extract on oxidative stress, cholinergic alterations, and proinflammatory cytokine in scopolamine-induced memory deficits in mice
Nutrire (2022)
