
Key Points
-
Aggressive cancers that do not respond to traditional chemotherapy regimes are often resistant owing to the lack of p53-dependent apoptosis. New therapies are being developed that aim to restore p53 tumour suppression to cancer cells.
-
50% of cancers lose p53 function as a result of mutations that produce single amino-acid substitutions in the core DNA-binding domain. In other cancers, wild-type p53 is targeted for degradation by cellular (MDM2) or viral (E6) oncoproteins.
-
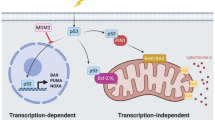
Mutant p53 is inactive for transcriptional transactivation. Cancer cells therefore accumulate mutant p53 (in the absence of MDM2) and fail to induce p53-dependent growth arrest, DNA repair or apoptosis. The mutant protein is a target for rescue by drugs that bind to the core domain to reverse the effects of mutation.
-
Mutations can reduce the DNA-binding affinity and thermodynamic stability of the core domain to varying extents, described by five mutant classes. The effect of a mutation is largely dictated by its location within the core-domain structure. Mutations in the β-sandwich induce unfolding, whereas mutations in the DNA-binding surface can have a greater effect on DNA binding.
-
The activity of certain p53 mutants can be restored by second-site suppressor mutations that introduce an additional DNA contact, correct local structural distortion or increase the stability of the core domain structure. Potential therapeutic drugs may rescue mutant p53 by the same mechanisms.
-
Small molecules, identified in a screen for protein folding, stabilize the wild-type and mutant p53 core domain, presumably by binding specifically to the native protein. Correction of mutant folding restores p53 function to cancer cells and inhibits tumour growth in mice.
-
These prototype compounds require increased potency for use at concentrations that are practical for cancer therapy. This may be achieved by further structure-based drug design once the binding site on the p53 core domain is characterized. Small molecules have also been discovered that relieve the inhibition of p53 by MDM2 and E6 in cancers that express wild-type p53.
Abstract
One protein — p53 — plays nemesis to most cancers by condemning damaged cells to death or quarantining them for repair. But the activity of p53 relies on its intact native conformation, which can be lost following mutation of a single nucleotide. With thousands of such mutations identified in patients, how can a future cancer drug buttress this fragile protein structure and restore the cell's natural defence?
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Finette, B. A., Homans, A. C. & Albertini, R. J. Emergence of genetic instability in children treated for leukemia. Science 288, 514–517 (2000).
Komarov, P. G. et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733–1737 (1999).Traditional cancer therapies are invasive. DNA damage can occur in healthy cells and induce p53-dependent apoptosis. This paper describes an inhibitor of p53 that protects against these effects.
Sherr, C. J. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 60, 3689–3695 (2000).
Hupp, T. R., Lane, D. P. & Ball, K. L. Strategies for manipulating the p53 pathway in the treatment of human cancer. Biochem. J. 352, 1–17 (2000). A recent review highlighting the progress of new approaches to cancer therapy based on restoring the p53 tumour-suppressor pathway.
Foster, B. A. et al. Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510 (1999).Reports the first screen for prototype pharmacological small molecules that stabilize the native structure of wild-type p53 conformation.
Lane, D. P. & Crawford, L. V. T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263 (1979).
Linzer, D. I. & Levine, A. J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52 (1979).
Lane, D. P. & Benchimol, S. p53: oncogene or anti-oncogene? Genes Dev. 4, 1–8 (1990).
Scheffner, M. et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136 (1990).
Kubbutat, M. H., Jones, S. N. & Vousden, K. H. Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997).
Haupt, Y. et al. Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 (1997).
Jones, S. N. et al. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208 (1995).
Montes de Oca Luna, R., Wagner, D. S. and Lozano, G. Rescue of early embryonic lethality in Mdm2-deficient mice by deletion of p53. Nature 378, 203–206 (1995).
de Rozieres, S. et al. The loss of Mdm2 induces p53-mediated apoptosis. Oncogene 19, 1691–1697 (2000).
Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53 network. Nature 408, 307–310 (2000).A good general introduction to p53 and its role in tumour suppression.
Nigro, J. M. et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708 (1989).
Zhang, Y. et al. Cell cycle inhibition by the anti-angiogenic agent TNP-470 is mediated by p53 and p21WAF1/CIP1. Proc. Natl Acad. Sci. USA 97, 6427–6432 (2000).
Hainaut, P. & Hollstein, M. p53 and human cancer: the first ten thousand mutations. Adv. Cancer Res. 77, 81–137 (2000).
Cho, Y. et al. Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science 265, 346–355 (1994).Presents the first crystal structure of p53 core domain. The location and structural consequences of hot-spot TP53 mutations are discussed.
Milner, J. Flexibility: the key to p53 function? Trends Biochem. Sci. 20, 49–51 (1995).
Rolley, N., Butcher, S. & Milner, J. Specific DNA binding by different classes of human p53 mutants. Oncogene 11, 763–770 (1995).
el-Deiry, W. S. et al. Definition of a consensus binding site for p53. Nature Genet. 1, 45–49 (1992).
Pavletich, N. P., Chambers, K. A. & Pabo, C. O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 7, 2556–2564 (1993).
Cohen, P. A. et al. Biochemical characterization of different conformational states of the Sf9 cell-purified p53 His175 mutant protein. FEBS Lett. 463, 179–184 (1999).
Hansen, S., Hupp, T. R. & Lane, D. P. Allosteric regulation of the thermostability and DNA binding activity of human p53 by specific interacting proteins. CRC Cell Transformation Group. J. Biol. Chem. 271, 3917–3924 (1996).
Bullock, A. N. et al. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl Acad. Sci. USA 94, 14338–14342 (1997).
Bullock, A. N., Henckel, J. & Fersht, A. R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256 (2000).Mutants of p53 are shown to have a wide range of stabilities and DNA-binding affinities. The mutant conformation is redefined as five mutant classes of differing severity that correlate with the site of mutation.
Wong, K. B. et al. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl Acad. Sci. USA 96, 8438–8442 (1999).
Wieczorek, A. M. et al. Structure-based rescue of common tumor-derived p53 mutants. Nature Med. 2, 1143–1146 (1996).
Brachmann, R. K. et al. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 17, 1847–1859 (1998).Second-site mutations are reported that rescue structural mutants of p53. Their action provides a model for small molecule drug therapy.
Nikolova, P. V. et al. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 19, 370–378 (2000).The effect of suppressor mutations is characterized by stability, DNA binding and NMR studies.
Nikolova, P. V. et al. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl Acad. Sci. USA 95, 14675–14680 (1998).
Milner, J. & Medcalf, E. A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65, 765–774 (1991).
Zhao, K. et al. Crystal structure of the mouse p53 core DNA-binding domain at 2.7 Å resolution. J. Biol. Chem. 276, 12120–12127 (2001).
Shaulian, E. et al. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol. Cell Biol. 12, 5581–5592 (1992).
Chene, P. In vitro analysis of the dominant negative effect of p53 mutants. J. Mol. Biol. 281, 205–209 (1998).
Nielsen, L. L. & Maneval, D. C. p53 tumor suppressor gene therapy for cancer. Cancer Gene Ther. 5, 52–63 (1998).
Ghaneh, P. et al. Adenovirus-mediated transfer of p53 and p16 (INK4a) results in pancreatic cancer regression in vitro and in vivo. Gene Ther. 8, 199–208 (2001).
Mateu, M. G. & Fersht, A. R. Mutually compensatory mutations during evolution of the tetramerization domain of tumor suppressor p53 lead to impaired hetero-oligomerization. Proc. Natl Acad. Sci. USA 96, 3595–3599 (1999).
Stavridi, E. S. et al. Change in oligomerization specificity of the p53 tetramerization domain by hydrophobic amino acid substitutions. Protein Sci. 8, 1773–1779 (1999).
Hupp, T. R. & Lane, D. P. Allosteric activation of latent p53 tetramers. Curr. Biol. 4, 865–875 (1994).
Anderson, M. E. et al. Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: implications for regulation. Mol. Cell Biol. 17, 6255–6264 (1997).
Kim, A. L. et al. Conformational and molecular basis for induction of apoptosis by a p53 C-terminal peptide in human cancer cells. J. Biol. Chem. 274, 34924–34931 (1999).
Hupp, T. R., Sparks, A. & Lane, D. P. Small peptides activate the latent sequence-specific DNA binding function of p53. Cell 83, 237–245 (1995).
Selivanova, G. et al. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nature Med. 3, 632–638 (1997).
Druker, B. J. et al. Efficacy and safety of a specific inhibitor of the BCR–ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037 (2001).
Hupp, T. R. Regulation of p53 protein function through alterations in protein-folding pathways. Cell. Mol. Life Sci. 55, 88–95 (1999).
Giannakakou, P. et al. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nature Cell Biol. 2, 709–717 (2000).
Gorre, M. E. et al. Clinical resistance to STI-571 cancer therapy caused by BCR–ABL gene mutation or amplification. Science 293, 876–880 (2001).
Stoll, R. et al. Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry 40, 336–344 (2001).
Grüschow, S. et al. Isolation and structure elucidation of chlorofusin, a novel p53-MDM2 antagonist from a Fusarium sp. J. Am. Chem. Soc. 123, 554–560 (2001).
Hietanen, S. et al. Activation of p53 in cervical carcinoma cells by small molecules. Proc. Natl Acad. Sci. USA 97, 8501–8506 (2000).
McCormick, F. Interactions between adenovirus proteins and the p53 pathway: the development of ONYX-015. Semin. Cancer Biol. 10, 453–459 (2000).
Gibbs, J. B. Mechanism-based target identification and drug discovery in cancer research. Science 287, 1969–1973 (2000).
Dewson, G. Small, but deadly: small-molecule inhibition of Bcl-2 homologue heterodimerization. Trends Biochem. Sci. 26, 218–219 (2001).
Sigal, A. & Rotter, V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 60, 6788–6793 (2000).
Gaiddon, C. et al. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell Biol. 21, 1874–1887 (2001).
Strano, S. et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J. Biol. Chem. 275, 29503–29512 (2000).
Koga, H. & Deppert, W. Identification of genomic DNA sequences bound by mutant p53 protein (Gly245→Ser) in vivo. Oncogene 19, 4178–4183 (2000).
Deppert, W. Binding of MAR-DNA elements by mutant p53: possible implications for its oncogenic functions. J. Cell Biochem. 62, 172–180 (1996).
Guo, A. et al. The function of PML in p53-dependent apoptosis. Nature Cell. Biol. 2, 730–736 (2000).
Fogal, V. et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19, 6185–6195 (2000).
Kussie, P. H. et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 (1996).
Jeffrey, P. D., Gorina, S. & Pavletich, N. P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267, 1498–1502 (1995).
Clore, G. M. et al. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nature Struct. Biol. 2, 321–333 (1995).
Lee, W. et al. Solution structure of the tetrameric minimum transforming domain of p53. Nature Struct. Biol. 1, 877–890 (1994).
Rustandi, R. R., Baldisseri, D. M. & Weber, D. J. Structure of the negative regulatory domain of p53 bound to S100B (betabeta). Nature Struct. Biol. 7, 570–574 (2000).
Koradi, R., Billeter, M. & Wuthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 29–32, 51–5 (1996).
Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11, 281–296 (1991).
Author information
Authors and Affiliations
Corresponding author
Related links
Glossary
- LARGE T ANTIGEN
-
Tumour-associated protein encoded by the DNA-transforming virus SV40. It binds RB and p53 to overcome inhibition of cell growth.
- DNA-DAMAGE CHECKPOINTS
-
The cell cycle can be arrested before the G1–S or G2–M phase transitions at checkpoints where DNA damage is detected.
- β-SANDWICH
-
Tertiary protein structure common to all immunoglobulins. Consists of β-strands arranged into two β-sheets that pack together as a sandwich.
- ENTHALPY OF DENATURATION
-
In order for a protein to be denatured as the temperature increases above the physiological level, many favourable stabilizing interactions within the protein have to be broken. The breaking of these requires input of energy from the surroundings — the 'enthalpy of denaturation'. The larger the enthalpy, the sharper the thermal transition.
- HETERONUCLEAR SINGLE QUANTUM CORRELATION
-
(HSQC). Magnetization is transferred between the nuclei of 1H and 15N atoms in amide groups. A plot of the 15N against 1H chemical shifts produces a single crosspeak for each amino acid.
- TWO-DIMENSIONAL NMR
-
NMR (nuclear magnetic resonance) spectroscopy measures the resonant absorption of radiofrequency radiation by magnetic nuclei in a magnetic field ('chemical shifts' that vary with environment; for example, with folding). Two-dimensional experiments use two magnetic nuclei, typically 1H and either 13C or 15N.
- DOMINANT NEGATIVE
-
A defective protein that retains interaction capabilities and so distorts or competes with normal proteins.
- E2F-1
-
A heterodimeric transcription factor that promotes cell cycle progression from G1 to S phase after phosphorylation and dissociation of RB. In tumours, deregulated E2F-1 promotes p53- and p73-dependent apoptosis.
Rights and permissions
About this article
Cite this article
Bullock, A., Fersht, A. Rescuing the function of mutant p53. Nat Rev Cancer 1, 68–76 (2001). https://doi.org/10.1038/35094077
Issue Date:
DOI: https://doi.org/10.1038/35094077
This article is cited by
-
Association of TP53 rs1042522 G > C, MDM2 rs2279744 T > G, and miR-34b/c rs4938723 T > C polymorphisms with aneuploidy pregnancy susceptibility
BMC Pregnancy and Childbirth (2023)
-
Arsenic trioxide extends survival of Li–Fraumeni syndrome mimicking mouse
Cell Death & Disease (2023)
-
Partial p53 reactivation is sufficient to induce cancer regression
Journal of Experimental & Clinical Cancer Research (2022)
-
Transgenic construction and functional miRNA analysis identify the role of miR-7 in prostate cancer suppression
Oncogene (2022)
-
Potential of rescue and reactivation of tumor suppressor p53 for cancer therapy
Biophysical Reviews (2022)
