


Background
Many vital biological transformations involve the incorporation of one (monooxygenases) or two (dioxygenases) O-atoms from molecular oxygen into organic substrates. Enzymes that utilize oxygen must coordinate the delivery of four protons and four electrons to O2 in order to prevent the formation of harmful molecular oxidants (O2 −, HO2 • H2O2, and HO•), collectively known as reactive oxygen species (ROS). It is our view that the risks posed by reactive intermediates are so great that oxygen-utilizing enzymes have protection mechanisms to help them avoid inactivation when the primary electron/proton transfer mechanism is disrupted.
The mechanism of O2 reduction by cytochrome c oxidase illustrates some of the challenges facing these enzymes (Wikström, Reference Wikström2012; Yu et al. Reference Yu, Egawa, Shinzawa-Itoh, Yoshikawa, Yeh, Rousseau and Gerfen2011, Reference Yu, Egawa, Shinzawa-Itoh, Yoshikawa, Guallar, Yeh, Rousseau and Gerfen2012). Reaction of the fully four-electron reduced enzyme (CuA II,I, FeII-heme a, FeII-heme a 3, and CuB I) with O2 generates an intermediate designated as PR. When the two-electron reduced, mixed valence enzyme (CuA II,II, FeIII-heme a, FeII-heme a 3, and CuB I) reacts with O2, the PM intermediate is formed. The O–O bond has been cleaved in both PR and PM to produce FeIV(O)-heme a 3 and CuB II in the binuclear site. The difference between PR and PM is in the source of the fourth electron: PM is thought to have a Tyr244 radical (bovine numbering), whereas the fourth electron in PR is provided by FeII-heme a. When PM is prepared using H2O2, the hole on (TyrO•)244 is believed to migrate through (Trp•+)236 to (TyrO•)129; the latter residue is suggested to participate in proton pumping (Yu et al. Reference Yu, Egawa, Shinzawa-Itoh, Yoshikawa, Guallar, Yeh, Rousseau and Gerfen2012). The key point is that Tyr244 is available to fill the gap when the fourth electron required for O2 reduction cannot be supplied by FeII-heme a (Wikström, Reference Wikström2012; Yu et al. Reference Yu, Egawa, Shinzawa-Itoh, Yoshikawa, Guallar, Yeh, Rousseau and Gerfen2012).
In many oxygenases, including the cytochromes P450 (P450) and the 2-oxo-glutarate-dependent nonheme iron oxygenases (2OG-Fe), the four electrons required for O2 reduction have different origins (Fig. 1). Typically, two electrons are delivered from a reductase (P450) or co-substrate (2OG), and the remaining two electrons are provided by the organic substrate (Denisov et al. Reference Denisov, Makris, Sligar and Schlichting2005; Hausinger, Reference Hausinger2004; Whitehouse et al. Reference Whitehouse, Bell and Wong2012). In the consensus mechanism for iron oxygenases, the first two electrons induce O–O bond cleavage, producing a powerfully oxidizing ferryl species. The ferryl complex abstracts a hydrogen atom from the substrate and HO• rebound leads to hydroxylated product (Denisov et al. Reference Denisov, Makris, Sligar and Schlichting2005; Hausinger, Reference Hausinger2004; Whitehouse et al. Reference Whitehouse, Bell and Wong2012). For enzymes with broad substrate specificities, or when operating in the presence of xenobiotic compounds, the fidelity of substrate oxidation is less than 100%, with potentially damaging consequences (Chen et al. Reference Chen, Comeaux, Herbst, Saban, Kennedy, Maroney and Knapp2008; De Matteis et al. Reference De Matteis, Ballou, Coon, Estabrook and Haines2012; Denisov et al. Reference Denisov, Baas, Grinkova and Sligar2007a ; Grinkova et al. Reference Grinkova, Denisov, McLean and Sligar2013; Saban et al. Reference Saban, Flagg and Knapp2011; Staudt et al. Reference Staudt, Lichtenb and Ullrich1974). This circumstance is manifested as an increased molar ratio of O2 consumption to substrate hydroxylation (uncoupling). We think it likely that organisms have evolved protection mechanisms to guard against deactivation of oxygenase enzymes in the event of uncoupled O2 consumption. In particular, we suggest that radical transfer pathways are employed to deliver strongly oxidizing holes (E°~1 V versus NHE) from ferryl complexes in active sites to less fragile regions of oxygenases.

Fig. 1. Schematic representation of the catalytic mechanisms of P450 and 2OG-Fe oxygenases: RH, substrate; 2OG, 2-oxoglutarate; Suc, succinate. Black arrows indicate the functional substrate hydroxylation pathways. Blue arrows indicate oxidase uncoupling pathways.
In this perspective, we will advance the hypothesis that there are potentially protective radical chains in P450 and 2OG-Fe; but first we will review what we know about the factors controlling hopping through aromatic amino acids in multistep electron tunneling constructs designed in azurin, a prototypal cupredoxin.
Radical transfer pathways in azurin
Azurin is a robust cupredoxin (128 residues) that is amenable to site-directed mutagenesis and surface-labeling with photosensitizers (Farver & Pecht, Reference Farver and Pecht2011; Gray & Winkler, Reference Gray and Winkler2010; Reece & Nocera, Reference Reece and Nocera2009; Wilson et al. Reference Wilson, Yu and Lu2013). Oxidized radicals of Trp and Tyr are substantially stronger acids than their neutral precursors (Trp, pK a > 14; Trp•+, pK a = 4; TyrOH, pK a = 10; TyrOH•+, pK a = −1) (Aubert et al. Reference Aubert, Vos, Mathis, Eker and Brettel2000; Bonin et al. Reference Bonin, Costentin, Louault, Robert, Routier and Saveant2010; Costentin et al. Reference Costentin, Louault, Robert and Saveant2009; Harriman, Reference Harriman1987; Jovanic et al. Reference Jovanic, Harriman and Simic1986); management of the acidic proton is a critically important factor controlling radical formation with these amino acids. Proton management is particularly challenging for buried amino acids and, thus far, we have not succeeded in detecting buried Trp or Tyr radicals as electron transfer (ET) intermediates. Our kinetics data indicate that surface exposed Trp•+ and NO2TyrO• radicals can, in appropriate constructs, accelerate CuI oxidation by distant Re- and Ru-diimine complexes (Shih et al. Reference Shih, Museth, Abrahamsson, Blanco-Rodriguez, Di Bilio, Sudhamsu, Crane, Ronayne, Towrie, Vlcek, Richards, Winkler and Gray2008; Warren et al. Reference Warren, Herrera, Hill, Winkler and Gray2013a ).
Multistep ET through Trp and Tyr radicals in azurin
We have used Pseudomonas aeruginosa azurin as a test bed for mechanistic investigations of Trp and Tyr radical formation in protein ET reactions (Blanco-Rodriguez et al. Reference Blanco-Rodriguez, Di Bilio, Shih, Museth, Clark, Towrie, Cannizzo, Sudhamsu, Crane, Sykora, Winkler, Gray, Zalis and Vlcek2011; Shih et al. Reference Shih, Museth, Abrahamsson, Blanco-Rodriguez, Di Bilio, Sudhamsu, Crane, Ronayne, Towrie, Vlcek, Richards, Winkler and Gray2008; Takematsu et al. Reference Takematsu, Williamson, Blanco-Rodríguez, Sokolová, Nikolovski, Kaiser, Towrie, Clark, Vlček, Winkler and Gray2013; Warren et al. Reference Warren, Ener, Vlček, Winkler and Gray2012, Reference Warren, Herrera, Hill, Winkler and Gray2013a ). Our initial investigation revealed that CuI oxidation by a photoexcited ReI–diimine complex (ReI(CO)3(4,7-dimethyl-1,10-phenanthroline)) covalently bound at His124 on a His124Gly123Trp122Met121 β-strand (ReHis124Trp122CuI-azurin) occurs in a few nanoseconds, fully two orders of magnitude faster than documented for single-step electron tunneling at a 19-Å donor–acceptor distance, owing to a two-step hopping mechanism involving a Trp•+ radical intermediate (Shih et al. Reference Shih, Museth, Abrahamsson, Blanco-Rodriguez, Di Bilio, Sudhamsu, Crane, Ronayne, Towrie, Vlcek, Richards, Winkler and Gray2008).
Our work on multistep ET in sensitizer-modified azurin is informed by semiclassical ET theory (Marcus & Sutin, Reference Marcus and Sutin1985). Given a particular spatial arrangement of redox cofactors, we can predict driving-force dependences of the relative time constants for single-step (τ ss = 1/k ss) and multistep (τ hop) electron transport (Warren et al. Reference Warren, Ener, Vlček, Winkler and Gray2012). Alternatively, given the redox and reorganization energetics, we can predict the hopping propensity for different cofactor arrangements (Warren et al. Reference Warren, Herrera, Hill, Winkler and Gray2013a ). We considered three Ru(2,2′-bipyridine)2(imidazole)(HisX)-labeled azurins (RuHis107, RuHis124, and RuHis126) and examined the hopping advantage (τ ss/τ hop) for a protein with a generalized intermediate (Int) situated between a diimine-RuIII oxidant and CuI (Warren et al. Reference Warren, Herrera, Hill, Winkler and Gray2013a ). In all cases, the greatest hopping advantage occurs in systems where the Int–RuIII distance is up to 5 Å shorter than the Int–CuI distance. The hopping advantage increases as systems orient nearer a linear Donor–Int–Acceptor configuration, owing to minimized intermediate tunneling distances. The smallest predicted hopping advantage is in RuHis124 azurin, which has the shortest Ru–Cu distance of the three proteins. The hopping advantage is nearly lost as ΔG° for the first step (RuIII ← Int) rises above +0.15 eV. Isoergic initial steps provide a wide distribution of arrangements, where advantages as great as 104 are possible (for a fixed donor–acceptor distance of 23.7 or 25.4 Å). A slightly exergonic Int → RuIII step provides an even larger distribution of arrangements for productive hopping, which will be the case as long as the driving force for the first step is not more favorable than that for overall transfer.
We tested these predictions experimentally in three Ru–His-labeled azurins using nitrotyrosinate (NO2TyrO–) as a redox intermediate (RuHis107(NO2TyrOH)109; RuHis124(NO2TyrOH)122; and RuHis126−(NO2TyrOH)122; E°′(NO2TyrO•/–) ≈ 1.02 V versus NHE) (Fig. 2) (Warren et al. Reference Warren, Herrera, Hill, Winkler and Gray2013a ). The first two systems have cofactor placements that are close to the predicted optimum; the last system has a larger first-step distance, which is predicted to decrease the hopping advantage. The phenol pK a of 3-nitrotyrosine (7.2) permitted us to work at near-neutral pH, rather than high pH (>10) required for hopping with tyrosinate. ET via nitrotyrosinate avoids the complexities associated with the proton-coupled redox reactions of tyrosine. We found specific rates of CuI oxidation more than 10 times greater than those of single-step ET in the corresponding azurins lacking NO2TyrOH, confirming that NO2TyrO– accelerates long-range ET. The results are in excellent agreement with hopping maps developed using semiclassical ET theory and parameters derived from our body of protein ET measurements (Gray & Winkler, Reference Gray and Winkler2010; Warren et al. Reference Warren, Ener, Vlček, Winkler and Gray2012, Reference Warren, Herrera, Hill, Winkler and Gray2013a ).
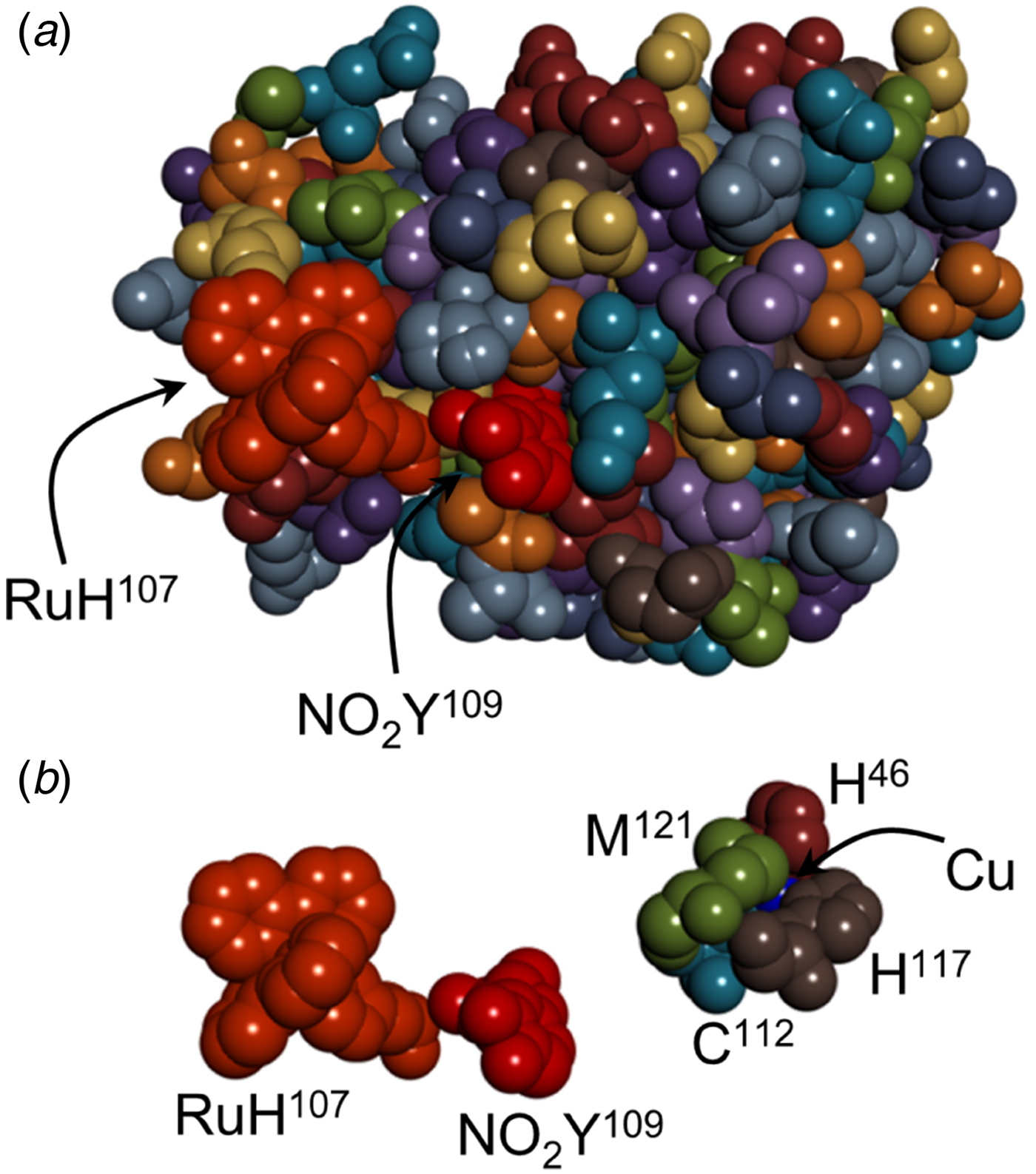
Fig. 2. (a) Space-filling structural model of RuHis107NO2TyrOH109Cu-azurin. (b) Space filling models of the residues comprising the hole-hopping pathway from Cu to RuHis107.
Potential radical transfer pathways in iron oxygenases
The cytochromes P450 are members of a superfamily of heme oxygenases that perform two broad functional roles: xenobiotic metabolism and biosynthesis (Denisov et al. Reference Denisov, Makris, Sligar and Schlichting2005; Johnson & Stout, Reference Johnson and Stout2013; Nebert et al. Reference Nebert, Wikvall and Miller2013; Orr et al. Reference Orr, Ripp, Ballard, Henderson, Scott, Obach, Sun and Kalgutkar2012; Whitehouse et al. Reference Whitehouse, Bell and Wong2012). The oxygenation chemistry catalyzed by some P450 enzymes is tightly coupled to substrate hydroxylation: one mole of product is produced for each mole of O2 consumed. In many enzymes, however, particularly the eukaryotic proteins with broad substrate specificities, hydroxylation is much less efficiently coupled to O2 consumption (frequently less than 10%) (Denisov et al. Reference Denisov, Baas, Grinkova and Sligar2007a ; Grinkova et al. Reference Grinkova, Denisov, McLean and Sligar2013; Staudt et al. Reference Staudt, Lichtenb and Ullrich1974). When the enzyme does not transfer an O-atom to substrate, it can produce ROS (O2 −, H2O2) or a second H2O molecule (Puntarulo & Cederbaum, Reference Puntarulo and Cederbaum1998). The production of ROS can lead to rapid degradation of the enzyme and other harmful chemistry. In the case of oxidase chemistry (formation of 2H2O from O2), two reducing equivalents must be delivered by sources other than the substrate. When a CYP enzyme binds a refractory substrate, ferryl formation is likely to proceed, but substrate hydroxylation is inhibited. Under these circumstances, chains of redox-active Tyr, Trp, Cys, and/or Met residues can direct the oxidizing hole to the protein periphery where it can react with intracellular antioxidants such as glutathione.
Enzymes from the 2OG-Fe superfamily use 2-oxoglutarate as a 2-electron donating co-substrate, Fe2+ as a cofactor, and O2 to effect the hydroxylation of organic substrates (Fig. 1). The 2OG-Fe enzymes exhibit a wide array of biological functions including collagen biosynthesis, lysyl hydroxylation of RNA splicing proteins, DNA repair, RNA modification, chromatin regulation, epidermal growth factor-like domain modification, hypoxia sensing, and fatty acid metabolism (Mantri et al. Reference Mantri, Zhang, McDonough and Schofield2012; Rose et al. Reference Rose, McDonough, King, Kawamura and Schofield2011). The 2OG-Fe oxygenase enzymes have conserved double-stranded β-helix folds with octahedral Fe-binding sites with the HXD/E…H triad providing two His imidazole ligands and one monodentate carboxylate ligand. The remaining three coordination sites in the resting enzyme are occupied by O-donors from 2OG and a water ligand.
Several 2OG-Fe enzymes have been reported to undergo autocatalyzed oxidative modifications of aromatic amino acids. In the taurine-2OG dioxygenase that catalyzes the conversion of taurine to bisulfite, EPR data indicate the transient formation of a Tyr73-based radical that converts to an FeIII-catecholate (Mantri et al. Reference Mantri, Zhang, McDonough and Schofield2012). In 2,4-dichlorophenoxyacetate oxygenase (TfdA) and factor-inhibiting hypoxia-inducible factor (FIH) there is evidence for Trp hydroxylation when substrate is unavailable (Mantri et al. Reference Mantri, Zhang, McDonough and Schofield2012). These aromatic amino acid oxidations lead to inactivation of the enzyme. As with P450, we suggest that radical chains of Trp, Tyr, Cys, and/or Met residues in 2OG-Fe hydroxylases protect the enzymes from damage in the event of slow or unsuccessful substrate hydroxylation by diverting the powerfully oxidizing hole from FeIV(O) to the protein surface, where it can react with intracellular reductants (e.g. glutathione). This diversion of oxidizing equivalents would extend the functional lifetime of an enzyme.
When considering the many remarkable transformations catalyzed by natural enzymes, it is easy to be left with the impression that these macromolecules are perfect catalysts that, after millions of years of tinkering, have solved the riddle of simultaneously maximizing speed, selectivity, and specificity. Upon closer inspection, however, heme and nonheme oxygenases are far from perfect catalysts, yet manage to accomplish their primary functions. Indeed, in many oxygenases, the coupling between oxygen consumption and substrate hydroxylation is extremely low. The most abundant P450 in human liver, CYP3A4, is a case in point (Denisov et al. Reference Denisov, Grinkova, McLean and Sligar2007b ; Grinkova et al. Reference Grinkova, Denisov, McLean and Sligar2013). For enzyme incorporated into nanodiscs (Grinkova et al. Reference Grinkova, Denisov and Sligar2010), the coupling of substrate hydroxylation to NADH consumption was ≤16% for testosterone as a substrate, ≤10% for bromocriptine, and 2% for tamoxifen (Grinkova et al. Reference Grinkova, Denisov, McLean and Sligar2013). It is fair to say that, although the primary CYP3A4 function may be substrate hydroxylation, the primary enzyme activity is distributed more or less equally between H2O2 and H2O production (Grinkova et al. Reference Grinkova, Denisov, McLean and Sligar2013). Indeed, it would not be inaccurate to characterize CYP3A4 as a flawed oxidase that occasionally oxygenates organic substrates. More importantly, unless the enzyme was protected from damage in the event of uncoupled turnover, CYP3A4 would function not as a catalyst but as a stoichiometric reagent. A similar situation exists for uncoupled turnover in the 2OG-Fe enzymes.
The active sites of heme and nonheme oxygenases often are deeply buried within a polypeptide matrix. Consequently, powerfully oxidizing active site holes cannot efficiently migrate in single-step tunneling reactions to the enzyme surface for reduction by external reagents (Winkler & Gray, Reference Winkler and Gray2014a , Reference Winkler and Gray b ). We have shown that multistep tunneling reactions can be hundreds to thousands of times faster than their single-step counterparts (Shih et al. Reference Shih, Museth, Abrahamsson, Blanco-Rodriguez, Di Bilio, Sudhamsu, Crane, Ronayne, Towrie, Vlcek, Richards, Winkler and Gray2008; Warren et al. Reference Warren, Ener, Vlček, Winkler and Gray2012, Reference Warren, Herrera, Hill, Winkler and Gray2013a , Reference Warren, Winkler and Gray b ). Radical transfer pathways composed of Tyr, Trp, Cys, and Met residues are ideally suited to deliver active-site oxygenase holes to enzyme surfaces when reaction with substrate is disrupted.
A biologically useful Fe-oxygenase protection mechanism requires that a fine balance be struck between substrate reaction and hole migration to the surface. Overly efficient hole migration would lower enzyme hydroxylation activity, while a sluggish pathway would be ineffective at protecting the enzyme. Active-site hole scavenging in P450 by the natural reductase may be possible, but the timing of this reaction would be extremely variable, owing to fluctuations in reductase concentration. In the 2OG-Fe enzymes, there is no reductase that could protect the enzyme. An intraprotein radical transfer mechanism can be tuned to provide the proper balance between enzyme protection and substrate reaction. We suggest that the first step in the hole-migration pathway is the critical determinant of ferryl survival time. Once a radical forms on the first residue in the pathway (the gateway residue), further migration to the surface is rapid. In the potential pathways that we have identified, the distance from the active site to the first pathway residue is often longer than subsequent steps. In addition to the longer distance, proton coupling and enzyme conformational changes could contribute to limiting the rate of the first step in the transfer chain.
CYP102A1
CYP102A1 from Bacillus megaterium (also known as P450 BM3) is a rare example of a bacterial Class II cytochrome P450 enzyme in which both reductase and heme domains are contained within a single polypeptide chain (Miura & Fulco, Reference Miura and Fulco1974; Narhi & Fulco, Reference Narhi and Fulco1986). The enzyme catalyzes the remarkably rapid hydroxylation of long-chain fatty acids using NAD(P)H and O2, without the presence of any other proteins or cofactors (Narhi & Fulco, Reference Narhi and Fulco1986). The full-length enzyme (CYP102A1HR) has been expressed in Escherichia coli, as have independent heme (CYP102A1H) and reductase (CYP102A1R) domains (Boddupalli et al. Reference Boddupalli, Estabrook and Peterson1990, Reference Boddupalli, Oster, Estabrook and Peterson1992; Li et al. Reference Li, Darwish and Poulos1991a ; Narhi et al. Reference Narhi, Wen and Fulco1988; Oster et al. Reference Oster, Boddupalli and Peterson1991). The individual domains, as well as an assembly between the heme domain and a flavin-containing reductase domain, have been structurally characterized (Girvan et al. Reference Girvan, Seward, Toogood, Cheesman, Leys and Munro2007; Sevrioukova et al. Reference Sevrioukova, Immoos, Poulos and Farmer2000; Warman et al. Reference Warman, Roitel, Neeli, Girvan, Seward, Murray, McLean, Joyce, Toogood, Holt, Leys, Scrutton and Munro2005). The soluble, 119 kDa CYP102A1H enzyme serves as a convenient model system for the more complex membrane-bound enzyme assemblies (Whitehouse et al. Reference Whitehouse, Bell and Wong2012).
Uncoupled substrate, O2, and NAD(P)H consumption in P450 catalysis is a well-recognized and relatively common phenomenon (De Matteis et al. Reference De Matteis, Dawson, Pons and Pipino2002, Reference De Matteis, Ballou, Coon, Estabrook and Haines2012; Denisov et al. Reference Denisov, Baas, Grinkova and Sligar2007a ; Grinkova et al. Reference Grinkova, Denisov, McLean and Sligar2013; Puntarulo & Cederbaum, Reference Puntarulo and Cederbaum1998; Staudt et al. Reference Staudt, Lichtenb and Ullrich1974). If two reducing equivalents are not delivered to O2 by the substrate, then alternative sources are necessary to avoid ROS production and/or enzyme degradation. In some cases, the extra equivalents can be delivered by NAD(P)H, leading to NAD(P)H : O2 molar consumption ratios greater than 1 (De Matteis et al. Reference De Matteis, Ballou, Coon, Estabrook and Haines2012). Exogenous reductants such as bilirubin and uroporphyrinogen have been shown to contribute reducing equivalents during NAD(P)H/O2 CYP102A1 turnover in the presence of halogenated (perfluorolaurate) substrates (De Matteis et al. Reference De Matteis, Ballou, Coon, Estabrook and Haines2012). Although it is possible that an active site hole could tunnel to the protein surface in a single step, a multistep radical transfer mechanism would be far more efficient. There are two attractive radical transfer pathways from the CYP102A1 heme to the protein surface (Fig. 3) (Girvan et al. Reference Girvan, Seward, Toogood, Cheesman, Leys and Munro2007). Pathway I is comprised of heme–Trp96–Trp90–Tyr334; pathway II is heme–Cys156–Tyr115–Met112–Tyr305.

Fig. 3. (a) Space-filling structural model of the heme domain of CYP102A1 (PDB #2IJ2) highlighting the surface locations of terminal residues in pathways I (Tyr334) and II (Tyr305). (b) Space-filling model of the residues comprising CYP102A1 radical transfer pathways I and II. Blue spheres represent structurally resolved water molecules.
CYP102A1 radical transfer pathway I
The shortest direct distance between aromatic atoms of CYP102A1 Trp96 and the heme is 7.3 Å and Trp(Nε)96 is hydrogen bonded to the heme propionate (Girvan et al. Reference Girvan, Seward, Toogood, Cheesman, Leys and Munro2007). Sequence alignment (UniProtKB) in the P450 family suggests that Trp is conserved at this position in >75% of the members of this group. Interestingly, of the 698 sequences with Trp at this position, all but 5 derive from eukaryotic sources, whereas about half of the proteins with His at this position derive from bacterial or archaeal sources. In this regard, it is noteworthy that archaeal CYP119 does not have a Trp residue at this site and is the only P450 in which Cmpd-1 has been characterized (Park et al. Reference Park, Yamane, Adachi, Shiro, Weiss, Maves and Sligar2002; Rittle & Green, Reference Rittle and Green2010). The strong conservation of the Trp96 residue has been noted previously (Munro et al. Reference Munro, Malarkey, McKnight, Thomson, Kelly, Price, Lindsay, Coggins and Miles1994). To the best of our knowledge, no role other than structural has been reported for this highly conserved Trp residue in P450 (Whitehouse et al. Reference Whitehouse, Bell and Wong2012).
We suggest that Trp96 is the gateway residue for hole transfer from the heme to the protein surface during uncoupled turnover. Studies of the reactions of substrate-free P450cam (CYP101) with peracids revealed that a second intermediate (Cmpd-ES) forms as a result of ET from a Tyr residue to Cmpd-1 (Schünemann et al. Reference Schünemann, Lendzian, Jung, Contzen, Barra, Sligar and Trautwein2004; Spolitak et al. Reference Spolitak, Dawson and Ballou2005, Reference Spolitak, Dawson and Ballou2006, Reference Spolitak, Dawson and Ballou2008). A Cmpd-ES intermediate has been detected in CYP102A1 and Trp96 has been implicated as one of the residues hosting the oxidized radical (Raner et al. Reference Raner, Thompson, Haddy, Tangham, Bynum, Reddy, Ballou and Dawson2006). Addition of NADPH to Cmpd-ES of the CYP102HR holoenzyme regenerates the ferric resting state; and formation of these radicals may play a protective role during uncoupled P450 catalysis (Spolitak et al. Reference Spolitak, Dawson and Ballou2006). A combined computational/experimental investigation of CYP102A1 implicated buried Trp96, Trp90, His92, and Tyr334 residues as components of an ET pathway that could deliver reducing equivalents to Cmpd-1 from the protein surface (Vidal-Limon et al. Reference Vidal-Limon, Aguila, Ayala, Batista and Vazquez-Duhalt2013). The shortest aromatic contacts in this chain are: Trp96–Trp90, 8.4 Å; Trp90–Tyr334, 4.4 Å (Girvan et al. Reference Girvan, Seward, Toogood, Cheesman, Leys and Munro2007). The environment around Tyr334 appears well-suited for radical formation: the phenol hydroxyl group is hydrogen-bonded to both a carboxylate (Asp68) and a water molecule (HOH1215).
Our prior studies of P450 ET reactions are consistent with involvement of Trp96 in a radical transfer pathway to the heme (Ener et al. Reference Ener, Lee, Winkler, Gray and Cheruzel2010). We have found that RuII(bpy)2(phen•––Cys97) can deliver an electron across 24 Å to the FeIII-heme in 20 μs, and RuIII(bpy)2(phen–Cys97)CYP102A1H can oxidize the heme to a porphyrin radical in under 2 μs (Ener et al. Reference Ener, Lee, Winkler, Gray and Cheruzel2010). The latter reaction is particularly rapid given the low driving force (<200 meV) expected for the transformation. We have prepared a Trp96His mutant and found that RuIII(bpy)2(phen–Cys97)(His96)CYP102A1H does not promote photochemical heme oxidation to Cmpd-2. Electron transfer to the FeIII-heme from RuII(bpy)2(phen•–−Cys97)(His96), however, is unaffected by the Trp96His mutation.
CYP102A1 radical transfer pathway II
The second potential radical transfer pathway in CYP102A1, heme–Cys156–Tyr115–Met112–Tyr305, does not appear as favorable as pathway I, due largely to a long distance between the heme and the first step in the path. The distance from Cys(Sγ)156 to the closest heme aromatic carbon atom (10.8 Å) is slightly longer than the shortest aromatic–aromatic contact between the heme and Tyr115 (10.2 Å). If a radical is formed on Tyr115, then hole transport to the surface Tyr305 via Met(Sδ)112 could provide a secondary protection route.
Potential radical transfer pathways in 2OG-Fe oxygenases
TauD
The 2-oxoglutarate nonheme iron oxygenases catalyze substrate hydroxylation reactions in a fashion that is reminiscent of the cytochromes P450, but with some critical distinctions (Fig. 1). The consensus mechanism for catalysis involves Fe2+ binding to the apo-enzyme followed by 2OG incorporation. Substrate binding induces loss of the water ligand from Fe2+, creating a vacant coordination site for O2 binding. Oxidation of 2OG produces CO2, succinate, and an FeIV(O) center that is thought to hydroxylate substrate via the usual H-atom abstraction, hydroxyl rebound cycle (Mantri et al. Reference Mantri, Zhang, McDonough and Schofield2012; Rose et al. Reference Rose, McDonough, King, Kawamura and Schofield2011). The 2OG-Fe hydroxylases differ from the P450 enzymes in that substrate hydroxylation proceeds from the FeIV(O) oxidation level (equivalent to P450 Cmpd-2). The E. coli 2OG-Fe enzyme TauD is synthesized under conditions of sulfur deprivation (Hausinger, Reference Hausinger2004); large quantities of TauD have been prepared by over expression in E. coli BL21(DE3)(pME4141) cells (Eichhorn et al. Reference Eichhorn, van der Ploeg, Kertesz and Leisinger1997; Ryle et al. Reference Ryle, Padmakumar and Hausinger1999). The enzyme catalyzes the hydroxylation of taurine (2-aminoethanesulfonate), producing an unstable species that decomposes into sulfite and aminoacetaldehyde (Hausinger, Reference Hausinger2004). In the absence of taurine, the enzyme will slowly consume O2 and become inactivated: protein analysis indicates hydroxylation of Tyr73 (Koehntop et al. Reference Koehntop, Marimanikkuppam, Ryle, Hausinger and Que2006; Ryle et al. Reference Ryle, Liu, Muthukumaran, Ho, Koehntop, McCracken, Que and Hausinger2003). Although with deuterated substrates coupling between oxygen consumption and substrate hydroxylation is diminished, 2OG oxidation is not, suggesting that FeIV(O) continues to be formed in the presence of refractory substrates; and bis-Tris buffer, a potential reducing agent, decreases coupling between O2 activation and C–H hydroxylation (McCusker & Klinman, Reference McCusker and Klinman2009). We suggest that when FeIV(O) is unable to effect substrate hydroxylation, the oxidizing hole is directed to the protein surface where it can be reduced by external reagents.
TauD radical transfer pathways
We have identified two possible radical transfer pathways in the structure of TauD: the most attractive pathway from Fe to the surface has four Trp residues: Fe–Trp248–Trp128–Trp240–Trp238; relevant distances are: Fe–Trp248, 4.8 Å; Trp248–Trp128, 3.1 Å; Trp128–Trp240, 3.7 Å; Trp240–Trp238, 3.7 Å (Fig. 4) (O'Brien et al. Reference O'Brien, Schuller, Yang, Dillard and Lanzilotta2003). The structure of this Trp chain compares favorably to that identified in E. coli DNA photolyase (4–5 Å separations) (Byrdin et al. Reference Byrdin, Eker, Vos and Brettel2003; Lukacs et al. Reference Lukacs, Eker, Byrdin, Villette, Pan, Brettel and Vos2006). The photolyase chain has just three Trp residues, and hole migration from FADH•* to Trp306 at the protein surface is complete in less than 10 ns (Byrdin et al. Reference Byrdin, Eker, Vos and Brettel2003; Lukacs et al. Reference Lukacs, Eker, Byrdin, Villette, Pan, Brettel and Vos2006). We anticipate that a hole injected by FeIV(O)-TauD into Trp248 should migrate to Trp238 at the surface in less than 1 μs. A secondary radical transfer pathway in TauD [Fe–Tyr73–Tyr164–(Trp174,Tyr162)] is of particular interest because hydroxylated Tyr73 has been found during turnover in the absence of taurine (Koehntop et al. Reference Koehntop, Marimanikkuppam, Ryle, Hausinger and Que2006; Ryle et al. Reference Ryle, Liu, Muthukumaran, Ho, Koehntop, McCracken, Que and Hausinger2003). Both Trp174 and Tyr162 are well-exposed at the enzyme surface and both (or just one) of these residues could be involved in a radical transfer pathway. Relevant distances are: Fe–Tyr73, 6.5 Å; Tyr73–Tyr164, 5.0 Å; Tyr164–Trp174, 4.2 Å; Tyr164–Tyr162, 7.6 Å; Trp174–Tyr162, 8.8 Å (O'Brien et al. Reference O'Brien, Schuller, Yang, Dillard and Lanzilotta2003).

Fig. 4. (a) Space-filling structural model of E. coli TauD (PDB #1OS7) highlighting the surface locations of terminal residues in postulated radical transfer pathways (Trp238, Trp174, and Tyr162). (b) Space-filling model of the residues comprising TauD radical transfer pathways.
Outlook
Functional radical transfer pathways have been identified in several enzymes, including ribonucleotide reductase (Argirevic et al. Reference Argirevic, Riplinger, Stubbe, Neese and Bennati2012; Holder et al. Reference Holder, Pizano, Anderson, Stubbe and Nocera2012; Offenbacher et al. Reference Offenbacher, Burns, Sherrill and Barry2013a , Reference Offenbacher, Minnihan, Stubbe and Barry b ; Sjöberg, Reference Sjöberg1997; Stubbe & van der Donk, Reference Stubbe and van der Donk1998; Stubbe et al. Reference Stubbe, Nocera, Yee and Chang2003; Worsdorfer et al. Reference Worsdorfer, Conner, Yokoyama, Livada, Seyedsayamdost, Jiang, Silakov, Stubbe, Bollinger and Krebs2013; Yokoyama et al. Reference Yokoyama, Smith, Corzilius, Griffin and Stubbe2011), photosystem II (Boussac et al. Reference Boussac, Rappaport, Brettel and Sugiura2013; Keough et al. Reference Keough, Zuniga, Jenson and Barry2013; Sjoholm et al. Reference Sjoholm, Styring, Havelius and Ho2012), DNA photolyase (Aubert et al. Reference Aubert, Mathis, Eker and Brettel1999, Reference Aubert, Vos, Mathis, Eker and Brettel2000; Byrdin et al. Reference Byrdin, Eker, Vos and Brettel2003; Kodali et al. Reference Kodali, Siddiqui and Stanley2009; Li et al. Reference Li, Heelis and Sancar1991b ; Lukacs et al. Reference Lukacs, Eker, Byrdin, Villette, Pan, Brettel and Vos2006; Sancar, Reference Sancar2003; Taylor, Reference Taylor1994; Woiczikowski et al. Reference Woiczikowski, Steinbrecher, Kubař and Elstner2011), and MauG (Davidson & Liu, Reference Davidson and Liu2012; Davidson & Wilmot, Reference Davidson and Wilmot2013; Geng et al. Reference Geng, Dornevil, Davidson and Liu2013; Yukl et al. Reference Yukl, Liu, Krzystek, Shin, Jensen, Davidson, Wilmot and Liu2013). If radical transfer pathways do indeed provide protection mechanisms for enzymes operating at high electrochemical potentials, then it is likely that they will be found in many more redox-active enzymes. A survey of oxidoreductases in the protein data bank reveals that nearly 80% of structurally characterized peroxidases, oxygenases, and dioxygenases (enzyme classes EC 1.11, 1.13, and 1.14; 587 structures with sequence identity less than 90%) contain chains of 2 or more redox-active residues (Tyr, Trp, heme, Fe, and Cu) separated by no more than 5 Å (Fig. 5). The fraction increases to almost 90% if the cutoff distance is increased to 8 Å. We think it very likely that hole hopping through these types of radical transfer chains greatly reduces the production of ROS that destroy enzymes and other molecules in living cells.

Fig. 5. Distributions of radical transfer chain lengths among structurally characterized oxidoreductases from enzyme sub-classes EC 1.11 (peroxidases, blue), 1.13 (oxygenases, green), and 1.14 (dioxygenases, red). Radical transfer chains are defined to be composed of Tyr, Trp, heme, Fe, and Cu residues. Tyr residues were included only if a carboxylate (Asp, Glu) oxygen atom, an imidazole (His) nitrogen atom, or a water molecule was within 4 Å of the Tyr hydroxyl oxygen atom.
Acknowledgments
We thank Maraia Ener, Jeff Warren, Lionel Cheruzel, Kana Takematsu, and Oliver Shafaat for helpful discussions.
Financial support
Research reported in this publication was supported by The National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number R01DK019038 to HBG and JRW. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the Arnold and Mabel Beckman Foundation.






 Open access
Open access