
Abstract
Grape production in China is significantly impacted by white rot disease, which is caused by Coniella diplodiella (Speg.) Sacc. This study analyzes the differences in leaf transcriptomes and phenotypes of two grape species, ‘Manicure Finger (Vitis vinifera L.)’ and ‘0940 (Vitis davidii Foex)’, following inoculation with C. diplodiella. Leaf anatomy and H2O2 content confirm the greater resistance of '0940' to C. diplodiella compared to 'Manicure Finger.' Comparative transcriptome analysis reveals that the defense mechanism of '0940' against C. diplodiella involves sesquiterpenoid and triterpenoid biosynthesis, plant-pathogen interactions, sulfur relay systems, suberin and wax biosynthesis, monoterpenoid biosynthesis, as well as flavonoid and flavonol biosynthesis pathways. Using Weighted Gene Co-expression Network Analysis (WGCNA), we identified three modules highly correlated with C. diplodiella resistance and 125 candidate genes, including resistant genes (R genes), pattern-recognition receptors (PRRs), and pathogenesis-related proteins genes (PR genes), which may play important roles in grape resistance to this disease.
Similar content being viewed by others

Introduction
Grapes represent a globally significant horticultural crop, serving both as fresh fruit and raw material for various products. According to the World Food and Agriculture Organization statistics, grapes are predominantly produced in China, with an impressive yield of 11.2 million tons (https://www.fao.org/faostat/en/#data/QCL). The primary grape cultivar in China is Vitis vinifera L., which exhibits high susceptibility to several fungal diseases (Li et al. 2008; Rex et al. 2014; Zhang et al. 2017; Vezzulli et al. 2019). This susceptibility arises from developmental stage of grapevines coinciding with the rainy season and warm summer temperatures, creating conditions conducive to pathogen proliferation (Chethana et al. 2017).
Grape white rot disease, attributed to Coniella diplodiella (Speg.) Sacc, can result in yield losses of up to 16.3% by infiltrating leaves, branches, and fruits through wounds (Li et al. 2008). Traditionally, controlling C. diplodiella in grape production relied on chemical methods, entailing significant costs and potential environmental and food safety concerns (Riaz et al. 2011). In light of molecular breeding advancements, selecting disease-resistant genes and breeding resistant varieties have emerged as practical and cost-effective strategies for white rot management (Li et al. 2008; Zhang et al. 2017). Previous research has identified Chinese wild grapes as a source of resistance, despite the high susceptibility of most Eurasian grape varieties to white rot. For example, resistance to white rot was investigated across 78 grape varieties, including European, American, and Chinese wild grapes. Among these, Chinese wild grapes, specifically V. davidii, exhibited the highest resistance, while 'Manicure Finger' (Vitis. vinifera) was proved to be the most susceptible (Zhang et al. 2017). Two quantitative trait loci (QTL) associated with grape white rot were identified on chromosomes 3 and 14 (Su et al. 2021; Li et al. 2023). Furthermore, several candidate genes for C. diplodiella resistance were discovered. Transcriptome analysis of grapevine varieties with varying white rot resistance levels revealed distinct modes of pathogenesis-related proteins 1 gene (PR1 gene) regulation (Su et al. 2019). Additionally, WRKY53 exhibited the highest expression in V. davidii after 12 h of C. diplodiella induction, and this gene was found to enhance resistance to C. diplodiella in Arabidopsis thaliana (Zhang et al. 2019a). Liu et al. (2021) conducted de novo assembly of the white rot strain WR01 genome and identified the effector Coniella diplodiella effector 1 (CDE1), which can inhibit programmed cell death induced by B cell lymphoma2-associated X.
Plant-pathogen interactions constitute a bidirectional communication process. Pathogens that are successfully infected can manipulate plant metabolism and biological functions, creating a favorable environment for their own growth and reproduction. Conversely, when plants encounter pathogen infections, recognition occurs, triggering various defense mechanisms for self-preservation. Two immune mechanisms have been identified in plants to defend against pathogen invasion. The first mechanism involves pathogen-associated molecular patterns (PAMPs), recognized by pattern-recognition receptors (PRRs), termed pattern-triggered immunity (PTI) (Boller and Felix 2009; Jones and Dangl 2006). Pathogens can secrete effectors to suppress PTI, but plants possess resistant (R) proteins capable of recognizing these effectors and trigger an immune response, known as effector-triggered immunity (ETI) (Jones and Dangl 2006; Spoel and Dong 2012). ETI induces the synthesis of methyl salicylic acid and glycerol 3-phosphate, promoting salicylic acid (SA) accumulation in uninfected tissues. This enables tissues to acquire systemic acquired resistance (SAR) (Fu and Dong 2013; Jones and Dangl 2006; Spoel and Dong 2012). RNA sequencing (RNA-seq) serves as a vital tool for studying plant-pathogen interactions, particularly in gene expression analysis, functional gene discovery, and the identification of key disease resistance pathways (Naidoo et al. 2018; Stark et al. 2019). It has been employed in pathogen interaction studies in grapes, apples, and pears. These research findings indicate that various pathways, including plant hormones, reactive oxygen species (ROS), hormone signaling, and a series of resistance (R) genes, are involved in plant defense. Previous research has primarily focused on European grape and American Vitis species (Ghaffari et al. 2020; Polesani et al. 2010), with limited investigation into the interaction mechanisms between Chinese wild grape and pathogens. V. davidii has shown high resistance to white rot, but our understanding of how C. diplodiella interacts with V. davidii at the molecular level is limited.
In this study, we conducted phenotypic and comparative transcriptomic analyses following C. diplodiella infection in the susceptible cultivar ‘Manicure Finger’ (V. vinifera) and the resistant cultivar ‘0940’ (V. davidii). Our results unveiled the key Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway responsible for ‘0940’ (V. davidii) resistance to white rot. Notably, Weighted Gene Co-expression Network Analysis (WGCNA) identified essential genes contributing to resistance against C. diplodiella in '0940.' This study establishes a foundational understanding for further exploration of grape resistance mechanisms to C. diplodiella.
Materials and methods
Plant material and inoculation treatment
The ‘0940’ (V. davidii, Vd0940) and ‘Manicure Finger’ (V. vinifera, VvMF) varieties were cultivated at the National Repository for Grapevine (Zhengzhou, China). Fungal strain WR01, C. diplodiella, was cultured on potato glucose agar (PDA) medium at 28°C in darkness. Leaves from both grape varieties, harvested from the vineyard, were punctured with a sterile needle to disrupt the upper epidermal layer. A PDA block containing fungal mycelium was then placed over the wounds. These prepared leaves were kept on moist filter paper in Petri dishes and incubated at 28°C. Sampling was done at two time points: 24 and 48 h post inoculation (hpi), with uninfected leaves at 0 h as the control. The experiment involved three biological replicates, and all samples were cryopreserved in liquid nitrogen for subsequent RNA extraction.
Leaf phenotype and anatomy
Lesion diameters on the leaves of both Vd0940 and VvMF were measured with a vernier caliper, using data from at least three distinct biological replicates. Microscopic damage was visualized through trypan blue and aniline blue staining after pathogen infection. Trypan blue staining of leaves followed the method outlined by Li et al. (Li et al. 2019a). Fragments of grape leaves (1 cm × 1 cm) were excised and immersed in trypan blue buffer (20 mL of lactic acid, 20 mL of glycerol, 20 mL of phenol, 20 mg of trypan blue, and 20 mL of distilled water) for 2 min. They were then decolorized overnight in a 2.5 g/mL chloral hydrate solution. After three rinses with water, the specimens were preserved in a 20% glycerol solution for examination under an optical microscope. Grape leaves were subjected to aniline blue staining, following the methodology outlined by Li et al. (Li et al. 2021). Paraffin-embedded grape leaf sections underwent deparaffinization in xylene I and xylene II for 20 min each, followed by destaining in absolute ethanol I, absolute ethanol II, and 75% alcohol (each for 5 min). Subsequently, the samples were rinsed extensively in tap water, repeated six to seven times, and then incubated in aniline blue solution for 5 min. Afterward, the rinsed specimens were delicately dried at 60°C, immersed in xylene for transparency, and sealed with a neutral adhesive. All samples were observed using an upright optical microscope, NIKON ECLIPSE E100 (Nikon, Japan).
Determination of H2O2
At 24 and 48 hpi, 0.1 g of freshly harvested leaf samples (including diseased pieces), were finely ground using liquid nitrogen for H2O2 extraction. H2O2 content was determined using the commercial kit (ADS-W-YH001, Jiangsu ads Biotechnology Co., Ltd., Jiangsu, China).
Isolation of RNA and construction of RNA-seq libraries
A total of 18 samples, including three replicates of two cultivars collected at different times, underwent RNA extraction with the Plant Total RNA Isolation Kit (DP441, Tiangen Biotech, China). RNA quality was initially assessed via 1% agarose gel electrophoresis and further evaluated using the Agilent Technologies 2100 bioanalyzer for integrity. RNA concentration was determined with the Qubit® 2.0 fluorescence quantifier. For transcriptome sequencing library construction, RNA sequencing was conducted on the DNBSEQ platform (MGI TECH, China). High-quality clean reads were obtained by filtering raw reads with Soapnuke (Chen et al. 2018). Subsequently, these reads were aligned to the grape 12X reference genome (https://www.ncbi.nlm.nih.gov/assembly/gcf_000003745.3) using HISAT software (Kim et al. 2015). Gene expression levels were quantified using RNA-Seq by Expectation Maximization (RSEM) and expressed as FPKM, which means "fragments per kilobase of transcript per million mapped reads" (Li and Dewey 2011). Significantly differentially expressed genes (DEGs, Genetype: mRNA) were identified by selecting genes with |log2foldchange|≥ 2 and a q-value ≤ 0.05 using DESeq2 software (Love et al. 2014). The associated datasets are available at NCBI under BioSample accession number PRJNA938012.
Enrichment analysis of differentially expressed genes
To explore the primary biological activities and biochemical pathways of DEGs, a study of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was conducted using the R package (Kanehisa et al. 2008; Yu et al. 2012). Heatmaps illustrating gene expression levels were generated using TBtools software (Chen et al. 2020).
WGCNA analysis
WGCNA is a widely used computational method that provides insights into gene expression patterns across diverse samples. In this study, the WGCNA package in R software was employed to identify related gene modules and key genes associated with resistance to white rot (Langfelder and Horvath 2008). The WGCNA network construction and module detection used an unsigned topological overlap matrix (TOM) with a power parameter (β) set to 9, a minimum module size of 30, a depth split of 2, and a minimum height threshold for merging modules of 0.25. Module eigenvalues were computed and used to assess the correlation between modules and lesion diameter. Notably, gene centrality within each module identified by WGCNA was determined based on an eigengene-based connectivity value (KME) > 0.9 (Ma et al. 2021).
qRT-PCR analysis
To validate the RNA-seq findings, quantitative Real-Time PCR (qRT-PCR) analysis was conducted on six putative disease-related genes. RNA was extracted using a previously described method, and cDNA synthesis was performed using the PrimeScript RT reagent Kit (KR116, Tiangen Biotech, China). qPCR primers were designed using Primer3 plus (https://www.primer3plus.com/) and gene expression levels were calculated using the 2−ΔΔCt method (Bustin et al. 2009). EF1γ (GenBank Accession No: AF176496) served as the reference gene for gene expression analysis. Specific primers used for this analysis are detailed in Supplementary Table S1.
Data analysis
Graphs were created using Excel and significant differences were assessed using SPSS 24.0 software (IBM Corp, Armonk, USA).
Results
Assessment of resistance to white rot
The leaves from two cultivars, VvMF and Vd0940, were infected with C. diplodiella. The resistance of the leaves to white rot was assessed by measuring the lesion diameter at 0 h post inoculation (hpi), 24 hpi, and 48 hpi. Distinct degrees of susceptibility to white rot infection were observed in the leaves of the two cultivars at 24 hpi and 48 hpi (Fig. 1a). Additionally, the lesion diameter in VvMF leaves significantly exceeded that in Vd0940 leaves (Supplementary Fig. 1). Trypan blue staining at 24 hpi and 48 hpi revealed pronounced cellular demise in VvMF leaves, whereas minimal cell death occurred in Vd0940 (Fig. 1b). Aniline blue staining of grapevine leaves showed that only conidia were present in Vd0940 at 24 and 48 hpi, with no mycelium invading leaf parenchyma cells. Conversely, mycelium production and invasion of leaf surface cells were already evident in VvMF at 24 hpi, progressing through the palisade organization and continuing to penetrate mesophyll cells at 48 hpi (Fig. 1c). Furthermore, assessment of H2O2 content revealed significant disparities between Vd0940 and VvMF leaves at both 24 hpi and 48 hpi, with the former displaying markedly higher H2O2 levels (P < 0.05) (Fig. 1d). These findings indicate that Vd0940 demonstrates higher resistance to white rot compared to VvMF.

VvMF and Vd0940 infected with C. diplodiella. (a) Leaf phenotypes after 0, 24, and 48 hpi. (b) Trypan blue staining of leaves after inoculation with C. diplodiella for 24 and 48 hpi. (c) Aniline blue staining of leaves. Arrows refer to conidia or hyphae. Ih, infection hyphae. (d) H2O2 content after 0, 24, and 48 h post inoculation
Transcriptome sequencing quality analysis
To comprehensively investigate transcriptional alterations in grapes infected by C. diplodiella, RNA sequencing was conducted on two grape cultivars (VvMF and Vd0940) at three different moments (0 h, 24 h, and 48 h). Each sample included three biological replicates, resulting in 18 transcriptome libraries and 822.02 million raw reads. After filtering out adapter sequences and low-quality reads, clean reads ranged from 44.48 to 45.45 million per sample, yielding 120.59 GB of clean bases. Clean reads exhibited high quality, with a Q30 ratio ranging from 92.52% to 93.91% across samples. On average, 86.38% of the high-quality clean reads successfully aligned to the grape reference genome (Supplementary Table S2). The biological replicates demonstrated high correlation through principal component analysis and correlation analysis of expression levels for each sample (Supplementary Fig. 2).
Differentially expressed genes in response to C.diplodiella infection
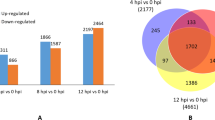
To elucidate the mechanism underlying grape resistance to C. diplodiella, DEGs of VvMF and Vd0940 were analyzed at 24 hpi and 48 hpi. The susceptible cultivar VvMF exhibited 2083 upregulated and 2803 downregulated genes at 24 hpi, and 2111 upregulated and 2409 downregulated genes at 48 hpi. In contrast, the resistant cultivar Vd0940 showed 1857 upregulated and 3120 downregulated genes at 24 hpi, and 1937 upregulated and 2312 downregulated genes at 48 hpi (Fig. 2a). Notably, 3274 and 3590 genes were coinduced in VvMF and Vd0940, respectively. A Venn diagram analysis revealed that 2138 genes were uniquely expressed in Vd0940, 2634 genes in VvMF, and 1625 genes were commonly involved in the response to C. diplodiella in both cultivars (Fig. 2d).

Differentially expressed genes (DEGs) between Vitis. davidii (Vd0940) and Vitis. Vinifera (VvMF) of grape in response to Coniella. diplodiella infection. S and R represent the grape species of VvMF and Vd0940, respectively. (a) Number of up- and down-regulation genes in Vd0940 and VvMF inoculated with C. diplodiella. (b) Differentially expressed genes (DEGs) at 24 h post inoculation (hpi) compared using a Venn diagram. (c) DEGs at 48 hpi compared using a Venn diagram. (d) DEGs at 24 hpi and 48 hpi in Vd0940 and VvMF were compared using a Venn diagram
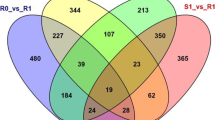
In the Venn diagram, a comparative analysis of differentially expressed genes revealed 2047 unique gene expressions in VvMF and 2138 specific to Vd0940 at 24 hpi by C. diplodiella (Fig. 2b). At 48 hpi by C. diplodiella, 2409 genes were specific to VvMF, and 2138 genes were specific to Vd0940 (Fig. 2c). Significant differences in enriched pathways were observed in KEGG enrichment analysis for these specifically induced genes in the two grape germplasms. Genes specifically expressed in VvMF at 24 hpi were enriched in pathways related to photosynthesis, flavonoid biosynthesis, porphyrin and chlorophyll metabolism, circadian rhythm, and glycolysis/gluconeogenesis. Conversely, genes specifically expressed in Vd0940 at 24 hpi were associated with pathways like sesquiterpenoid and triterpenoid biosynthesis, the mitogen-activated protein kinase (MAPK) signaling pathway, glutathione metabolism, and cutin, suberin, and wax biosynthesis (Fig. 3a). Moreover, genes specifically expressed at 48 hpi in VvMF were mainly enriched in pathways including photosynthesis, alpha-Linolenic acid metabolism, carbon fixation in photosynthetic organics, carbon metabolism, and phenylpropanoid biosynthesis. Meanwhile, genes specifically expressed in Vd0940 at 48 hpi were primarily associated with pathways related to sesquiterpenoid and triterpenoid biosynthesis, flavone and flavonol biosynthesis, sulfur metabolism, and stilbenoid, diarylheptanoid, and gingerol biosynthesis (Fig. 3b). These findings highlight significant differences in response mechanisms between the two grape varieties following infestation by C. diplodiella.

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially expressed genes (DEGs) in Vitis. Vinifera (VvMF) and Vitis. davidii (Vd0940) after infection with Coniella. diplodiella. (a) KEGG significantly enriched DEGs in VvMF and Vd0940 inoculated with C. diplodiella at 24 hpi. (b) KEGG significantly enriched DEGs in VvMF and Vd0940 inoculated with C. diplodiella at 48 hpi. S and R represent the grape species of VvMF and Vd0940, respectively
Enrichment analysis highlights the pathways related to resistance in resistant cultivars
A comparative analysis was conducted to examine differentially expressed genes between VvMF and Vd0940 in response to C. diplodiella invasion. Vd0940 revealed 2138 unique genes responsive to C. diplodiella. After 24 h of C. diplodiella induction, Vd0940 exhibited 607 DEGs, and this number decreased to 327 after 48 h. Notably, 1,204 DEGs responded to C. diplodiella invasion at both time points (Fig. 2d). KEGG analysis of these DEGs showed significant enrichment in disease resistance-related pathways, including sesquiterpenoid and triterpenoid biosynthesis, plant-pathogen interactions, flavonoid and flavonol biosynthesis, sulfur relay system, suberin and wax biosynthesis, and monoterpenoid biosynthesis (P < 0.05). These findings suggest the importance of these pathways in Vd0940's defense against C. diplodiella (Fig. 4).

KEGG analysis of specific differentially expressed genes in Vd0940 post-C. diplodiella inoculation
To validate the transcriptome data's reliability, we selected six DEGs from these pathways and analyzed their relative expression levels in both species at different infection stages using qRT-PCR. The expression patterns of these DEGs were consistent with the RNA-seq results, confirming the RNA-seq data's validity (Supplementary Fig. 3, Supplementary data 1).
Co-expression network modules identified based on WGCNA
To identify potential key genes associated with C. diplodiella resistance, we performed differential gene expression analysis using WGCNA. Co-expression modules were constructed for all samples based on FPKM values, resulting in the identification of 11 gene modules (Supplementary data 2). Correlation analysis between gene expression modules and disease resistance traits revealed significant positive correlations between the MElightyellow, MEpink, and MElightgreen modules and disease resistance traits (P < 0.05) (Fig. 5). Specifically, the MEpink module exhibited significant positive correlations with disease resistance at both R24 hpi and R48 hpi, while the MElightyellow and MElightgreen modules were significantly positively correlated with disease resistance at R48 hpi. Among the three modules significantly associated with disease resistance, the pink module contained 747 genes, the light yellow module contained 37 genes, and the light green module contained 34 genes (Supplementary data 3). Furthermore, 125 genes related to the disease showed higher expression in Vd0940 than in VvMF at 24 h and 48 h after C. diplodiella infection (Fig. 6, Supplementary data 4), indicating their significant role in grape white rot resistance.

WGCNA analysis of key modules related to C. diplodiella resistance in grapes. (a) Co-expression module hierarchical cluster diagram according to WGCNA. (b) Relationships between modules and traits. The correlation and p-value are displayed in each cell. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940)

Heatmap of candidate gene expression in Vitis. Vinifera (VvMF) and Vitis. davidii (Vd0940) inoculated with Coniella. diplodiella. S and R represent the grape species of VvMF and Vd0940, respectively. R genes, resistant genes; PRRs, pattern-recognition receptors; PR, pathogenesis-related proteins; MAPK, mitogen-activated protein kinase; TFs, transcription factors; 24 hpi, 24 h post inoculation
Discussion
Grape white rot occurrence has been widely reported in viticulture areas across many countries. Surveys in major Chinese grape-growing regions have revealed its widespread presence (Chethana et al. 2017; Niekerk et al. 2004). Furthermore, C. diplodiella strongly affects V. vinifera, highlighting the need for disease-resistant grape varieties for disease management (Chen et al. 2018). While numerous studies have noted the strong resistance of Chinese wild grape germplasm to white rot, the mechanisms behind this resistance remain unclear. This study aims to compare grape varieties' resistance and susceptibility to white rot, thus providing a theoretical basis for selecting and breeding disease-resistant grape varieties.
Leaf cells contain various organelles, including chloroplasts and mitochondria, crucial for photosynthesis and ROS production (Wang et al. 2013). Trypan blue staining revealed that post-C. diplodiella leaf inoculation, VvMF exhibited more dead cells than Vd0940, thereby impacting VvMF leaf physiology. Additionally, increased H2O2 content reportedly enhanced plant disease resistance (Li et al. 2019b). When infected by C. diplodiella, the H2O2 content of Vd0940 significantly exceeded that of VvMF, possibly contributing to its greater resistance to C. diplodiella. Aniline blue staining demonstrated that Vd0940 only showed conidia infestation when infected by C. diplodiella, whereas VvMF showed mycelium development at 24 hpi, which entered the mesophyll cells at 48 hpi. Similarly, ‘V. quinquangularis (Shang-24)’ a resistant grape anthracnose cultivar, only showed conidial development after 24 and 48 h of Elsinoë ampelina induction, while the susceptible variety ‘V. vinifera (Thompson Seedless)’ not only showed spores but also hyphae (Han et al. 2021).
RNA-seq technology has been utilized to examine differential gene expression in various crop-resistant strains infected with pathogens, including wheat, rice, tomato, grape, watermelon, and apple (Dorostkar et al. 2022; Kamber et al. 2016; Kumar and Dasgupta 2020; Li et al. 2017; Tan et al. 2015). Key pathways and disease resistance genes have been identified. In this study, KEGG enrichment analysis of DEGs resulting from inoculation with C. diplodiella in VvMF and Vd0940, as determined by RNA-seq, revealed 10 pathways, including sesquiterpenoid and triterpenoid biosynthesis, plant-pathogen interactions, flavonoid and flavonol biosynthesis, suberin and wax biosynthesis, and monoterpenoid biosynthesis pathways, that may be involved in Vd0940's defense mechanism against C. diplodiella. These pathways have been associated with plant disease resistance in multiple plant-pathogen interaction studies (Chen et al. 2016; Liu et al. 2022). For example, Zhang conducted a transcriptome analysis of grapevine green shoots infected by Lasiodiplodia. theobromae and identified numerous DEGs related to plant-pathogen interactions and the hormone signal transduction pathway (Zhang et al. 2019b).
The integrity of the plant cell wall, acting as an initial physical barrier against pathogens, is vital for triggering and regulating plant defense responses (Vaahtera et al. 2019; Wan et al. 2021). Pathogen infection induces cell wall pectin degradation, causing changes in Ca2+ signaling and ROS levels. Wall-associated kinases (WAKs) are involved in Ca2+-dependent interactions with pectin, playing a key role in mediating intracellular immune signaling during plant defense (Denoux et al. 2008; Benedetti et al. 2015; De Lorenzo et al. 2019). Recent studies show that WAKs boost plant resistance by inducing defense gene expression, promoting cellulose synthesis, and strengthening the cell wall (Yang et al. 2019). In this experiment, Wall-associated kinase 5 (WAK5) (Gene ID: 100852951), WAK8 (Gene ID: 100241421; Gene ID: 100247390), and WAK11 (Gene ID: 100264549) gene expression levels were higher in Vd0940 after C. diplodiella infestation compared to VvMF. This suggests that they may recognize pectin residues from the damaged Vd0940 cell wall following C. diplodiella infection, thereby bolstering the cell wall and enhancing intracellular defense responses, potentially limiting pathogenic bacteria invasion.
The primary immune system, swiftly detects pathogens and initiates various defense responses. Significantly, higher expression levels of multiple receptor kinase genes were observed in Vd0940 compared to VvMF following C. diplodiella infestation. This implies the presence of multiple receptors in Vd0940 for recognizing microbial structures released by C. diplodiella. Lectin receptor kinases (LecRKs), identified as PRRs capable of recognizing glycoproteins and glycolipids in pathogenic bacteria, play a pivotal role in countering pathogenic invasion (Lannoo and Van Damme 2014).
For successful infestation, C. diplodiella strains secrete several effectors, including Coniella diplodiella effector 1 (CDE1), to suppress the pattern-triggered immunity (PTI)-mediated immune response in grapes. However, grape resistant (R) proteins can detect these effectors, triggering the immune response known as effector-triggered immunity (ETI) (Spoel and Dong 2012; Liu et al. 2021). Typically, R genes exhibit low expression levels under normal conditions but significantly increase upon pathogen infection (Gu et al. 2005). For instance,resistant genes (R genes) identified in riparian grapes (V. riparia) maintained low expression under normal conditions but exhibited substantial upregulation 12 h after inoculation with downy mildew (Kortekamp et al. 2008).
In this study, 26 R genes in Vd0940 exhibited significant upregulation in response to C. diplodiella, whereas their expression levels were lower in VvMF. Both PTI and ETI can activate downstream responses, including cell wall thickening, ROS burst, the MAPK cascade pathway, and the production of disease-related proteins (Jones and Dangl 2006; Monaghan and Cyril 2012). Genes associated with these pathways, such as respiratory burst oxidase (Gene ID: 100252158) and MKS1 (Gene ID: 100854780), showed high expression levels in Vd0940.
SA, an endogenously synthesized plant hormone, plays a crucial role in the response to pathogen invasion (Kumar et al. 2015). After white rot infestation, Vd0940 exhibited an increase in endogenous SA levels, peaking at 48 h, significantly surpassing VvMF's SA content following white rot infestation (Rahman et al. 2022). SA receptor, SA-binding protein 2 (SABP2), required for plant immune responses, catalyzes the conversion of MeSA to SA (Chen and Klessig 1991; Kumar and Klessig 2003). SABP2 (Gene ID: 100852648) and PR1 (Gene ID: 100246419), markers of the SA pathway, exhibited higher expression levels in Vd0940 after C. diplodiella infestation compared to VvMF, underscoring the pivotal role of the SA pathway in grape resistance to C. diplodiella.
Secondary metabolites, including resveratrol and flavonoids, enhance plant disease resistance (Qiao et al. 2013; Jeandet et al. 2017). Resveratrol, a naturally occurring compound synthesized de novo by plants, is a low molecular weight antimicrobial compound that restricts pathogen invasion. Resveratrol synthesis depends on stilbene synthase (STS). Studies have revealed that overexpressing the STS gene enhances plant resistance to pathogenic bacteria (Leckband and Lörz 1998; Wang et al. 2017; Xu et al. 2019). In grapes, phenylpropanoids, flavonoids, and stilbenes have been implicated in downy mildew resistance promotion (Malacarne et al. 2011). Elevated expression of several secondary metabolism-related genes was observed in Vd0940 following C. diplodiella infestation. For instance, STS1 (Gene ID: 104877274) exhibited higher expression levels in Vd0940 compared to VvMF at various C. diplodiella induction time points, suggesting a significant role of secondary metabolism in white rot resistance.
Conclusions
In this study, significant differences were detected between C. diplodiella-inoculated VvMF and Vd0940 leaves. Vd0940 displayed minimal cell death, markedly higher H2O2 levels than VvMF, and only produced conidia when infected with C. diplodiella. In contrast, C. diplodiella-infected VvMF exhibited extensive cell death and showed conidial and mycelial infestation. Subsequent comparative transcriptome analysis identified potential involvement of pathways such as sesquiterpenoid and triterpenoid biosynthesis, plant-pathogen interactions, flavonoid and flavonol biosynthesis, sulfur relay system, suberin and wax biosynthesis and monoterpenoid biosynthesis in Vd0940's white rot defense mechanism. Ultimately, WGCNA analysis led to the identification of 125 candidate genes associated with grape white rot resistance.
Availability of data and materials
Analysis of all data from the current study is available from the corresponding author.
References
Benedetti M, Pontiggia D, Raggi S, Cheng Z, Scaloni F, Ferrari S, et al. Plant immunity triggered by engineered in vivo release of oligogalacturonides, damage-associated molecular patterns. Plant Biol. 2015;112:5533–8. https://doi.org/10.1073/pnas.1504154112.
Boller T, Felix G. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol. 2009;60:379–406. https://doi.org/10.1146/annurev.arplant.57.032905.105346.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin Chem. 2009;55:611–22. https://doi.org/10.1373/clinchem.2008.112797.
Chen Z, Klessig DF. Identification of a soluble salicylic acid-binding protein that may function in signal transduction in the plant disease-resistance response. Proc Natl Acad Sci USA. 1991;88:8179–83. https://doi.org/10.1073/pnas.88.18.8179.
Chen J, Zhang H, Feng M, Zuo D, Hu Y, Jiang T. Transcriptome analysis of woodland strawberry (Fragaria vesca) response to the infection by Strawberry vein banding virus (SVBV). Virol J. 2016;13:128. https://doi.org/10.1186/s12985-016-0584-5.
Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 2018;7:gix120. https://doi.org/10.1093/gigascience/gix120.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202. https://doi.org/10.1016/j.molp.2020.06.009.
Chethana KWT, Zhou Y, Zhang W, Liu M, Xing QK, Li XH, et al. Coniella vitis sp. nov. is the common pathogen of white rot in Chinese vineyards. Plant Dis. 2017;101:2123–36. https://doi.org/10.1094/PDIS-12-16-1741-RE.
De Lorenzo G, Ferrari S, Giovannoni M, Mattei B, Cervone F. Cell wall traits that influence plant development, immunity, and bioconversion. Plant J. 2019;97:134–47. https://doi.org/10.1111/tpj.14196.
Denoux C, Galletti R, Mammarella N, Gopalan S, Werck D, De Lorenzo G, et al. Activation of defense response pathways by OGs and Flg22 elicitors in Arabidopsis seedlings. Mol Plant. 2008;1:423–45. https://doi.org/10.1093/mp/ssn019.
Dorostkar S, Dadkhodaie A, Ebrahimie E, Heidari B, Ahmadi-Kordshooli M. Comparative transcriptome analysis of two contrasting resistant and susceptible Aegilops tauschii accessions to wheat leaf rust (Puccinia triticina) using RNA-sequencing. Sci Rep. 2022;12:821. https://doi.org/10.1038/s41598-021-04329-x.
Fu ZQ, Dong X. Systemic acquired resistance: turning local infection into global defense. Annu Rev Plant Biol. 2013;64:839–63. https://doi.org/10.1146/annurev-arplant-042811-105606.
Ghaffari S, Reynard JS, Rienth M. Single berry reconstitution prior to RNA-sequencing reveals novel insights into transcriptomic remodeling by leafroll virus infections in grapevines. Sci Rep. 2020;10:12905. https://doi.org/10.1038/s41598-020-69779-1.
Gu K, Yang B, Tian D, Wu LF, Wang DJ, Sreekala C, et al. R gene expression induced by a type-III effector triggers disease resistance in rice. Nature. 2005;435:1122–5. https://doi.org/10.1038/nature03630.
Han R, Yin W, Ahmad B, Gao P, Li Z, Wang X. Pathogenesis and immune response in resistant and susceptible cultivars of grapevine (Vitis spp.) against Elsinoë ampelina infection. Phytopathol. 2021;111:799–807. https://doi.org/10.1094/PHYTO-03-20-0079-R.
Jeandet P, Courot E, Clément C, Ricord S, Crouzet J, Aziz A, et al. Molecular engineering of phytoalexins in plants: benefits and limitations for food and agriculture. J Agric Food Chem. 2017;65:2643–4. https://doi.org/10.1021/acs.jafc.7b00936.
Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–9. https://doi.org/10.1038/nature05286.
Kamber T, Buchmann JP, Pothier JF, Smits THM, Wicker T, Duffy B, et al. Fire blight disease reactome: RNA-seq transcriptional profile of apple host plant defense responses to Erwinia amylovora pathogen infection. Sci Rep. 2016;6:21600. https://doi.org/10.1038/srep21600.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4. https://doi.org/10.1093/nar/gkm882.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. https://doi.org/10.1038/nmeth.3317.
Kortekamp A, Welter L, Vogt S, Knoll A, Schwander F, Töpfer R, et al. Identification, isolation and characterization of a CC-NBS-LRR candidate disease resistance gene family in grapevine. Mol Breeding. 2008;22:421–32. https://doi.org/10.1007/s11032-008-9186-2.
Kumar G, Dasgupta I. Comprehensive molecular insights into the stress response dynamics of rice (Oryza sativa L.) during rice tungro disease by RNA-seq-based comparative whole transcriptome analysis. J Biosci. 2020;45:27. https://doi.org/10.1007/s12038-020-9996-x.
Kumar D, Klessig DF. High-affinity salicylic acid-binding protein 2 is required for plant innate immunity and has salicylic acid-stimulated lipase activity. Proc Natl Acad Sci USA. 2003;100:16101–6. https://doi.org/10.1073/pnas.0307162100.
Kumar D, Chapagai D, Dean P, Davenport M. Biotic and abiotic stress signaling mediated by salicylic acid. Funct Genomics Perspect. 2015. https://doi.org/10.1007/978-1-4939-2211-6_12.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. https://doi.org/10.1186/1471-2105-9-559.
Lannoo N, Van Damme EJM. Lectin domains at the frontiers of plant defense. Front Plant Sci. 2014;5:397. https://doi.org/10.3389/fpls.2014.00397.
Leckband G, Lörz H. Transformation and expression of a stilbene synthase gene of Vitis vinifera L. in barley and wheat for increased fungal resistance. Theor Appl Genet. 1998;96:1004–12. https://doi.org/10.1007/s001220050832.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. https://doi.org/10.1186/1471-2105-12-323.
Li D, Wan Y, Wang Y, He P. Relatedness of resistance to anthracnose and to white rot in Chinese wild grapes. Vitis. 2008;47:213–5. https://doi.org/10.5073/vitis.2008.47.213-215.
Li X, An M, Xia Z, Bai X, Wu Y. Transcriptome analysis of watermelon (Citrullus lanatus) fruits in response to Cucumber green mottle mosaic virus (CGMMV) infection. Sci Rep. 2017;7:16747. https://doi.org/10.1038/s41598-017-17140-4.
Li Z, Zhang S, Han R, Zhang H, Li K, Wang X. Infection process and host responses to Elsinoë ampelina, the causal organism of grapevine anthracnose. Eur J Plant Pathol. 2019a;155:571–82. https://doi.org/10.1007/s10658-019-01793-0.
Li Y, Cao XL, Zhu Y, Yang XM, Zhang KN, Xiao ZY, et al. Osa-miR398b boosts H2O2 production and rice blast disease-resistance via multiple superoxide dismutase. New Phytol. 2019b;222:1507–22. https://doi.org/10.1111/nph.15678.
Li Y, Lv J, Zhao Q, Chen L, Dong Y, Dong K. Wheat/faba bean intercropping improves physiological and structural resistance of faba bean to fusaric acid stress. Plant Pathol. 2021;70:827–40. https://doi.org/10.1111/ppa.13331.
Li P, Tan X, Liu R, Rahman FU, Jiang J, Sun L, et al. QTL detection and candidate gene analysis of grape white rot resistance by interspecific grape (Vitis vinifera L.× Vitis davidii Foex.) crossing. Hortic Res. 2023;10:uhad063. https://doi.org/10.1093/hr/uhad063.
Liu R, Wang Y, Li P, et al. Genome assembly and transcriptome analysis of the fungus Coniella diplodiella during infection on grapevine (Vitis vinifera L.). Front Microbiol. 2021;11:599150. https://doi.org/10.3389/fmicb.2020.599150.
Liu X, Fang P, Wang Z, Cao X, Yu Z, Chen X, et al. Comparative RNA-seq analysis reveals a critical role for ethylene in rose (Rosa hybrida) susceptible response to Podosphera pannosa. Front Plant Sci. 2022;13:1018427. https://doi.org/10.3389/fpls.2022.1018427.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. https://doi.org/10.1186/s13059-014-0550-8.
Ma L, Zhang M, Chen J, Qing C, He S, Zou C, et al. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor Appl Genet. 2021;134:3305–18. https://doi.org/10.1007/s00122-021-03897-w.
Malacarne G, Vrhovsek U, Zulini L, Cestaro A, Stefanini M, Mattivi F, et al. Resistance to Plasmopara viticola in a grapevine segregating population is associated with stilbenoid accumulation and with specific host transcriptional responses. BMC Plant Biol. 2011;11:114. https://doi.org/10.1186/1471-2229-11-114.
Monaghan J, Cyril Z. Plant pattern recognition receptor complexes at the plasma membrane. Curr Opin Plant Biol. 2012;15:349–57. https://doi.org/10.1016/j.pbi.2012.05.006.
Naidoo S, Visser EA, Zwart L, Dutoit Y, Bhadauria V, Shuey LS. Dual RNA-seq to elucidate the plant–pathogen duel. Curr Issues Mol Biol. 2018;27:127–42. https://doi.org/10.21775/cimb.027.127.
Polesani M, Bortesi L, Ferrarini A, Zamboni A, Fasoli M, Zadra C, et al. General and species-specific transcriptional responses to downy mildew infection in a susceptible (Vitis vinifera) and a resistant (V. riparia) grapevine species. BMC Genomics. 2010;11:117. https://doi.org/10.1186/1471-2164-11-117.
Qiao Y, Meng X, Jin X, Ding G. Genes expression of key enzymes in phenylpropanes metabolism pathway in cucumber with RT-PCR. Adv Mater Res. 2013;746:53–7. https://doi.org/10.4028/www.scientific.net/AMR.746.53.
Rahman FU, Khan IA, Aslam A, Liu R, Sun L, Wu Y, et al. Transcriptome analysis reveals pathogenesis-related gene 1 pathway against salicylic acid treatment in grapevine (Vitis vinifera L). Front Genet. 2022;13:1033288. https://doi.org/10.3389/fgene.2022.1033288.
Rex F, Fechter I, Hausmann L, Töepfer R. QTL mapping of black rot (Guignardia bidwellii) resistance in the grapevine rootstock ‘Börner’ (V. riparia Gm183× V. cinerea Arnold). Theor Appl Genet. 2014;127:1667–77. https://doi.org/10.1007/s00122-014-2329-4.
Riaz S, Tenscher AC, Ramming DW, Walker MA. Using a limited mapping strategy to identify major QTLs for resistance to grapevine powdery mildew (Erysiphe necator) and their use in marker-assisted breeding. Theor Appl Genet. 2011;122:1059–73. https://doi.org/10.1007/s00122-010-1511-6.
Spoel SH, Dong X. How do plants achieve immunity? Defence without specialized immune cells. Nat Rev Immunol. 2012;12:89–100. https://doi.org/10.1038/nri3141.
Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20:631–56. https://doi.org/10.1038/s41576-019-0150-2.
Su K, Guo Y, Zhao Y, Gao H, Liu Z, Li K, et al. Candidate genes for grape white rot resistance based on SMRT and Illumina sequencing. BMC Plant Biol. 2019;19:501. https://doi.org/10.1186/s12870-019-2119-x.
Su K, Guo Y, Zhong W, Lin H, Liu Z, Li K, et al. High-density genetic linkage map construction and white rot resistance quantitative trait loci mapping for genus Vitis based on restriction site-associated DNA sequencing. Phytopathology. 2021;111:659–70. https://doi.org/10.1094/PHYTO-12-19-0480-R.
Tan G, Liu K, Kang J, Xu K, Zhang Y, Hu L, et al. Transcriptome analysis of the compatible interaction of tomato with Verticillium dahliae using RNA-sequencing. Front Plant Sci. 2015;6:428. https://doi.org/10.3389/fpls.2015.00428.
Vaahtera L, Schulz J, Hamann T. Cell wall integrity maintenance during plant development and interaction with the environment. Nat Plants. 2019;5:924–32. https://doi.org/10.1038/s41477-019-0502-0.
Van Niekerk JM, Groenewwald JZE, Verkley GJM, Fourie PH, Wingfield MJ, Crous PW. Systematic reappraisal of Coniella and Pilidiella, with specific reference to species occurring on Eucalyptus and Vitis in South Africa. Mycol Res. 2004;108:283–303. https://doi.org/10.1017/S0953756204009268.
Vezzulli S, Doligez A, Bellin D. Molecular mapping of grapevine genes. Compendium Plant Genomes. 2019. https://doi.org/10.1007/978-3-030-18601-2_7.
Wan J, He M, Hou Q, Zou L, Yang Y, Wei Y, et al. Cell wall associated immunity in plants. Stress Biol. 2021;1:3. https://doi.org/10.1007/s44154-021-00003-4.
Wang Y, Lin A, Loake GJ, Chu C. H2O2-induced leaf cell death and the crosstalk of reactive nitric/oxygen species. J Integr Plant Biol. 2013;55:202–8. https://doi.org/10.1111/jipb.12032.
Wang Y, Wang D, Wang F, Huang L, Tian X, Van Nocker S, et al. Expression of the grape VaSTS19 gene in Arabidopsis improves resistance to powdery mildew and Botrytis cinerea but increases susceptibility to Pseudomonas syringe pv tomato DC3000. Int J Mol Sci. 2017;18:2000. https://doi.org/10.3390/ijms18092000.
Xu W, Ma F, Li R, Zhou Q, Yao W, Jiao Y, et al. VpSTS29/STS2 enhances fungal tolerance in grapevine through a positive feedback loop. Plant Cell Environ. 2019;42:2979–98. https://doi.org/10.1111/pce.13600.
Yang P, Praz C, Li B, Singla J, Robert CAM, Kessel B, et al. Fungal resistance mediated by maize wall-associated kinase ZmWAK-RLK1 correlates with reduced benzoxazinoid content. New Phytol. 2019;221:976–87. https://doi.org/10.1111/nph.15419.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. https://doi.org/10.1089/omi.2011.0118.
Zhang Y, Fan X, Sun H, Jiang JF, Liu CH. Identification and evaluation of resistance to white rot in grape resources. J Fruit Sci. 2017;34:1095–105. https://doi.org/10.13925/j.cnki.gsxb.20160281.
Zhang Y, Yao JL, Feng H, Jiang J, Fan X, Jia YF, et al. Identification of the defense-related gene VdWRKY53 from the wild grapevine Vitis davidii using RNA sequencing and ectopic expression analysis in Arabidopsis. Hereditas. 2019a;156:14. https://doi.org/10.1186/s41065-019-0089-5.
Zhang W, Yan J, Li X, Xing Q, Chethana KWT, Zhao W. Transcriptional response of grapevine to infection with the fungal pathogen Lasiodiplodia theobromae. Sci Rep. 2019b;9:5387. https://doi.org/10.1038/s41598-019-41796-9.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Key Research and Development Program of China (No. 2021YFD1200200), the National Natural Science Foundation of China (No. 31872057), the China Agriculture Research System (No. CARS-29) and The Agricultural Science and Technology Innovation Program (No. CAAS-ASTIP-2021-ZFRI).
Author information
Authors and Affiliations
Contributions
YZ designed the research and experimental protocols. PL, WH, and XT provided support in the execution of phenotyping experiments. JJ, LS and XF helped in the extraction of RNA. PL assumed responsibility for data analysis and drafted the article. YZ and CL made significant contributions to manuscript revision. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Table S1. Primers used in this study.
Additional file 2:
Supplementary Table S2. Transcriptome data and quality assessment.
Additional file 3: Supplementary Fig. 1.
Lesion diameter of leaves. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940).
Additional file 4: Supplementary Fig. 2.
Correlation Analysis of Different Samples. (a) Pearson correlation coefficient analysis based on gene expression. (b) Principal component analysis of different samples. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940); 24h, at 24 h post inoculation.
Additional file 5: Supplementary Fig. 3.
Validation of RNA-seq by qRT-PCR. The column chart and principal longitudinal coordinate indicate the relative expression of quantitative real-time PCR (qRT-PCR), whereas the broken line diagram and secondary longitudinal coordinate show the FPKM value of RNA-seq. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940); 24h, at 24 h post inoculation; RPM1, Pseudomonas syringae pv. maculicola 1; FPKM, Fragments per kilobase of transcript per million fragments mapped.
Additional file 6: Supplementary data 1.
Results data of qRT-PCR. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940); 24h, at 24 h post inoculation.
Additional file 7: Supplementary data 2.
All gene of module.
Additional file 8: Supplementary data 3.
All gene in three module. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940); 24h, at 24 h post inoculation.
Additional file 9: Supplementary data 4.
List of candidate genes. S, the grape specie of Vitis. Vinifera (VvMF); R, the grape specie of Vitis. davidii (Vd0940); 24h, at 24 h post inoculation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, P., Tan, X., Wanghao et al. Transcriptome analysis of resistant and susceptible grapes reveals molecular mechanisms underlying resistance of white rot disease. HORTIC. ADV. 1, 9 (2023). https://doi.org/10.1007/s44281-023-00011-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-023-00011-6