
Abstract
Pancreatic cancer is a highly aggressive disease, which is often diagnosed late. Consequently, metastasis is common among newly diagnosed patients, leading to a poor prognosis and high mortality rates. The tumor microenvironment of pancreatic cancer, which comprises pancreatic cancer cells, stromal cells, and immune cells, as well as a multitude of extracellular components, plays a pivotal role in cancer progression and metastasis. Conventional immunotherapies focused on targeting the adaptive immune response have achieved suboptimal outcomes in patients with pancreatic cancer. Thus, the focus has shifted toward targeting innate immune cells, which can infiltrate the pancreatic tumor and contribute to the development and maintenance of the immunosuppressive microenvironment to promote tumor growth and metastasis. This review focuses on the roles of innate immune cells and their interactions in the shaping of an immunosuppressive tumor microenvironment to promote the metastasis of pancreatic cancer. In addition, we review strategies that target innate immune cells to remodel the immunosuppressive tumor microenvironment and improve the prognosis of pancreatic cancer.
Similar content being viewed by others


Background
Despite decades of research, pancreatic cancer remains one of the most lethal cancers. In the U.S., the 5-year survival rate for pancreatic cancer is only 10%, which is primarily attributed to its aggressive nature and the late stage of diagnosis [1]. Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer, is frequently diagnosed at the advanced stage after has metastasis occurred, which contributes to the high motility rates observed. PDAC is characterized by a highly immunosuppressive tumor microenvironment (TME), which lowers the efficacy of currently available immunotherapies. Thus, there is an urgent need to investigate how the TME contributes to PDAC development and metastasis,this information will help in the identification of potential targets to improve the outcomes of patients with PDAC.
Immune cells play indispensable roles in tumor recognition and eradication. However, tumors often develop mechanisms to evade immune surveillance and re-educate immune cells to form an immunosuppressive TME, which is beneficial to tumor survival and progression. While residing in tissues, innate immune cells can be activated to participate in either tumoricidal or tumorigenic processes. Among these cells, neutrophils, macrophages, and dendritic cells (DCs), have been extensively studied due to their high heterogeneity and plasticity in the TME.
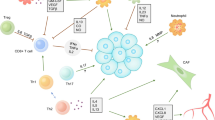
The TME of PDAC consists of tumor cells, stromal cells, immune cells, and extracellular components (Fig. 1). Tumor cells, stromal cells, and immune cells, which include tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), regulatory T cells (Tregs), and DCs, secrete extracellular components. These extracellular components, such as the extracellular matrix (ECM), growth factors, and chemokines, are essential for maintaining an immunosuppressive TME, which facilitates tumor progression and metastasis.

The tumor microenvironment of PDAC. The PDAC tumor microenvironment (TME) is characterized by desmoplasia and immunosuppression. Extracellular matrix proteins, including collagen and laminin, are secreted by pancreatic stellate cells (PSCs). In addition, the PDAC TME contains immunosuppressive cells such as tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), natural killer (NK) cells, cancer stem cells (CSCs), and cancer-associated fibroblasts (CAFs). Figure generated using Biorender
PDAC forms ‘cold tumors’, which are characterized by low effector T cell infiltration and immunogenicity. Thus, PDAC tumors respond poorly to currently available immunotherapies, which primarily focus on the action of adaptive immune cells such as T cells. Consequently, research focus is shifting toward the therapeutic potential of innate immune cells, which are considerably more abundant in the PDAC TME than adaptive immune cells [2]. This review briefly summarizes the roles of various innate immune cells in the PDAC TME. Specifically, we describe the mechanisms used by innate immune cells to contribute to PDAC metastasis and how they interact in the TME, before discussing the potential clinical implications of targeting these immune cells.
Innate immune cells in the PDAC TME
The unsatisfactory response of PDAC to immunotherapy can be attributed to the PDAC TME, which is characterized by immunosuppression and desmoplasia (the excessive deposition of connective tissue). Tumor cells activate PSCs to promote fibrosis of the tissue surrounding the tumor. This creates a mechanical barrier around the tumor, limiting the infiltration of immune cells or exposure to chemotherapeutic drugs [3, 4]. Cytokines, such as tumor growth factor-β (TGF-β) and fibroblast growth factor 2 secreted in the ECM, can differentiate fibroblasts into cancer-associated fibroblasts (CAFs), promoting desmoplasia in the PDAC TME [5]. In addition to PSCs, immune cells are crucial components of the immunosuppressive PDAC TME,their roles are discussed in the following sections. The interactions between innate immune cells and tumor cells are summarized in Fig. 2.

Interaction between tumor and innate immune cells in the PDAC microenvironment. Interactions between tumor and tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), and natural killer (NK) cells are depicted. Arrows depict interactions between tumor cells and each immune cell type. Image generated using Biorender
TAMs
Macrophages are innate phagocytic cells, which have broad functions in the process of inflammation; they exist as a heterogeneous population with distinct functional characteristics [6]. Macrophages are commonly classified as either the pro-inflammatory M1 type or the anti-inflammatory M2 type, which represent opposing extremes of a continuous functional spectrum [7]. Colony stimulating factor 1 (CSF-1), interleukin (IL)-4, and IL-13, which are abundant in the TME, promote the recruitment of monocytes and polarize TAMs toward the M2 type [8]. Thus, in solid tumors, including PDAC, TAMs have similar functions and characteristics to M2-type macrophages and their accumulation is associated with a poor prognosis [9,10,11]. However, the roles of TAMs appear to be more complex than initially thought. Several single-cell RNA-sequencing (scRNA-seq) studies have demonstrated that TAMs express a mixture of M1 and M2 markers in colorectal [12], liver [13], and renal [14] cancers. Another scRNA-seq study discovered that TAMs in PDAC could be subdivided into two groups: the SPP1+TAMs and the C1QC+TAMs [15]. The SPP1+TAMs are enriched in genes involved in epithelial-mesenchymal transition (EMT), glucose metabolism, and hypoxia, whereas the C1QC+TAMs are enriched in genes associated with the interferon (IFN) response and antigen presentation. Intriguingly, both the SPP1+ TAMs and C1QC+ TAMs also have an M2-like signature, which prevents them from being classified into the M1 or M2 categories [15].
TAMs contribute to the immunosuppressive TME by secreting cytokines, chemokines, and enzymes, such as TGF-β, IL-10, prostaglandin E2 (PGE2), and arginase-1 (Arg-1) [16]. For instance, the secretion of C–C chemokine ligand 5 (CCL5) and tumor necrosis factor-α (TNF-α) by TAMs induces pancreatic acinar-to-ductal metaplasia through NF-κB pathway activation [17]. TAMs polarized by tumor-secreted granulocyte macrophage (GM)-CSF, inhibited CD8+ T cells in a pancreatic mouse model [18]. Moreover, TAMs have been shown to express high levels of C-X-C motif chemokine receptor 2 (CXCR2,these CXCR2+TAMs trafficked to the PDAC tumor site in response to the tumor-derived C-X-C motif ligand 8 (CXCL8), impairing the efficacy of anti-PD1 therapy [19]. The galectin-9-mediated activation of dectin-1 on TAM also leads to tolerogenic T cell program induction in PDAC [20].
MDSCs
Although their distribution and functions are still under debate, MDSCs are an important component of the PDAC TME [21]. MDSCs are a heterogeneous population of immature myeloid cells, which can be classified into two subtypes: granulocytic or polymorphonuclear (PMN-MDSCs) and monocytic (M-MDSCs) MDSCs. PMN-MDSCs are phenotypically and morphologically similar to neutrophils, with CD11b+Gr-1+Ly6GhighLy6Clow and HLA-DR−CD33+CD11b+CD15+CD14− signatures in mice and humans, respectively. Meanwhile, M-MDSCs are related to monocytes and have a CD11b+Gr-1+Ly6GlowLy6Chigh signature in mice and a HLA-DRlowCD11b+CD15−CD14+ signature in humans [22,23,24].
MDSCs help shape the immunosuppressive TME by suppressing CD4+ and CD8+ T cells, stimulating Tregs expansion, and promoting M2 phenotype polarization [25, 26]. Moreover, the TGF-β and IL-10 in the TME cause MDSCs to release reactive oxygen species (ROS), which promote oxidative stress and further impair T cell function [8, 27]. Interactions between MDSCs and activated T cells lead to STAT3 activation in MDSCs and an increase in PD-1 expression in T cells, resulting in the suppression of T-cell activation [28]. MDSCs can be recruited to the tumor sites via chemokines and cytokines, such as CXCL12, CCL2, and IL-6 [29, 30]. Inflammatory CAFs use the FAP-STAT3 signaling pathway to release CCL2, which recruits MDSCs and ultimately dampens the activity of CD8+ T cells in the TME; these events further promote tumor progression [31, 32]. The expression of CXCL12 by CAFs facilitates the migration of MDSCs, which can be suppressed by using the poly ADP ribose polymerase inhibitor olaparib (R. [30]. Direct physical interactions between MDSCs and Tregs were observed in both the murine PDAC model and the tissues of PDAC patients, whereby MDSCs induced Treg proliferation [33].
TANs
Similar to TAMs, TANs can be divided into the pro-inflammatory N1 and the anti-inflammatory N2 types [34]. The TME can influence the polarization of TANs. For instance, IFN-γ and TGF-β in the TME polarize TANs toward the N1 or N2 types, respectively [34, 35]. N2 neutrophils have a strong immunosuppressive function. They recruit Tregs and macrophages to the TME and secrete factors such as matrix metalloproteinases (MMPs), hepatocyte growth factor (HGF), and neutrophil elastase (NE) [26, 36]. scRNA-seq analysis has revealed that TANs can be classified into four subpopulations: a terminally differentiated pro-tumor subtype (TAN-1), an inflammatory subpopulation (TAN-2), a transitional stage population (TAN-3), and a subtype that preferentially expresses IFN-γ-associated genes (TAN-4) [37]. The infiltration of TANs into the PDAC TME is associated with a poor prognosis [38]. Moreover, in clinical studies of PDAC patients, the neutrophil to lymphocyte ratio was suggested as a predictor of prognosis [39,40,41]. After being recruited to the tumor site by adipocyte-secreted IL-1β, TANs induce the activation of PSCs, which leads to further IL-1β section and contributes to PDAC progression [42]. Mutations in genes such as SETD2 and TP53 promote the recruitment of TANs, and consequently, PDAC tumorigenesis [43, 44]. SETD2-deficient PDAC tumor cells recruit neutrophils to the tumor site and reprogram them toward the N2 type,this causes the neutrophils to upregulate genes such as IL10 and MRC1 via the activation of AKT signaling [43]. The gain-of-function TP53R172H mutation promotes TAN infiltration into the tumor in response to tumor-cell-derived chemokines; these TANs subsequently render the tumor resistant to chemotherapy and CD40 combination immunotherapy [44]. scRNA-seq analysis has shown that, in liver cancer, CCL4+TANs and PD-L1+ TANs recruit TAMs to the tumor site and suppress T cell cytotoxicity, respectively [45].
Neutrophil extracellular traps (NETs) were first discovered in the context of inflammation. NETs are web-like structures composed of DNA, histones, and various proteins, which are extruded by neutrophils in a process termed NETosis [46]. In the TME, NETs promote tumor cell proliferation and metastasis, as well as inducing hypercoagulation [47]. Moreover, IL-17 production in the PDAC TME leads to the recruitment of neutrophils and triggers NET formation [48]. A recent study reported that the binding of TIMP1 (an MMP inhibitor) to its receptor CD63 induced NET formation in neutrophils via ERK signaling [49]. Moreover, KDM6A depletion from PDAC cells induced neutrophil recruitment to the tumor site and NET formation via the CXCL1-CXCR2 axis [50]. Emerging evidence suggests that NETs directly or indirectly foster tumor proliferation, shield tumor cells from cytotoxic lymphocytes, and promote tumor angiogenesis [51].
TANs and MDSCs have common origins and share a differentiation pathway, which raises questions about whether they are indeed distinct cell types. Moreover, there is currently no standardized nomenclature or methods for accurately differentiating these cells. MDSCs were named on the basis of their immunosuppressive function, whereas TANs described a group of neutrophils modulated by the tumor. Although TANs and MDSCs express similar surface markers (e.g., CD66b+, CD11b+, and HLA-DR−), unlike MDSCs, TANs exhibit high chemokine secretion and low ROS production, suggesting that TANs and MDSCs are different types of cells [52]. In addition, despite some sample processing limitations, neutrophils and MDSCs can be separated by gradient centrifugation [53]. However, Shaul et al. suggested that MDSCs are a subset of neutrophils with a unique activation state rather than a separate cellular entity. Furthermore, the existence of MDSCs as a population of myeloid cells with an entirely immunosuppressive function contradicts the typical plasticity and dynamics of myeloid cells [54]. Given the challenges of differentiating between these myeloid cell types, TANs and MDSCs will be discussed alongside each other in this review,however, we prefer the term TANs over MDSCs as it better describes the plasticity of these cells and their interplay with various other cell types.
DCs
Dendritic cells bridge the gap between innate and adaptive immunity. Generally, DCs can be classified into three populations: conventional DCs (cDCs), plasmacytoid DCs (pDCs), and monocyte-derived DCs (moDCs) [55]. cDCs can be further divided into two subsets: the CD8α+ and/or CD103+ cDC1 subset, which presents antigens and recruits cytotoxic T cells, and the CD103+ cDC2 subset, which activates CD4+ T cells such as T helper type 17 (Th17) cells. pDCs are less adept at antigen presentation than cDCs and instead play a dominant role in IFN-γ secretion during viral infection. moDCs are mainly generated under inflammatory conditions and are involved in Treg generation during cancer pathogenesis [55, 56].
The tumor nests of pancreatic cancer contain fewer cDCs than those of lung cancer; moreover, these cDCs exhibit reduced antigen presentation capacity [57]. Moreover, the level of cDC infiltration into PDAC tumors is correlated with increased patient survival [58]. The recruitment of cDCs into early pancreatic lesions leads to a decrease in the number of immunosuppressive Th17 cells and an increase in that of cytotoxic CD8+ T cells. This remodeling of the TME activates the antitumor Th1 response and promotes tumor eradication [57]. DCs lacking heat shock proteins 70 (Hsp70) express higher TNF-α and MHC-II levels and are more effective at reducing the tumor burden than wildtype DCs in KPC mice models [59]. Moreover, the stimulator of IFN genes (STING) agonist induced DC activation and maturation both in vivo and in vitro, which also increased the DC-mediated secretion of the proinflammatory cytokines IL-6 and TNF-α [60]. Collectively, these findings suggest that DCs are promising targets in the treatment of pancreatic cancer.
Innate lymphoid cells (ILCs)
Despite representing a small population of immune cells, ILCs are crucial players in the progression and prognosis of cancers. ILCs are divided into five subsets: natural killer (NK) cells, ILC1, ILC2, NCR+ILC3, and NCR−ILC3, based on their lineage-specific progenitor populations [61].
NK cells, which have similar functions to CD8+ cytotoxic T cells, have been the most extensively investigated of all the ILCs in the tumor. The function of NK cells is impaired by the accumulation of tumor-derived factors such as TGF-β, IL-10, indoleamine 2,3-dioxygenase (IDO), in the TME; moreover, the characteristics of NK cells are influenced by the tumor type [62]. For instance, in PDAC, NK cells exhibit impaired cytotoxicity, while expressing low levels of IFN-γ and high levels of IL-10 [63]. The cytotoxicity of NK cells can also be impaired by the overexpression of UQCRC1, a key component of mitochondrial complex III, in response to elevated extracellular ATP concentrations [64]. The roles of other ILCs in PDAC remain unclear, largely due to their low frequencies in the PDAC TME. While the IL-33-mediated activation of ILC2s, which led to the recruitment of CD103+ DCs into the PDAC TME and subsequently CD8+ T cell activation, was associated with better PDAC prognosis in one study, it was associated with poor prognosis in another [65, 66].
Compared with other immune cells, our understanding of ILCs remains limited, especially considering the complexity of the TME and the low ILC frequencies. Therefore, strategies such as scRNA-seq will be instrumental in investigating ILC function in PDAC.
Role of innate immune cells in PDAC metastasis
Approximately 80% of PDAC patients present with unresectable or metastatic cancer; thus, metastasis is a leading cause of death among newly diagnosed PDAC patients [1]. Common PDAC metastatic pathways are local invasion and lymphatic metastasis, with distal metastasis typically occurring in the liver, lung, and bone. Metastasis involves several sequential steps: angiogenesis, lymphangiogenesis, EMT, migration, invasion of surrounding tissues, formation of the pre-metastatic niche, and growth at the metastasis site. The crosstalk between tumor cells and stromal cells, which contributes to PDAC metastasis, is outlined in Fig. 3.

Innate immune cells participate in PDAC metastasis. Innate immune cells participate in multiple steps of PDAC metastasis. Cytokines, such as vascular endothelial growth factor (VEGF), IL-1β, and CCL5, secreted by innate immune cells promote angiogenesis and tumor cell proliferation. Meanwhile, signaling pathways such as NF-κB induce the invasion and migration of tumor cells. Exosomes and cytokines derived from TAMs and TANs promote EMT and growth of PDAC tumors at metastatic sites. Image generated using Biorender
TAMs
Interplay among TAMs, tumor cells, and stromal cells through various pathways is pivotal in PDAC metastasis. A recent study has shown that TAM numbers are positively correlated with the microvessel density of PDAC tissues and that exosomes derived from TAMs promote the angiogenesis of endothelial cells in vitro [67]. TAMs are involved in angiogenesis and lymphangiogenesis through the direct secretion of vascular endothelial growth factor (VEGF) [68] or the activation of several signaling pathways. Moreover, ANXA-1, contained in the extracellular vesicles of PDAC cells, regulates M2 phenotype polarization, which in turn activates endothelial cells and fibroblasts and contributes to angiogenesis and ECM degradation [69]. In addition, the frequency of M2-polarized TAMs in the regional lymph nodes of PDAC patients is strongly associated with nodal lymphatic vessel density, suggesting that TAMs are capable of lymphangiogenesis [70].
Several other PDAC metastasis mechanisms implicating TAMs have been described. For instance, the exosomal microRNA-301a-3p expressed by hypoxic pancreatic cancer cells induced M2 polarization of macrophages via the PTEN-PI3Kγ signaling axis, promoting PDAC cell migration, invasion, and EMT [71]. Notch signaling triggered by microRNA-124 in PDAC cells promoted M2 polarization, leading to STAT3 pathway activation, which facilitated tumor cell EMT and invasion [72]. EMT is also triggered when TAM-secreted oncostatin M (OSM) activates the LOXL2-mediated metastatic cascade [73]. In addition, debris and IgG derived from PDAC cells can induce IL-1β secretion from TAMs via the TLR4/TRIF/NF-κB signaling pathway, resulting in EMT, and consequently, PDAC metastasis [74]. Moreover, blocking, growth-arrest-specific 6 (GAS6), which is produced by TAMs and CAFs in the PDAC TME, partially reversed EMT and supported NK activation [75]. TAMs can also enhance PDAC cell migration by inducing EMT via the TGF-β/SMAD/SNAIL signaling axis [76].
TAM-derived exosomal micro-RNA-501-3p activates the TGF-β signaling pathway, promoting PDAC cell migration and invasion by inhibiting the tumor suppressor gene TGFBR3 [77]. The STAT3/NF-κB pathway is activated by PDAC-derived exosomal FDG5-AS1, polarizing the formation of M2 macrophages, which in turn stimulates PDAC cell proliferation and metastasis [78]. The expression of TNFSF9, an immune checkpoint marker originally shown to be expressed on antigen-presenting cells, on PDAC cells was associated with a poor prognosis [79]. A recent study revealed that TNFSF9 promoted the metastasis of PDAC by inducing M2 polarization via Src/FAK/p-Akt/IL-1β signaling [79]. TAMs can also contribute to liver fibrosis and sustain the growth of tumor cells by secreting granulin to activate resident hepatic stellate cells; these events contribute to the development of PDAC liver metastases [80].
Besides tumor cells, TAMs interact with CAFs to promote PDAC metastasis. The binding of OSM secreted by TAMs to its receptor (OSMR) on CAFs induces inflammatory gene expression in CAFs. Thus, OSM depletion creates a more immunogenic environment, in which CAFs exhibit reduced inflammation gene expression, M2-like TAM numbers decline, and T cell function increases (evidenced by elevated CD44 and CD127 expression) [81]. Crosstalk between PDAC cells, TAMs, and CAFs is mediated via the IL-33/ST2/CXCL3/CXCR2 signaling pathway. The activation of the IL-33/ST2 pathway in TAMs induces them to express CXCL3, which in turn converts CAFs into myoblast CAFs,because these myoblast CAFs express the cell surface matrix protein collagen III, they can form clusters with PDAC cells to promote PDAC metastasis [82].
MDSCs and TANs
An increasing number of studies have revealed that TANs and their NETs are involved in the progression and metastasis of PDAC via diverse signaling pathways. GAS6 expressed by TANs activates the AXL receptor on PDAC cells, enabling their regrowth after chemotherapy [83]. Gap junction protein beta 3 (GJB3), a protein which forms gap junctions (channels for the transportation of small molecules between adjacent cells), was found to facilitate PDAC liver metastasis by promoting neutrophil accumulation and N2 polarization by transferring cAMP [84], GJB3 depletion consequently suppressed PDAC liver metastasis in vivo. It was also observed that circulating tumor cells (CTCs were surrounded by neutrophils in tumor-adjacent vessels of PDAC tumors [85],thus, TANs may assist in distant metastasis formation through their direct interaction with CTCs. A population of immunosuppressive P2RX-1− neutrophils, which promoted metastatic tumor growth by upregulating PD-L1 expression on tumor cells, was identified in a murine PDAC liver metastasis model and in clinical PDAC samples [86]. NRF2, a ROS-sensitive transcription factor, may promote PDAC liver metastasis by increasing PD-L1 expression on PDAC cells after boosting intracellular ROS production by P2RX-1− neutrophils [86].
NETs also contribute to PDAC metastasis via a variety of signaling pathways. The peptidylarginine deiminase 4 (PAD4)-mediated release of DNA from NETs was shown to activate PSCs by interacting with receptors for advanced glycation end products (RAGE) and promoting the proliferation and metastasis of PDAC cells [87]. In addition, PDAC cells express collagen-induced discoid domain receptor (DDR1). In response NF-κB signaling, DDR1 stimulates CXCL5 production from tumor cells, leading to TAN recruitment, NET formation, and eventually, the invasion and metastasis of PDAC cells [88]. In PDAC, NETs facilitate EMT, as well as tumor cell migration and invasion, via the IL-1β/EGFR/ERK pathway [89]. NETs can also promote PDAC liver metastasis by enhancing the migration of hepatic stellate cells [90].
Although less investigated, the interactions among MDSCs and other cells in the PDAC TME may also have important roles in the metastatic process. For instance, MDSCs significantly increased CSC numbers in a mouse model of PDAC, which was accompanied by a significant upregulation of genes related to EMT in tumor cells [91].
Other innate immune cells
The roles of NK cells, DCs, and other innate immune cells in PDAC metastasis are not as well-studied as those of TAMs or TANs. An immunosuppressive DC subset was found to express PD-L2 in the metastatic site and induce the expansion of Tregs in vitro, suggesting that they played a role in shaping the immunosuppressive PDAC TME [92]. NK cells are the only innate immune cells with direct tumoricidal function. Moreover, NK-cell-derived exosomal miR-3607-3p inhibits the migration and invasion of PDAC cells in vitro by targeting IL-26 [93].
Crosstalk between innate immune cells
In the TME, innate immune cells communicate via the secretion of soluble factors (e.g., cytokines and chemokines) or interactions between surface molecule and their receptors, which regulate various signaling pathways (Fig. 4). The levels of cytokines such as IL-1β, IL-10, VEGF, and TNF were reported to be increased in patients with PDAC [94]. IL-10 is an anti-inflammatory cytokine secreted by most immune and tumor cells. A study of breast cancer revealed that macrophages were a major source of IL-10 and that DCs were responsive to this cytokine as they expressed high levels of the IL-10 receptor (IL-10R). Thus, high IL-10 levels reduced the capacity of DCs to produce IL-12, which in turn impaired the recruitment of cytotoxic T cells to the tumor site [95]. In addition, IL-1β, which promotes NETosis, can be secreted by TAMs, DCs, and tumor cells, suggesting possible interactions among these cell types [96, 97]. Moreover, TAMs release IL-8 in response to tumor cells, which leads to the recruitment of neutrophils and MDSCs to the tumor site [98]. In addition, innate immune cells may mutually recruit each other via the secretion of specific chemokines. For instance, scRNA-seq analysis predicted that CCL4+TANs recruited macrophages via the CCL4/CCR5 axis; this result was validated in an in vitro chemotactic assay in which liver tumor cells were co-cultured with TANs [45]. In chronic pancreatitis, macrophages were recruited via the CCL2/CCR2 axis,however, the disruption of this axis inhibited not only macrophage but also neutrophil recruitment, implying that chemokines are important for crosstalk between macrophages and neutrophils [99]. Gas6 is a TAM receptor ligand, and binding of Gas6 to its receptor AXL causes phosphorylation and activation of AXL. Interestingly, blocking GAS6 binding to AXL activated NK cells and partially reverse the EMT of tumor cells [75]. In the PDAC TME, TAMs express high levels of apolipoprotein E (ApoE), which drives tumor cells to secrete CXCL1 and leads to the recruitment of more immunosuppressive myeloid cells [100].

Crosstalk between innate immune cells in the PDAC TME. Innate immune cells, such as tumor-associated macrophages, tumor-associated neutrophils, dendritic cells, and natural killer cells, secrete cytokines, chemokines, and molecules to interact with each other, forming an immunosuppressive microenvironment that promotes tumor progression and proliferation. Image generated using Biorender
Despite being well documented in the context of inflammation, the crosstalk between innate immune cells and tumor cells in cancer is not well characterized. The arrival of state-of-the-art technologies such as scRNA-seq and spatial transcriptomics, will enable a more comprehensive and detailed exploration of the intricate interactions among innate immune cells in the TME. A deeper understanding of the PDAC TME will pave the way for the development for promising therapeutic strategies.
Targeting immune cells to remodel the PDAC TME
The PDAC TME comprises diverse interactions among cells, cytokines, chemokines, and other factors; this complex and dynamic nature of the TME poses challenges for the development of strategies aimed at targeting its specific components. The studies discussed above provide strong evidence that TAMs, TANs, MDSCs, and other innate immune cells play vital roles in PDAC metastasis. Targeting these cells can be a potential therapeutic strategy against PDAC. Here, we discuss present and potential future strategies related to the targeting of innate immune cells in PDAC TME. Among these strategies, the targeting of macrophages, neutrophils, and MDSCs has been most extensively investigated (Table 1); however, as most of the evidence has been gathered at the preclinical stage, the efficacy of these strategies will need to be validated in larger trials.
Targeting TAMs
Reprogramming TAMs is one of the most popular immunotherapeutic strategies being developed for PDAC. For instance, IFN-γ was reported to re-educate TAMs into M1-type macrophages, which released higher levels of the pro-inflammatory cytokine IL-12 and lower levels of the pro-tumorigenic factors IL-10, MMP9, and VEGF [101]. CD40 can also be targeted to reprogram TAMs. Treatment with an agonistic anti-CD40 antibody mAb CP-870,893 led to partial tumor regression in both mice and humans, via a mechanism in which the CD40-activated macrophages became tumoricidal and contributed to the degradation of the tumor stroma [102]. A phase Ib, multicenter study combining a monoclonal, agonistic anti-CD40 antibody with chemotherapy, showed that this combination had promising clinical activity and tolerable adverse effects [103]. The use of selicrelumab, another agonistic anti-CD40 antibody as a neoadjuvant therapy, activated T cells, increased the production of the inflammatory factors CXCL10 and CCL22 by several cell types, and decreased TAM numbers in the PDAC TME [104]. The PI3K-γ and CSF1-R signaling pathways have also been targeted to enhance the response of T cells to checkpoint immunotherapy and reprogram TAMs in pancreatic cancer mouse models (clinical trial number: NCT02777710) [105, 106]. Specifically, a PI3K-γ inhibitor and a CSF1-R-siRNA were simultaneously administered to PDAC model mice [105]. Increased numbers of M1 type macrophages and a reduction in the M2 type macrophages were observed in mouse tumors, which was associated with a significant decrease in tumor weight [105].
Macrophage depletion is another research direction being explored. Several studies [109, 110, 112, 129] have targeted the CCL2/CCR2 axis, which is vital in the recruitment of TAMs to the TME [130]. Using a CCR2 inhibitor in combination with FOLFIRINOX chemotherapy, achieved local tumor control with tolerable adverse effects [109]. CCL2 inhibition increased effector T cell responses, enhanced chemotherapeutic efficacy, and inhibited metastasis [110]. The specific depletion of TAMs with lurbinectedin increased the extent of gemcitabine-mediated DNA damage in a PDAC mouse model, thus improving the efficacy of gemcitabine therapy [111].
CD47 is a transmembrane glycoprotein, which binds to signal regulatory protein α (SIRPα) to send a “don’t eat me” signal to macrophages, reducing their phagocytic ability [131]. Therefore, blocking the CD47/SIRPα interaction has emerged as a promising next-generation immune checkpoint disruption strategy [131]. Indeed, administering a blocking anti-CD47 antibody to PDAC model mice increased the numbers of CD4+ and CD8+ T cells in the tumor, while decreasing those of monocytes/macrophages [132]. A phase I trial of an anti-CD47 antibody has demonstrated that it is well tolerated and associated with objective responses in multiple tumor types [107, 108].
Targeting TANs and MDSCs
Similar to targeting TAMs, the reprogramming or depletion of TANs are potential PDAC strategies being investigated in clinical trials. The protumor N2-like TANs have the potential to transform into N1-like TANs via IFN signaling pathway activation. Indeed, β-glucan administered to a subcutaneous mouse model of melanoma activated the IFN signaling pathway and promoted neutrophils to exhibit long-term antitumor effects [133]. Endogenous IFN-β inhibits tumor angiogenesis through the repression of genes encoding VEGF, MMP9, and CXCR4 in TANs [35]. Blocking CXCR2 signaling on TANs improved antitumor immunity in a murine PDAC model by enhancing the chemotherapeutic efficacy [112]. Lorlatinib treatment inhibited PDAC progression in a PDAC mouse model by specifically targeting Ly6G+ neutrophils and suppressing their development, mobilization, and infiltration, as well as improving the efficacy of immune checkpoint blockade [113].
The depletion of neutrophils or the inhibition of NETosis are valid strategies aimed at remodeling the PDAC TME. NETosis inhibition by DNase, chloroquine, IL-17/IL-17R blockade, or the PAD4 inhibitor have been reported [48, 87, 114,115,116]. DNase treatment of PDAC model mice inhibited tumor growth and stromal activation within the PDAC TME [87]. In a phase II clinical trial, the administration of chloroquine to preoperative patients with PDAC in combination with chemotherapy resulted in greater antitumor responses and autophagy inhibition [114]. IL-17 recruits neutrophils and triggers NET release. Accordingly, disrupting the IL-17/IL-17R interaction increased immune checkpoint blockade sensitivity in a PDAC model [48]. Thus, IL-17 and checkpoint blockade may be combined to enhance the efficacy of existing PD-1-targeting therapies for the treatment of metastatic PDAC. Inhibiting PAD4, an enzyme with a central role in NET formation, reduced the NET forming capacity of both murine and human neutrophils,moreover, this PAD4 inhibitor can be used in combination with other therapeutic agents such as an IL-17 inhibitor [116]. CXCL1 inhibition significantly reduced TAN infiltration and NETosis, and ultimately attenuated the tumor growth in KDM6A-deficient PDAC mice model in vivo [50].
CCR5 and CXCR2, two chemokine receptors involved in the recruitment or maturation of neutrophils, are also potential targets for remodeling the PDAC TME [117, 119]. In other cancers, the inhibition of CCR5 reduced MDSC recruitment to the tumor and prevented tumor metastasis [117]. In PDAC, CXCR2 signaling is predominantly upregulated in TANs and MDSCs. Thus, treatment of a PDAC model mice with a CXCR2 inhibitor significantly decreased their intratumoral MDSC numbers, while increasing the infiltration of CD8+T cell into the PDAC TME [120]. Moreover, CXCR2 blockade suppressed PDAC metastasis and, to some degree, inhibited tumorigenesis [119].
The CD11b/CD18 integrin heterodimer is expressed on the membranes of MDSCs, TANs, and TAMs, where it mediates myeloid adhesion, migration, tissue recruitment, phagocytosis, and survival. Given these functions, CD11b/CD18 blockade promises to reduce the infiltration of most myeloid subsets into the tumor [134]. Indeed, the CD11b modulator GB1275 reduced the tumor infiltration of CD11b+ MDSCs and prolonged the survival of KPC model mice [118].
Targeting DCs
Emerging DC-targeting treatment methods have been developed. These include DC vaccines and the use of CD40 agonists to promote cDC1 maturation [121, 122]. A phase I clinical trial was conducted by injecting autologous, tumor-lysate-loaded moDCs into patients with resected PDAC. After a median follow-up of 25 months, seven out of ten patients did not experience PDAC recurrence or progression and had no vaccine-related serious adverse effects, suggesting the favorable safety and feasibility of this DC vaccine [121]. A clinical trial combining DC agonists with allogeneic tumor-lysate-loaded DCs is ongoing, with treatment safety and tolerability as the primary endpoints and the magnitude of the antitumor immune response as the secondary endpoint [122]. In the context of lung cancer, combining neoantigen-presenting DCs with an anti-CD38 antibody has been shown to reduce Treg infiltration, thereby reshaping the immunosuppressive TME [123].
As professional antigen-presenting cells, DCs prime and stimulate T cells to eliminate cancer cells. Hence, boosting DC function represents a promising immunotherapeutic strategy in cancer treatment. Growth factors, such as FMS-like tyrosine kinase 3 ligand (FLT3L) have attracted increasing attention due to their ability to expand and activate DCs [135]. Administration of FLT3L expanded cDC1s in both lymphoid and peripheral tissues, while also enhancing tumor-specific T cell responses in conditions such as breast cancer and melanoma [124, 136]. Notably, in the case of melanoma, the administration of FLT3L activates CD103+ DC progenitors, thereby increasing the efficacy of BRAF and PD-L1 blockade [124].
Targeting NK cells
NK cells can be likened to the innate immune equivalent of T cells; as such, NK cells represent promising targets for the treatment of PDAC. Inspired by the successes of CAR-T cell therapy, CAR-NK cell therapy has emerged as a budding immunotherapy, with several notable advantages over CAR-T cells (e.g., a better safety profile) [137]. Treating a PDAC subcutaneous mouse model with CAR-NK cells engineered to target folate receptor alpha and death receptor 4, both highly expressed in tumor cells, increased NK infiltration into the tumor tissue and promoted tumor cell apoptosis [138]. CAR-NK cells displayed even greater antitumor efficacy when used in combination with a STING agonist, as evidenced by marked tumor growth inhibition in PDAC model mice and their prolonged survival [125]. Robo1-specific CAR-NK cell immunotherapy enhanced the efficacy of 125I seed treatment. Higher greyscale values and a significant reduction in tumor size were observed in the orthotopic PDAC mouse model treated with the combination therapy compared with either monotherapy [126]. Given that NK cells are less likely to induce graft-versus-host disease than CD8+ T cells, clinical trials of allogenic NK cells have also been carried out [127, 137]. Patients with stage III PDAC being treated with a combination of irreversible electroporation and allogenic NK cell immunotherapy had a higher median overall survival and progression-free survival than the control group, and few adverse events [127].
Induced pluripotent stem cells (iPSCs) provide an “off-the-shelf” supply of lymphocytes, which can be used a source of NK cells for immunotherapy. NK cells generated from human iPSCs, express NK-defining markers such as CD56, CD16, and death-inducing ligands, and exhibit cytotoxicity through cytokine secretion or antibody-dependent cell-mediated cytotoxicity [128]. The iPSC-derived NK cell product FT500 is being administrated in a phase I clinical trial to target solid tumors, including pancreatic cancer (clinical trial number: NCT03841110). FT516 and FT576 are other allogeneic NK cells being trialed in the treatment of ovarian cancer and multiple myeloma, respectively (clinical trial number: NCT04630769, NCT05182073).
Conclusions and future perspectives
Despite the emergence of advanced therapies, pancreatic cancer remains one of the most lethal cancers, which is largely due to its complex immunosuppressive TME. Innate immune cells such as TAMs, TANs, and MDSCs play critical roles in shaping the immunosuppressive PDAC TME, which also impacts tumor progression and metastasis.
Despite our growing understanding of the PDAC TME and the development of many innovative TME remodeling strategies, various challenges still exist. Owing to the heterogeneity of tumors, the TME of orthotopic and metastatic sites may be different. This may explain the unsatisfactory response observed to currently available treatments. Moreover, innate immune cells, especially TAMs and TANs, exhibit considerable plasticity, which is greatly influenced by the TME. Thus, techniques such as spatial transcriptomics and multiplex phenotyping are needed to accurately characterize the subtypes of immunosuppressive cells in the TME. These cells can then either be eliminated or repolarized to attack tumor cells and enhance the efficacy of other immunotherapies. Given the complexity of the TME, interactions among its various components, especially among innate immune cells, are not fully understood. In addition, PDAC patients are usually diagnosed at a late stage after metastasis has occurred. Thus, finding more sensitive diagnostic biomarkers for PDAC remains a priority.
In light of these challenges, we believe that a deeper understanding of the PDAC TME is needed to successfully and safely target this cancer. For instance, recent studies have used scRNA-seq to explore the differences in the TME between orthotopic and liver metastasis in PDAC. However, the primary focus of these studies was on cancer cells, CAFs, and T cells, with less emphasis on innate immune cells such as TAMs and TANs [139]. Consequently, the specific roles and mechanisms through which innate immune cells, such as macrophages and neutrophils, may influence these TME differences are unclear. Moreover, the intricacies of interactions among innate immune cells in the PDAC TME remain a mystery. At present, the hunt for potential biomarkers and drug targets expressed by cells in the PDAC TME continues. What is evident, however, is that by gaining a deeper understanding of the PDAC TME, we are moving closer to developing more effective, targeted therapeutics for this aggressive cancer.
Availability of data and materials
Not applicable.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33. https://doi.org/10.3322/caac.21708.
Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, Lazarus J, Amir ED, Gunchick V, Espinoza C, Bell S, Harris L, Lima F, Irizarry-Negron V, Paglia D, Macchia J, Chu AKY, Schofield H, Wamsteker EJ, . . . Pasca di Magliano, M. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat Cancer. 2020;1(11):1097–1112. https://doi.org/10.1038/s43018-020-00121-4
Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, Apte MV. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68(7):2085–93. https://doi.org/10.1158/0008-5472.Can-07-2477.
Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer — clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17(9):527–40. https://doi.org/10.1038/s41571-020-0363-5.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA. IL1-Induced JAK/STAT signaling is antagonized by TGFβ to Shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9(2):282–301. https://doi.org/10.1158/2159-8290.Cd-18-0710.
Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64. https://doi.org/10.1038/nri1733.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. https://doi.org/10.1016/j.cell.2010.03.014.
Ren B, Cui M, Yang G, Wang H, Feng M, You L, Zhao Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer. 2018;17(1):108. https://doi.org/10.1186/s12943-018-0858-1.
Habtezion A, Edderkaoui M, Pandol SJ. Macrophages and pancreatic ductal adenocarcinoma. Cancer Lett. 2016;381(1):211–6. https://doi.org/10.1016/j.canlet.2015.11.049.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, Alizadeh AA. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–45. https://doi.org/10.1038/nm.3909.
Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. 2016;37(7):8657–64. https://doi.org/10.1007/s13277-015-4741-z.
Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, Gao R, Orf J, Wang T, Sawant D, Kang J, Bhatt D, Lu D, Li CM, Rapaport AS, Yu X. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181(2):442-459.e429. https://doi.org/10.1016/j.cell.2020.03.048.
Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, Peet GW, Zhong G, Lu S, Zhu W, Mao Y, Xiao M, Bergmann M, Hu X, Kerkar SP, Zhang Z. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829-845.e820. https://doi.org/10.1016/j.cell.2019.10.003.
Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, van den Broek M, Beisel C, Stadler MB, Gedye C, Reis B, Pe’er D, Bodenmiller B. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169(4):736-749.e718. https://doi.org/10.1016/j.cell.2017.04.016.
Werba G, Weissinger D, Kawaler EA, Zhao E, Kalfakakou D, Dhara S, Wang L, Lim HB, Oh G, Jing X, Beri N, Khanna L, Gonda T, Oberstein P, Hajdu C, Loomis C, Heguy A, Sherman MH, Lund AW, Simeone DM. Single-cell RNA sequencing reveals the effects of chemotherapy on human pancreatic adenocarcinoma and its tumor microenvironment. Nat Commun. 2023;14(1):797. https://doi.org/10.1038/s41467-023-36296-4.
Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36(4):229–39. https://doi.org/10.1016/j.it.2015.02.004.
Liou GY, Döppler H, Necela B, Krishna M, Crawford HC, Raimondo M, Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J Cell Biol. 2013;202(3):563–77. https://doi.org/10.1083/jcb.201301001.
Boyer S, Lee HJ, Steele N, Zhang L, Sajjakulnukit P, Andren A, Ward MH, Singh R, Basrur V, Zhang Y, Nesvizhskii AI, Pasca di Magliano M, Halbrook CJ, Lyssiotis CA. Multiomic characterization of pancreatic cancer-associated macrophage polarization reveals deregulated metabolic programs driven by the GM-CSF-PI3K pathway. Elife. 2022;11. https://doi.org/10.7554/eLife.73796
Zhang M, Huang L, Ding G, Huang H, Cao G, Sun X, Lou N, Wei Q, Shen T, Xu X, Cao L, Yan Q. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J Immunother Cancer. 2020;8(1):e000308. https://doi.org/10.1136/jitc-2019-000308.
Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, Lee KB, Zambirinis CP, Pandian GSB, Savadkar S, Torres-Hernandez A, Nayak S, Wang D, Hundeyin M, Diskin B, Aykut B, Werba G, Barilla RM, Rodriguez R, Miller G. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. 2017;23(5):556–67. https://doi.org/10.1038/nm.4314.
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47. https://doi.org/10.1016/j.ccr.2012.04.024.
Thyagarajan A, Alshehri MSA, Miller KLR, Sherwin CM, Travers JB, Sahu RP. Myeloid-derived suppressor cells and pancreatic cancer: implications in novel therapeutic approaches. Cancers (Basel). 2019;11(11):1627. https://doi.org/10.3390/cancers11111627.
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. https://doi.org/10.1038/ncomms12150.
Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–81. https://doi.org/10.1136/gutjnl-2013-306271.
Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22(4):275–81. https://doi.org/10.1016/j.semcancer.2012.01.011.
Huber M, Brehm CU, Gress TM, Buchholz M, Alashkar Alhamwe B, von Strandmann EP, Slater EP, Bartsch JW, Bauer C, Lauth M. The immune microenvironment in pancreatic cancer. Int J Mol Sci. 2020;21(19):7307. https://doi.org/10.3390/ijms21197307.
Torroella-Kouri M, Rodríguez D, Caso R. Alterations in macrophages and monocytes from tumor-bearing mice: evidence of local and systemic immune impairment. Immunol Res. 2013;57(1–3):86–98. https://doi.org/10.1007/s12026-013-8438-3.
Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, Mocellin S, Rossi CR, Bronte V, Mandruzzato S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. 2016;7(2):1168–84. https://doi.org/10.18632/oncotarget.6662.
Lin Y, Cai Q, Chen Y, Shi T, Liu W, Mao L, Deng B, Ying Z, Gao Y, Luo H, Yang X, Huang X, Shi Y, He R. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology. 2022;75(1):28–42. https://doi.org/10.1002/hep.32099.
Sun R, Luo H, Su J, Di S, Zhou M, Shi B, Sun Y, Du G, Zhang H, Jiang H, Li Z. Olaparib suppresses MDSC recruitment via SDF1α/CXCR4 Axis to improve the anti-tumor efficacy of CAR-T cells on breast cancer in mice. Mol Ther. 2021;29(1):60–74. https://doi.org/10.1016/j.ymthe.2020.09.034.
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, Dang Y, Chu Y, Fan J, He R. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124–35. https://doi.org/10.1158/0008-5472.Can-15-2973.
Liu Y, Sun Y, Wang P, Li S, Dong Y, Zhou M, Shi B, Jiang H, Sun R, Li Z. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. J Transl Med. 2023;21(1):255. https://doi.org/10.1186/s12967-023-04080-z.
Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, André P, Rigot V, Iovanna J, van de Pavert S, Lombardo D, Mas E, Martirosyan A. Deciphering the crosstalk between myeloid-derived suppressor cells and regulatory t cells in pancreatic ductal adenocarcinoma. Front Immunol. 2019;10:3070. https://doi.org/10.3389/fimmu.2019.03070.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94. https://doi.org/10.1016/j.ccr.2009.06.017.
Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120(4):1151–64. https://doi.org/10.1172/jci37223.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-associated neutrophils recruit macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150(7):1646-1658.e1617. https://doi.org/10.1053/j.gastro.2016.02.040.
Wang L, Liu Y, Dai Y, Tang X, Yin T, Wang C, Wang T, Dong L, Shi M, Qin J, Xue M, Cao Y, Liu J, Liu P, Huang J, Wen C, Zhang J, Xu Z, Bai F, . . . Shen B. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut. 2022. https://doi.org/10.1136/gutjnl-2021-326070
Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, Altinel D, Adsay V. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol. 2011;24(12):1612–9. https://doi.org/10.1038/modpathol.2011.113.
Toledano-Fonseca M, Cano MT, Inga E, Gómez-España A, Guil-Luna S, García-Ortiz MV, Mena-Osuna R, De la Haba-Rodriguez JR, Rodríguez-Ariza A, Aranda E. The combination of neutrophil-lymphocyte ratio and platelet-lymphocyte ratio with liquid biopsy biomarkers improves prognosis prediction in metastatic pancreatic cancer. Cancers (Basel). 2021;13(6):1210. https://doi.org/10.3390/cancers13061210.
Ben Q, An W, Wang L, Wang W, Yu L, Yuan Y. Validation of the pretreatment neutrophil-lymphocyte ratio as a predictor of overall survival in a cohort of patients with pancreatic ductal adenocarcinoma. Pancreas. 2015;44(3):471–7. https://doi.org/10.1097/mpa.0000000000000271.
Arima K, Okabe H, Hashimoto D, Chikamoto A, Tsuji A, Yamamura K, Kitano Y, Inoue R, Kaida T, Higashi T, Taki K, Imai K, Komohara Y, Beppu T, Takeya M, Baba H. The diagnostic role of the neutrophil-to-lymphocyte ratio in predicting pancreatic ductal adenocarcinoma in patients with pancreatic diseases. Int J Clin Oncol. 2016;21(5):940–5. https://doi.org/10.1007/s10147-016-0975-z.
Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, Babykutty S, Huang Y, Jung K, Rahbari NN, Han X, Chauhan VP, Martin JD, Kahn J, Huang P, Jain RK. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016;6(8):852–69. https://doi.org/10.1158/2159-8290.Cd-15-1177.
Niu N, Shen X, Zhang L, Chen Y, Lu P, Yang W, Liu M, Shi J, Xu D, Tang Y, Yang X, Weng Y, Zhao X, Wu LM, Sun Y, Xue J. Tumor cell-intrinsic SETD2 deficiency reprograms neutrophils to foster immune escape in pancreatic tumorigenesis. Adv Sci (Weinh). 2023;10(2): e2202937. https://doi.org/10.1002/advs.202202937.
Siolas D, Vucic E, Kurz E, Hajdu C, Bar-Sagi D. Gain-of-function p53(R172H) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep. 2021;36(8): 109578. https://doi.org/10.1016/j.celrep.2021.109578.
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, Feng M, Wang F, Cheng J, Li Z, Zhan Q, Deng M, Zhu J, Zhang Z, Zhang N. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612(7938):141–7. https://doi.org/10.1038/s41586-022-05400-x.
Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of Neutrophil Extracellular Traps (NETs) in tumor progression and metastasis. Front Immunol. 2020;11:1749. https://doi.org/10.3389/fimmu.2020.01749.
Jung HS, Gu J, Kim JE, Nam Y, Song JW, Kim HK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS One. 2019;14(4): e0216055. https://doi.org/10.1371/journal.pone.0216055.
Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, Zoltan M, Arora N, Baydogan S, Horne W, Burks J, Xu H, Hussain P, Wang H, Gupta S, Maitra A, Bailey JM, Moghaddam SJ, Banerjee S, McAllister F. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020;217(12):e20190354. https://doi.org/10.1084/jem.20190354.
Schoeps B, Eckfeld C, Prokopchuk O, Böttcher J, Häußler D, Steiger K, Demir IE, Knolle P, Soehnlein O, Jenne DE, Hermann CD, Krüger A. TIMP1 triggers neutrophil extracellular trap formation in pancreatic cancer. Cancer Res. 2021;81(13):3568–79. https://doi.org/10.1158/0008-5472.Can-20-4125.
Yang J, Jin L, Kim HS, Tian F, Yi Z, Bedi K, Ljungman M, Pasca di Magliano M, Crawford H, Shi J. KDM6A loss recruits tumor-associated neutrophils and promotes neutrophil extracellular trap formation in pancreatic cancer. Cancer Res. 2022;82(22):4247–60. https://doi.org/10.1158/0008-5472.Can-22-0968.
Cristinziano L, Modestino L, Antonelli A, Marone G, Simon HU, Varricchi G, Galdiero MR. Neutrophil extracellular traps in cancer. Semin Cancer Biol. 2022;79:91–104. https://doi.org/10.1016/j.semcancer.2021.07.011.
McKenna E, Mhaonaigh AU, Wubben R, Dwivedi A, Hurley T, Kelly LA, Stevenson NJ, Little MA, Molloy EJ. Neutrophils: need for standardized nomenclature. Front Immunol. 2021;12: 602963. https://doi.org/10.3389/fimmu.2021.602963.
Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19–28. https://doi.org/10.1016/j.smim.2017.12.004.
Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16(10):601–20. https://doi.org/10.1038/s41571-019-0222-4.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24. https://doi.org/10.1038/s41577-019-0210-z.
Chen B, Zhu L, Yang S, Su W. Unraveling the heterogeneity and ontogeny of dendritic cells using single-cell RNA sequencing. Front Immunol. 2021;12: 711329. https://doi.org/10.3389/fimmu.2021.711329.
Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, Hogg GD, Tang JP, Baer JM, Mpoy C, Lee KB, Alexander KA, Rogers BE, Murphy KM, Hawkins WG, Fields RC, DeSelm CJ, Schwarz JK, DeNardo DG. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell. 2020;37(3):289-307.e289. https://doi.org/10.1016/j.ccell.2020.02.008.
Plesca I, Benešová I, Beer C, Sommer U, Müller L, Wehner R, Heiduk M, Aust D, Baretton G, Bachmann MP, Feldmann A, Weitz J, Seifert L, Seifert AM, Schmitz M. Clinical significance of tumor-infiltrating conventional and plasmacytoid dendritic cells in pancreatic ductal adenocarcinoma. Cancers (Basel). 2022;14(5):1216. https://doi.org/10.3390/cancers14051216.
Giri B, Sharma P, Jain T, Ferrantella A, Vaish U, Mehra S, Garg B, Iyer S, Sethi V, Malchiodi Z, Signorelli R, Jacob HKC, George J, Sahay P, Bava EP, Dawra R, Ramakrishnan S, Saluja A, Dudeja V. Hsp70 modulates immune response in pancreatic cancer through dendritic cells. Oncoimmunology. 2021;10(1):1976952. https://doi.org/10.1080/2162402x.2021.1976952.
Jing W, McAllister D, Vonderhaar EP, Palen K, Riese MJ, Gershan J, Johnson BD, Dwinell MB. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immunother Cancer. 2019;7(1):115. https://doi.org/10.1186/s40425-019-0573-5.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H. Innate lymphoid cells: 10 years on. Cell. 2018;174(5):1054–66. https://doi.org/10.1016/j.cell.2018.07.017.
Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–66. https://doi.org/10.1158/0008-5472.Can-18-3962.
Marcon F, Zuo J, Pearce H, Nicol S, Margielewska-Davies S, Farhat M, Mahon B, Middleton G, Brown R, Roberts KJ, Moss P. NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype. Oncoimmunology. 2020;9(1):1845424. https://doi.org/10.1080/2162402x.2020.1845424.
Cong H, Gao J, Wang Q, Du M, Li H, Li Q, Li J, Liang Y, Zhao D, Yang H, Gan Y, Tu H. Increased expression of mitochondrial uqcrc1 in pancreatic cancer impairs antitumor immunity of natural killer cells via elevating extracellular ATP. Front Oncol. 2022;12: 872017. https://doi.org/10.3389/fonc.2022.872017.
Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, Ramnarain A, Gasmi B, Gururajan M, Redmond D, Askan G, Bhanot U, Elyada E, Park Y, Tuveson DA, Gönen M, Leach SD, Wolchok JD, DeMatteo RP, Balachandran VP. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature. 2020;579(7797):130–5. https://doi.org/10.1038/s41586-020-2015-4.
Alam A, Levanduski E, Denz P, Villavicencio HS, Bhatta M, Alhorebi L, Zhang Y, Gomez EC, Morreale B, Senchanthisai S, Li J, Turowski SG, Sexton S, Sait SJ, Singh PK, Wang J, Maitra A, Kalinski P, DePinho RA, Dey P. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell. 2022;40(2):153-167.e111. https://doi.org/10.1016/j.ccell.2022.01.003.
Yang Y, Guo Z, Chen W, Wang X, Cao M, Han X, Zhang K, Teng B, Cao J, Wu W, Cao P, Huang C, Qiu Z. M2 Macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2. Mol Ther. 2021;29(3):1226–38. https://doi.org/10.1016/j.ymthe.2020.11.024.
Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57(6):630–6. https://doi.org/10.1136/jcp.2003.014498.
Novizio N, Belvedere R, Pessolano E, Morello S, Tosco A, Campiglia P, Filippelli A, Petrella A. ANXA1 contained in EVs regulates macrophage polarization in tumor microenvironment and promotes pancreatic cancer progression and metastasis. Int J Mol Sci. 2021;22(20):11018. https://doi.org/10.3390/ijms222011018.
Kurahara H, Takao S, Maemura K, Mataki Y, Kuwahata T, Maeda K, Sakoda M, Iino S, Ishigami S, Ueno S, Shinchi H, Natsugoe S. M2-polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pN0 pancreatic cancer. Pancreas. 2013;42(1):155–9. https://doi.org/10.1097/MPA.0b013e318254f2d1.
Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, Liu B, Su L, Qiu Z. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis. Cancer Res. 2018;78(16):4586–98. https://doi.org/10.1158/0008-5472.Can-17-3841.
Geng Y, Fan J, Chen L, Zhang C, Qu C, Qian L, Chen K, Meng Z, Chen Z, Wang P. A notch-dependent inflammatory feedback circuit between macrophages and cancer cells regulates pancreatic cancer metastasis. Cancer Res. 2021;81(1):64–76. https://doi.org/10.1158/0008-5472.Can-20-0256.
Alonso-Nocelo M, Ruiz-Cañas L, Sancho P, Görgülü K, Alcalá S, Pedrero C, Vallespinos M, López-Gil JC, Ochando M, García-García E, David Trabulo SM, Martinelli P, Sánchez-Tomero P, Sánchez-Palomo C, Gonzalez-Santamaría P, Yuste L, Wörmann SM, Kabacaoğlu D, Earl J, . . . Sainz B, Jr. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut. 2022. https://doi.org/10.1136/gutjnl-2021-325564
Chen Q, Wang J, Zhang Q, Zhang J, Lou Y, Yang J, Chen Y, Wei T, Zhang J, Fu Q, Ye M, Zhang X, Dang X, Liang T, Bai X. Tumour cell-derived debris and IgG synergistically promote metastasis of pancreatic cancer by inducing inflammation via tumour-associated macrophages. Br J Cancer. 2019;121(9):786–95. https://doi.org/10.1038/s41416-019-0595-2.
Ireland L, Luckett T, Schmid MC, Mielgo A. Blockade of stromal Gas6 alters cancer cell plasticity, activates NK cells, and inhibits pancreatic cancer metastasis. Front Immunol. 2020;11:297. https://doi.org/10.3389/fimmu.2020.00297.
Xiong C, Zhu Y, Xue M, Jiang Y, Zhong Y, Jiang L, Shi M, Chen H. Tumor-associated macrophages promote pancreatic ductal adenocarcinoma progression by inducing epithelial-to-mesenchymal transition. Aging (Albany NY). 2021;13(3):3386–404. https://doi.org/10.18632/aging.202264.
Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, Zou Y, Chen S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J Exp Clin Cancer Res. 2019;38(1):310. https://doi.org/10.1186/s13046-019-1313-x.
He Z, Wang J, Zhu C, Xu J, Chen P, Jiang X, Chen Y, Jiang J, Sun C. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. Cancer Lett. 2022;548: 215751. https://doi.org/10.1016/j.canlet.2022.215751.
Wu J, Wang Y, Yang Y, Liu F, Jiang Z, Jiang Z. TNFSF9 promotes metastasis of pancreatic cancer by regulating M2 polarization of macrophages through Src/FAK/p-Akt/IL-1β signaling. Int Immunopharmacol. 2022;102: 108429. https://doi.org/10.1016/j.intimp.2021.108429.
Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, Ireland L, Sakai T, Sakai K, Kim YS, Engle D, Campbell F, Palmer D, Ko JH, Tuveson DA, Hirsch E, Mielgo A, Schmid MC. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18(5):549–60. https://doi.org/10.1038/ncb3340.
Lee BY, Hogg EKJ, Below CR, Kononov A, Blanco-Gomez A, Heider F, Xu J, Hutton C, Zhang X, Scheidt T, Beattie K, Lamarca A, McNamara M, Valle JW, Jørgensen C. Heterocellular OSM-OSMR signalling reprograms fibroblasts to promote pancreatic cancer growth and metastasis. Nat Commun. 2021;12(1):7336. https://doi.org/10.1038/s41467-021-27607-8.
Sun X, He X, Zhang Y, Hosaka K, Andersson P, Wu J, Wu J, Jing X, Du Q, Hui X, Ding B, Guo Z, Hong A, Liu X, Wang Y, Ji Q, Beyaert R, Yang Y, Li Q, Cao Y. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut. 2022;71(1):129–47. https://doi.org/10.1136/gutjnl-2020-322744.
Bellomo G, Rainer C, Quaranta V, Astuti Y, Raymant M, Boyd E, Stafferton R, Campbell F, Ghaneh P, Halloran CM, Hammond DE, Morton JP, Palmer D, Vimalachandran D, Jones R, Mielgo A, Schmid MC. Chemotherapy-induced infiltration of neutrophils promotes pancreatic cancer metastasis via Gas6/AXL signalling axis. Gut. 2022;71(11):2284–99. https://doi.org/10.1136/gutjnl-2021-325272.
Huo Y, Zhou Y, Zheng J, Jin G, Tao L, Yao H, Zhang J, Sun Y, Liu Y, Hu LP. GJB3 promotes pancreatic cancer liver metastasis by enhancing the polarization and survival of neutrophil. Front Immunol. 2022;13: 983116. https://doi.org/10.3389/fimmu.2022.983116.
Tao L, Zhang L, Peng Y, Tao M, Li L, Xiu D, Yuan C, Ma Z, Jiang B. Neutrophils assist the metastasis of circulating tumor cells in pancreatic ductal adenocarcinoma: a new hypothesis and a new predictor for distant metastasis. Medicine (Baltimore). 2016;95(39): e4932. https://doi.org/10.1097/md.0000000000004932.
Wang X, Hu LP, Qin WT, Yang Q, Chen DY, Li Q, Zhou KX, Huang PQ, Xu CJ, Li J, Yao LL, Wang YH, Tian GA, Yang JY, Yang MW, Liu DJ, Sun YW, Jiang SH, Zhang XL, Zhang ZG. Identification of a subset of immunosuppressive P2RX1-negative neutrophils in pancreatic cancer liver metastasis. Nat Commun. 2021;12(1):174. https://doi.org/10.1038/s41467-020-20447-y.
Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D, Kang R, Lotze MT, Zeh HJ 3rd. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology. 2019;8(9): e1605822. https://doi.org/10.1080/2162402x.2019.1605822.
Deng J, Kang Y, Cheng CC, Li X, Dai B, Katz MH, Men T, Kim MP, Koay EA, Huang H, Brekken RA, Fleming JB. DDR1-induced neutrophil extracellular traps drive pancreatic cancer metastasis. JCI Insight. 2021;6(17):e146133. https://doi.org/10.1172/jci.insight.146133.
Jin W, Yin H, Li H, Yu XJ, Xu HX, Liu L. Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J Cell Mol Med. 2021;25(12):5443–56. https://doi.org/10.1111/jcmm.16555.
Takesue S, Ohuchida K, Shinkawa T, Otsubo Y, Matsumoto S, Sagara A, Yonenaga A, Ando Y, Kibe S, Nakayama H, Iwamoto C, Shindo K, Moriyama T, Nakata K, Miyasaka Y, Ohtsuka T, Toma H, Tominaga Y, Mizumoto K, Nakamura M. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int J Oncol. 2020;56(2):596–605. https://doi.org/10.3892/ijo.2019.4951.
Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, Mukherjee P, Wang-Gillam A, Link DC, Denardo DG, Goedegebuure SP, Linehan DC. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63(5):513–28. https://doi.org/10.1007/s00262-014-1527-x.
Kenkel JA, Tseng WW, Davidson MG, Tolentino LL, Choi O, Bhattacharya N, Seeley ES, Winer DA, Reticker-Flynn NE, Engleman EG. An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res. 2017;77(15):4158–70. https://doi.org/10.1158/0008-5472.Can-16-2212.
Sun H, Shi K, Qi K, Kong H, Zhang J, Dai S, Ye W, Deng T, He Q, Zhou M. Natural killer cell-derived exosomal miR-3607-3p inhibits pancreatic cancer progression by targeting IL-26. Front Immunol. 2019;10:2819. https://doi.org/10.3389/fimmu.2019.02819.
Yako YY, Kruger D, Smith M, Brand M. Cytokines as biomarkers of pancreatic ductal adenocarcinoma: a systematic review. PLoS One. 2016;11(5): e0154016. https://doi.org/10.1371/journal.pone.0154016.
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–37. https://doi.org/10.1016/j.ccell.2014.09.006.
Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, Serbulea V, Leitinger N, Ailawadi G, Upchurch GR Jr. Novel role of IL (Interleukin)-1β in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2018;38(4):843–53. https://doi.org/10.1161/atvbaha.117.309897.
Padoan A, Plebani M, Basso D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int J Mol Sci. 2019;20(3):676. https://doi.org/10.3390/ijms20030676.
David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines (Basel). 2016;4(3):22. https://doi.org/10.3390/vaccines4030022.
Frossard JL, Lenglet S, Montecucco F, Steffens S, Galan K, Pelli G, Spahr L, Mach F, Hadengue A. Role of CCL-2, CCR-2 and CCR-4 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. J Clin Pathol. 2011;64(5):387–93. https://doi.org/10.1136/jcp.2010.088500.
Kemp SB, Carpenter ES, Steele NG, Donahue KL, Nwosu ZC, Pacheco A, Velez-Delgado A, Menjivar RE, Lima F, The S, Espinoza CE, Brown K, Long D, Lyssiotis CA, Rao A, Zhang Y, Pasca di Magliano M, Crawford HC. Apolipoprotein E promotes immune suppression in pancreatic cancer through NF-κB-Mediated production of CXCL1. Cancer Res. 2021;81(16):4305–18. https://doi.org/10.1158/0008-5472.Can-20-3929.
Duluc D, Corvaisier M, Blanchard S, Catala L, Descamps P, Gamelin E, Ponsoda S, Delneste Y, Hebbar M, Jeannin P. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. 2009;125(2):367–73. https://doi.org/10.1002/ijc.24401.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. https://doi.org/10.1126/science.1198443.
O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, Fisher G, Rahma O, Lyman JP, Cabanski CR, Mick R, Gherardini PF, Kitch LJ, Xu J, Samuel T, Karakunnel J, Fairchild J, Bucktrout S, LaVallee TM, Vonderheide RH. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021;22(1):118–31. https://doi.org/10.1016/s1470-2045(20)30532-5.
Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA, Chiorean EG, O’Hara MH, Liudahl SM, Newcomb C, Alanio C, Ferreira AP, Park BS, Ohtani T, Huffman AP, Väyrynen SA, Dias Costa A, Kaiser JC, Lacroix AM, Vonderheide RH. Neoadjuvant selicrelumab, an agonist CD40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin Cancer Res. 2021;27(16):4574–86. https://doi.org/10.1158/1078-0432.Ccr-21-1047.
Li M, Li M, Yang Y, Liu Y, Xie H, Yu Q, Tian L, Tang X, Ren K, Li J, Zhang Z, He Q. Remodeling tumor immune microenvironment via targeted blockade of PI3K-γ and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J Control Release. 2020;321:23–35. https://doi.org/10.1016/j.jconrel.2020.02.011.
Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69. https://doi.org/10.1158/0008-5472.Can-13-3723.
Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, Colevas AD, O’Rourke T, Narayanan S, Papadopoulos K, Fisher GA, Villalobos V, Prohaska SS, Howard M, Beeram M, Chao MP, Agoram B, Chen JY, Huang J, Padda SK. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37(12):946–53. https://doi.org/10.1200/jco.18.02018.
Roohullah A, Ganju V, Zhang F, Zhang L, Yu T, Wilkinson K, Cooper A, Souza PD. First-in-human phase 1 dose escalation study of HX009, a novel recombinant humanized anti-PD-1 and CD47 bispecific antibody, in patients with advanced malignancies. J Clin Oncol. 2021;39(15):2517–2517. https://doi.org/10.1200/JCO.2021.39.15_suppl.2517.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, Fowler KJ, Lockhart AC, Suresh R, Tan BR, Lim KH, Fields RC, Strasberg SM, Hawkins WG, DeNardo DG, Linehan DC. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62. https://doi.org/10.1016/s1470-2045(16)00078-4.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang-Gillam A, Goedegebuure P, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41. https://doi.org/10.1158/0008-5472.Can-12-2731.
Céspedes MV, Guillén MJ, López-Casas PP, Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM, Allavena P, Avilés P, Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech. 2016;9(12):1461–71. https://doi.org/10.1242/dmm.026369.
Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, Fields RC, DeNardo DG, Hawkins WG, Goedegebuure P, Linehan DC. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112–23. https://doi.org/10.1136/gutjnl-2017-313738.
Nielsen SR, Strøbech JE, Horton ER, Jackstadt R, Laitala A, Bravo MC, Maltese G, Jensen ARD, Reuten R, Rafaeva M, Karim SA, Hwang CI, Arnes L, Tuveson DA, Sansom OJ, Morton JP, Erler JT. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat Commun. 2021;12(1):3414. https://doi.org/10.1038/s41467-021-23731-7.
Zeh HJ, Bahary N, Boone BA, Singhi AD, Miller-Ocuin JL, Normolle DP, Zureikat AH, Hogg ME, Bartlett DL, Lee KK, Tsung A, Marsh JW, Murthy P, Tang D, Seiser N, Amaravadi RK, Espina V, Liotta L, Lotze MT. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin Cancer Res. 2020;26(13):3126–34. https://doi.org/10.1158/1078-0432.Ccr-19-4042.
Karasic TB, O’Hara MH, Loaiza-Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, De Jesus-Acosta A, Redlinger C, Burrell JA, Laheru DA, Von Hoff DD, Amaravadi RK, Drebin JA, O’Dwyer PJ. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5(7):993–8. https://doi.org/10.1001/jamaoncol.2019.0684.
Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, Chung CW, Craggs PD, Davis RP, Eberhard D, Joberty G, Lind KE, Locke K, Maller C, Martinod K, Wilson DM. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11(3):189–91. https://doi.org/10.1038/nchembio.1735.
Velasco-Velázquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, Pestell RG. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012;72(15):3839–50. https://doi.org/10.1158/0008-5472.Can-11-3917.
Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, Breden MA, Li X, Krisnawan VE, Khan SQ, Schwarz JK, Rogers BE, Fields RC, Hawkins WG, Gupta V, DeNardo DG. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med. 2019;11(499):eaau9240. https://doi.org/10.1126/scitranslmed.aau9240.
Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, Eberlein C, Candido JB, Clarke M, Nixon C, Connelly J, Jamieson N, Carter CR, Balkwill F, Chang DK, Morton JP. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29(6):832–45. https://doi.org/10.1016/j.ccell.2016.04.014.
Gulhati P, Schalck A, Jiang S, Shang X, Wu CJ, Hou P, Ruiz SH, Soto LS, Parra E, Ying H, Han J, Dey P, Li J, Deng P, Sei E, Maeda DY, Zebala JA, Spring DJ, Kim M, DePinho RA. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat Cancer. 2023;4(1):62–80. https://doi.org/10.1038/s43018-022-00500-z.
Lau SP, Klaase L, Vink M, Dumas J, Bezemer K, van Krimpen A, van der Breggen R, Wismans LV, Doukas M, de Koning W, Stubbs AP, Mustafa DAM, Vroman H, Stadhouders R, Nunes JB, Stingl C, de Miranda N, Luider TM, van der Burg SH, van Eijck CHJ. Autologous dendritic cells pulsed with allogeneic tumour cell lysate induce tumour-reactive T-cell responses in patients with pancreatic cancer: a phase I study. Eur J Cancer. 2022;169:20–31. https://doi.org/10.1016/j.ejca.2022.03.015.
Lau SP, van’t Land FR, van der Burg SH, Homs MYV, Lolkema MP, Aerts J, van Eijck CHJ. Safety and tumour-specific immunological responses of combined dendritic cell vaccination and anti-CD40 agonistic antibody treatment for patients with metastatic pancreatic cancer: protocol for a phase I, open-label, single-arm, dose-escalation study (REACtiVe-2 trial). BMJ Open. 2022;12(6):e060431. https://doi.org/10.1136/bmjopen-2021-060431.
Sun C, Nagaoka K, Kobayashi Y, Nakagawa H, Kakimi K, Nakajima J. Neoantigen dendritic cell vaccination combined with anti-CD38 and CpG elicits anti-tumor immunity against the immune checkpoint therapy-resistant murine lung cancer cell line LLC1. Cancers (Basel). 2021;13(21):5508. https://doi.org/10.3390/cancers13215508.
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Merad M. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44(4):924–38. https://doi.org/10.1016/j.immuni.2016.03.012.
Da Y, Liu Y, Hu Y, Liu W, Ma J, Lu N, Zhang C, Zhang C. STING agonist cGAMP enhances anti-tumor activity of CAR-NK cells against pancreatic cancer. Oncoimmunology. 2022;11(1):2054105. https://doi.org/10.1080/2162402x.2022.2054105.
Xia N, Haopeng P, Gong JU, Lu J, Chen Z, Zheng Y, Wang Z, Sun YU, Yang Z, Hoffman RM, Liu F. Robo1-specific CAR-NK immunotherapy enhances efficacy of (125)I seed brachytherapy in an orthotopic mouse model of human pancreatic carcinoma. Anticancer Res. 2019;39(11):5919–25. https://doi.org/10.21873/anticanres.13796.
Lin M, Alnaggar M, Liang S, Wang X, Liang Y, Zhang M, Chen J, Niu L, Xu K. An important discovery on combination of irreversible electroporation and allogeneic natural killer cell immunotherapy for unresectable pancreatic cancer. Oncotarget. 2017;8(60):101795–807. https://doi.org/10.18632/oncotarget.21974.
Nianias A, Themeli M. Induced pluripotent stem cell (iPSC)-derived lymphocytes for adoptive cell immunotherapy: recent advances and challenges. Curr Hematol Malig Rep. 2019;14(4):261–8. https://doi.org/10.1007/s11899-019-00528-6.
Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, Ben-Josef E, Beatty GL. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2017;23(1):137–48. https://doi.org/10.1158/1078-0432.Ccr-16-0870.
Korbecki J, Kojder K, Simińska D, Bohatyrewicz R, Gutowska I, Chlubek D, Baranowska-Bosiacka I. CC Chemokines in a tumor: a review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int J Mol Sci. 2020;21(21):8412. https://doi.org/10.3390/ijms21218412.
Jiang Z, Sun H, Yu J, Tian W, Song Y. Targeting CD47 for cancer immunotherapy. J Hematol Oncol. 2021;14(1):180. https://doi.org/10.1186/s13045-021-01197-w.
Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, Dang Y, Hou Z, Lin W, Lin X, Zhang Z, Pan M, Huang H. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol. 2019;12(1):124. https://doi.org/10.1186/s13045-019-0822-6.
Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T, Hagag E, Sinha A, Has C, Dietz S, de Jesus Domingues AM, Nati M, Sormendi S, Neuwirth A, Chatzigeorgiou A, Ziogas A, Lesche M, Dahl A, Henry I, Chavakis T. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell. 2020;183(3):771-785.e712. https://doi.org/10.1016/j.cell.2020.09.058.
De Nardo DG, Galkin A, Dupont J, Zhou L, Bendell J. GB1275, a first-in-class CD11b modulator: rationale for immunotherapeutic combinations in solid tumors. J Immunother Cancer. 2021;9(8):e003005. https://doi.org/10.1136/jitc-2021-003005.
Cueto FJ, Sancho D. The Flt3L/Flt3 axis in dendritic cell biology and cancer immunotherapy. Cancers (Basel). 2021;13(7):1525. https://doi.org/10.3390/cancers13071525.
Chen K, Braun S, Lyman S, Fan Y, Traycoff CM, Wiebke EA, Gaddy J, Sledge G, Broxmeyer HE, Cornetta K. Antitumor activity and immunotherapeutic properties of Flt3-ligand in a murine breast cancer model. Cancer Res. 1997;57(16):3511–6.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59: 102975. https://doi.org/10.1016/j.ebiom.2020.102975.
Lee YE, Ju A, Choi HW, Kim JC, Kim EE, Kim TS, Kang HJ, Kim SY, Jang JY, Ku JL, Kim SC, Jun E, Jang M. Rationally designed redirection of natural killer cells anchoring a cytotoxic ligand for pancreatic cancer treatment. J Control Release. 2020;326:310–23. https://doi.org/10.1016/j.jconrel.2020.07.016.
Zhang S, Fang W, Zhou S, Zhu D, Chen R, Gao X, Li Z, Fu Y, Zhang Y, Yang F, Zhao J, Wu H, Wang P, Shen Y, Shen S, Xu G, Wang L, Yan C, Zou X, Lv Y. Single cell transcriptomic analyses implicate an immunosuppressive tumor microenvironment in pancreatic cancer liver metastasis. Nat Commun. 2023;14(1):5123. https://doi.org/10.1038/s41467-023-40727-7.
Acknowledgements
Not applicable.
Funding
This work was supported by project grants from The National Natural Science Foundation of China (82271883, 82073195), the Hospital-pharma Integration Project on Innovative Achievement Translation (SHDC2022CRD043), the Special Funds for Technological Innovation of Shanghai Baoshan Science and Technology Commission (No.20-E-32), the Clinical Research Innovation Cultivation Fund of Baoshan Branch of Renji Hospital, Shanghai Jiaotong University School of Medicine (2019-rbcxjj-001), and the Shanghai Municipal Education Commission Gaofeng Clinical Medicine Grant Support.
Author information
Authors and Affiliations
Contributions
Qing Xia provided direction and guidance throughout the preparation of this manuscript. Tianyi Zhu and Xiuqi Wu wrote the main manuscript text. Yuan Liao and Yidan Yan collected the relevant papers and designed Figs. 1, 2, 3 and 4. Minhao Yu, Liwei Wang, and Qing Xia reviewed and significantly revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, T., Wu, X., Liao, Y. et al. The role of innate immune cells as modulators of the tumor microenvironment in the metastasis and treatment of pancreatic cancer. CCB 2, 2 (2023). https://doi.org/10.1007/s44272-023-00005-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44272-023-00005-5