
Abstract
Droughts threaten crop yields worldwide. Compared to other major staple cereal crops, maize (Zea mays) is especially sensitive to drought, which can cause dramatic fluctuations in its yield potential. Natural maize populations contain many superior alleles that can enhance drought resistance through complex regulatory mechanisms. We recently de novo assembled the genome of a prominent drought-resistant maize germplasm, CIMBL55, and systematically dissected the genetic basis for its drought resistance on the genome, transcriptome, and epigenome levels. These analyses revealed 65 favorable drought resistance alleles in CIMBL55. Subsequently, we genetically verified the functions of the drought resistance genes ZmABF4, ZmNAC075, and ZmRtn16 and unraveled the function of ZmRtn16 on a molecular level.
Similar content being viewed by others
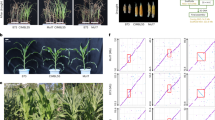

This article highlights our recent study (Tian et al. 2023), in which we assembled the genome of a prominent drought-resistant maize (Zea mays) germplasm, CIMBL55, and identified several superior drought resistance alleles. Crops are subjected to numerous environmental stresses throughout their life cycle (Hirt et al. 2023). Maize, as one of the most widely cultivated crops worldwide, suffers severe or even total production losses due to drought stress. Maize originated in Mexico ~ 9,000 years ago, and as new varieties were selected, its cultivation region expanded from low to high latitudes (Liang et al. 2021). Interestingly, varieties with a tropical or subtropical pedigree tend to have higher drought resistance than varieties from temperate areas. Drought resistance is a quantitative trait with a complex genetic basis. Population genetics studies have helped identify superior drought resistance alleles and uncover the molecular mechanisms of drought resistance, contributing to the breeding of drought-resistant maize varieties with enhanced yield potential. A genome-wide association study of maize seedling survival rate after drought treatment, using 368 lines of an association panel, identified 83 genetic variants surrounding 42 candidate genes that explained approximately 55.2% of the phenotypic variation (Wang et al. 2016). In a subsequent study, expression quantitative trait locus (eQTL) analysis of seedlings exposed to well-watered, moderate drought (70% relative leaf water content), and severe drought (58% relative leaf water content) conditions identified 19,566 (26.6%) static and 54,007 (73.4%) dynamic eQTLs, encompassing 97 genes associated with drought resistance (Liu et al. 2020). Later studies identified superior alleles of several candidate genes, such as ZmVPP1, ZmNAC111, ZmTIP1, ZmSRO1, and ZmABH2, in the maize inbred line CIMBL55, which directly or indirectly function in the regulation of water uptake and water loss to maintain viability under water deficit (Rodrigues et al. 2019; Yang and Qin 2023). However, many other drought resistance loci remain to be characterized in maize.
Third-generation sequencing technologies have produced high-quality genome assemblies for maize and other plants with complex genomes. The 5th edition of the maize reference genome, B73 has fewer gaps than previous versions, and has a contig N50 greater than 50 Mb (Hufford et al. 2021). Since the release of the B73 reference genome, dozens of additional genome assemblies have been released, including Mo17 (Sun et al. 2018), small kernel (SK) (Yang et al. 2019), A188 (Lin et al. 2021), and K0326Y (Li et al. 2020), along with 26 nested association population founders (Hufford et al. 2021), 7 wild relatives of maize (Chen et al. 2022), and 12 maize germplasms from different heterotic groups (Wang et al. 2023). These assemblies have enhanced genomics-based breeding and the analysis of adaptive evolution. For example, the genome for maize inbred line SK, which has small kernels and low productivity, was assembled to analyze yield traits. An 8.9-kb insertion upstream of ZmBAM1d was identified in the B73 genome that promotes its expression and increases hundred-kernel weight, possibly due to altered chromatin interactions and methylation levels (Yang et al. 2019). Furthermore, the assembly A188 revealed that a copy number variation in carotenoid cleavage dioxygenase 1 (ccd1) between A188 and B73 alters carotenoid accumulation and results in different seed colors (Lin et al. 2021). Finally, Huang et al. used a high-quality teosinte haplotype assembly to identify a superior allele of TEOSINTE HIGH PROTEIN 9 (THP9) in the wild ancestor of maize that confers increased seed protein content (Huang et al. 2022).
These high-quality genomes counteract the bias inherent in using a single reference genome and provide an extensive allele resource repository. However, these genomes do not include any germplasm noted for drought resistance. The maize inbred line CIMBL55 has a tropical and subtropical pedigree and is a well-known drought-resistant germplasm (Wang et al. 2016). Using 160 × PacBio single-molecule real-time sequencing, 160 × BioNano single-molecule optical mapping, and 35 × Hi-C sequencing technologies, along with state-of-the-art assembly strategies and annotation pipelines, we generated a contiguous CIMBL55 genome assembly containing 38,439 protein-coding genes and 83.95% repeats (Tian et al. 2023). We identified large structural variations between CIMBL55 and 30 other high-quality assembled maize genomes, including the reference genome B73; these variations may contribute to the drought resistance of this unique germplasm.
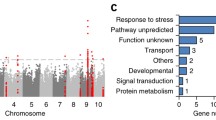
We analyzed gene synteny, structural variations, and epigenetic differences. First, we compared the gene synteny maps of CIMBL55 and B73 or Mo17 and found differences in gene order and gene presence/absence. Genes within syntenic blocks on homologous chromosomes tended to share a common order, while non-syntenic regions had undergone massive changes, such as chromosome rearrangements and transposon hopping (Wang et al. 2012). We found a special class of genes named Class2 (29% of total maize genes), which are syntenic as well as duplicated and include a higher percentage of abscisic acid signaling and stress response genes compared to other gene classes. This phenomenon is consistent with the trend of evolutionary conservation of key signaling factors and hub genes (Madan Babu and Teichmann 2003). Next, we used four state-of-the-art strategies, including assembled contig-based (MUMmer + SyRi and blasr + smartie-sv) and clean read-based (pbmm + pbsv and ngmlr + sniffle) strategies, to identify DNA sequence variations, including single nucleotide variations, small insertions/deletions (indels), large insertions, large deletions, inversions, translocations, and duplications between CIMBL55 and the 30 other maize genome assemblies. We obtained some overlaps from different strategies and programs, highlighting the importance of combining multi-layer algorithms to identify genomic variation in complex genomes. In total, we identified 127,742,577 single nucleotide polymorphisms (SNPs), 14,499,458 indels, and 3,081,556 structural variants (SVs) on a pan-genome level.
Next, we developed a bi-directional SV identification strategy to accurately identify SVs between two genomes, which is especially important for predicting their potential regulatory roles. Insertion variations within the flanking region of genes may introduce or disrupt cis-acting elements or change the chromatin status. Furthermore, a translocation within a coding region may abolish gene function. To investigate epigenomic differences in conserved and variable regions, we conducted whole-genome bisulfite sequencing of three maize inbred lines (CIMBL55, B73, and Mo17). In conserved regions, we found many regions that were hypomethylated (11,019) and hypermethylated (12,066) in CIMBL55 compared to B73 or Mo17 and identified 7,170 genes near those differentially methylated regions. However, these genes showed no significant differences in gene expression or DNA methylation among the three inbred lines. Interestingly, we found that DNA methylation variations were more common in variable regions than in conserved regions. About 80% of inserted sequences were located in regions containing at least one type of DNA methylation. We found that clusters of inserted sequences with higher CHH (where “H” is A, C, or T) methylation levels were enriched in DNA transposon of Harbinger (DTH), which may impact the expression of proximal genes. Some members of the NAC (NAM, ATAF, and CUC) family of transcription factors function as positive regulatory factors of drought resistance, and their overexpression leads to increased yield under water deficit conditions (Al Abdallat et al. 2014). Two insertional sequences upstream of ZmNAC075, S-9041 (182 bp) and S-1425 (5,123 bp), were hypermethylated in B73, but not in CIMBL55, significantly reducing ZmNAC075 expression in B73. Furthermore, when the CHH methylation level at these two insertional sequences in B73 was diminished by knocking out Zmdrd1, which functions in RNA-directed de novo DNA methylation, the expression of ZmNAC075 was enhanced. These results suggested that hypermethylation of the two inserted sequences in B73 inhibits ZmNAC075 expression and compromises drought resistance.
We identified 208,036 B73 reference-based and 336,817 CIMBL55 reference-based SVs and genotyped them in an association panel consisting of 368 inbred lines. Analysis of the association of these newly identified SVs with drought resistance provided a comprehensive and accurate genetic dissection for the trait. Several newly identified SVs showed strong linkage disequilibrium with SNPs previously significantly associated with drought resistance, suggesting that low-density SNPs can be used to pinpoint the potential causative region, and high-density variations, especially SVs, are beneficial to unravel the causal variants for gene expression regulation. For example, Zhang et al. established that ZmRtn16 encodes a reticulon-like protein that is associated with the endoplasmic reticulum (ER) and functions in protein trafficking from the ER to the Golgi apparatus as part of autophagic flux in endosperm aleurone cells during seed germination (Zhang et al. 2020). In this research, ZmRtn16 interacts with the A and E3 subunits of the tonoplast proton pump (the vacuolar H+-ATPase) and plays an essential role in directing the two subunits to the tonoplast. Loss of function of ZmRtn16 resulted in reduced vacuolar H+-ATPase activity and compromised drought resistance. Our study identified a deletion, S-2290 (28 bp), in the 3′ untranslated region of ZmRtn16CIMBL55 compared with ZmRtn16B73; this deletion strongly enhanced its expression and is in strong linkage disequilibrium with a previously identified SNP associated with drought resistance. Removal of this 28-bp fragment from the ZmRtn16B73 allele dramatically increased its transcript abundance to a level similar to that of the ZmRtn16CIMBL55 allele. An RNA binding motif within the 28-bp fragment was predicted based on a search of the online CISBP-RNA (Catalog of Inferred Sequence Binding Proteins of RNA) database (http://cisbp-rna.ccbr.utoronto.ca), which contains a collection of experimentally identified and inferred RNA binding motifs. The increased expression of ZmRtn16 CIMBL55 relative to ZmRtn16B73 may be mediated by RNA binding proteins at the post-transcriptional level.
High-quality genome assemblies enable accurate and efficient identification of key components of genomes, such as exact gene-coding sequences, transcriptional regulatory elements, transcript isoforms, non-coding RNAs, and transposon structures. Efficient and accurate identification of complex structural variations and prediction of their associations or contributions to interesting traits demand further innovations in algorithms and their integration with deep neural network frameworks of artificial intelligence. Furthermore, high-throughput phenotyping technology for temporal and spatial developmental data are necessary for mining superior drought resistance alleles from the huge genetic resource of maize. Finally, exploiting these drought resistance alleles via gene editing will help to develop robust crops that are more resilient to the stresses caused by global climate change.
Fundings
This research was supported by Beijing Outstanding Young Scientist Program (BJJWZYJH01201910019026) and Chinese Postdoctoral Science Foundation (2019M660874, 2021T140714).
Availability of data and material
Not applicable.
References
Al Abdallat AM, Ayad JY, Abu Elenein JM, Al Ajlouni Z, Harwood WA (2014) Overexpression of the transcription factor HvSNAC1 improves drought tolerance in barley (Hordeum vulgare L.). Mol Breeding 33:401–414. https://doi.org/10.1007/s11032-013-9958-1
Chen L, Luo J, Jin M, Yang N, Liu X, Peng Y, Li W, Phillips A, Cameron B, Bernal JS et al (2022) Genome sequencing reveals evidence of adaptive variation in the genus Zea. Nat Gen 54:1736. https://doi.org/10.1038/s41588-022-01184-y
Hirt H, Al-Babili S, Almeida-Trapp M, Antoine M, Aranda M, Bartels D, Bennett M, Blilou I, Boer D, Boulouis A et al (2023) PlantACT! - how to tackle the climate crisis. Trends Plant Sci 28:537. https://doi.org/10.1016/j.tplants.2023.01.005
Huang Y, Wang H, Zhu Y, Huang X, Li S, Wu X, Zhao Y, Bao Z, Qin L, Jin Y et al (2022) THP9 enhances seed protein content and nitrogen-use efficiency in maize. Nature 2022(612):292. https://doi.org/10.1038/s41586-022-05441-2
Hufford MB, Seetharam AS, Woodhouse MR, Chougule KM, Ou S, Liu J, Ricci WA, Guo T, Olson A, Qiu Y et al (2021) De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 373:655–662. https://doi.org/10.1126/science.abg5289
Li C, Xiang X, Huang Y, Zhou Y, An D, Dong J, Zhao C, Liu H, Li Y, Wang Q et al (2020) Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nat Commun 11:17. https://doi.org/10.1038/s41467-019-14023-2
Liang Y, Liu HJ, Yan J, Tian F (2021) Natural variation in crops: realized understanding, continuing promise. Annu Rev Plant Biol 72:357–385. https://doi.org/10.1146/annurev-arplant-080720-090632
Lin G, He C, Zheng J, Koo DH, Le H, Zheng H, Tamang TM, Lin J, Liu Y, Zhao M et al (2021) Chromosome-level genome assembly of a regenerable maize inbred line A188. Genome Biol 22:175. https://doi.org/10.1186/s13059-021-02396-x
Liu S, Li C, Wang H, Wang S, Yang S, Liu X, Yan J, Li B, Beatty M, Zastrow-Hayes G et al (2020) Mapping regulatory variants controlling gene expression in drought response and tolerance in maize. Genome Biol 21:163. https://doi.org/10.1186/s13059-020-02069-1
Madan Babu M, Teichmann SA (2003) Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res 31:1234–1244. https://doi.org/10.1093/nar/gkg210
Rodrigues J, Inze D, Nelissen H, Saibo NJM (2019) Source-sink regulation in crops under water deficit. Trends Plant Sci 24:652–663. https://doi.org/10.1016/j.tplants.2019.04.005
Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, Song W, Zhang M, Cui Y, Dong X et al (2018) Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat Genet 50:1289–1295. https://doi.org/10.1038/s41588-018-0182-0
Tian T, Wang S, Yang S, Yang Z, Liu S, Wang Y, Gao H, Zhang S, Yang X, Jiang C et al (2023) Genome assembly and genetic dissection of a prominent drought-resistant maize germplasm. Nat Gen 55:496. https://doi.org/10.1038/s41588-023-01297-y
Wang B, Hou M, Shi J, Ku L, Song W, Li C, Ning Q, Li X, Li C, Zhao B et al (2023) De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat Genet 55:312–323. https://doi.org/10.1038/s41588-022-01283-w
Wang X, Wang H, Liu S, Ferjani A, Li J, Yan J, Yang X, Qin F (2016) Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet 48:1233–1241. https://doi.org/10.1038/ng.3636
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H et al (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40:e49. https://doi.org/10.1093/nar/gkr1293
Yang N, Liu J, Gao Q, Gui S, Chen L, Yang L, Huang J, Deng T, Luo J, He L et al (2019) Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet 51:1052–1059. https://doi.org/10.1038/s41588-019-0427-6
Yang Z, Qin F (2023) The battle of crops against drought: Genetic dissection and improvement. J Integr Plant Biol 65:496–525. https://doi.org/10.1111/jipb.13451
Zhang X, Ding X, Marshall RS, Paez-Valencia J, Lacey P, Vierstra RD, Otegui MS (2020) Reticulon proteins modulate autophagy of the endoplasmic reticulum in maize endosperm. Elife 9:e51918. https://doi.org/10.7554/elife.51918
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
Contributions
T.T. & F.Q. wrote the paper. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Handling editor: Huazhong Shi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, T., Qin, F. CIMBL55: a repository for maize drought resistance alleles. Stress Biology 3, 13 (2023). https://doi.org/10.1007/s44154-023-00091-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44154-023-00091-4