
Abstract
To maintain, develop and rationally utilize marine organisms, understanding their genetic structure and habitat adaptation pattern is necessary. Konosirus punctatus, which is a commercial fish species inhabiting the Indo-west Pacific Ocean, has shown an obvious annual global capture and aquaculture production decline due to climate changes and human activities. In the present study, restriction-site associated DNA sequencing (RAD-seq) was used to describe its genome-wide single nucleotide polymorphisms panel (SNPs). Among 146 individuals collected at nine locations scattered in China, Korea and Japan, a set of 632,090 SNPs were identified. Population genetic analysis showed that K. punctatus individuals were divided into two significant genetic clusters. Meanwhile, potential genetic differentiation between northern and southern population of K. punctatus was found. Treemix results indicated that gene flow existed among sampling locations of K. punctatus, especially from southern Japan to others. Moreover, candidate genes associated with habitat adaptations of K. punctatus were identified, which are involved in diverse physiological processes of K. punctatus including growth and development (e.g., KIDINS220, PAN3), substance metabolism (e.g., PGM5) and immune response (e.g., VAV3, CCT7, HSPA12B). Our findings may aid in understanding the possible mechanisms for the population genetic structure and local adaptation of K. punctatus, which is beneficial to establish the management and conservation units of K. punctatus, guiding the rational use of resources, with reference significance for a profound understanding of the adaptative mechanisms of other marine organisms to the environment.
Similar content being viewed by others

Introduction
Population genetic studies of marine organisms are helpful to understand their distribution, which is of special importance to managing and protecting marine population resources scientifically. Marine fish populations are rarely segregated, especially in large populations, as water mobility reduces barriers to genetic exchange between populations (Cano et al. 2008; Conover et al. 2006; Shanks et al. 2003). However, the complex geographic dynamics and hydrographic systems of the oceans have also influenced the isolation or dispersal of various marine populations, ultimately altering the genetic structure (Fernández et al. 2011; Hellberg et al. 2002). As environmental factors in marine habitats become more heterogeneous over time, the role of natural selection will shift, ultimately promoting adaptive habitat differentiation among marine populations that exhibit significant genetic differences (Nielsen et al. 2009). Moreover, climate warming and habitat destruction have now affected the genetic diversity of some small and isolated marine populations, which are more vulnerable to genetic drift and inbreeding (Ouborg et al. 2010). In short, understanding the level of genetic diversity and the mechanisms of genetic differentiation in marine organisms may contribute to the conservation and management of marine organism resources. Furthermore, it is necessary to clarify the adaptive evolution potential of different populations of marine organisms for their habitat adaptation.
Konosirus punctatus (Temminck and Schlegel 1846), known as the dotted gizzard shad or spotted sardine, is a suitable subject for molecular phylogeographic studies. K. punctatus belongs to the herring family Clupeidae (Zhang 2001). It is not only a commercial species, but is also taken as a by-catch, and is widely distributed along the coasts of China, Japan, and Korea (Liu et al. 2020). As a marine fish, K. punctatus can withstand freshwater conditions (Lee 1983; Myoung and Kim 2014; Song et al. 2016). The high energy conversion efficiency and decent ecological conversion efficiency make it greatly adaptable to environmental changes, ensuring the stability of the K. punctatus population, which plays an essential role in marine ecosystems (Yao et al. 1999). However, the annual global catch of K. punctatus is declining, indicating that there are some threats to its existence, particularly the degradation of estuarine ecosystems (Liu et al. 2020). Therefore, management and recovery plans are needed to prevent the collapse of K. punctatus populations.
With the recognition of the importance of K. punctatus, there has been a growing number of molecular genetic studies of its population structure. K. punctatus has a long pelagic larval duration (24–28 days) which may give K. punctatus a potential dispersal capacity and enhance the gene flow among different populations (Kong et al. 2004; Okamura and Yamada 1986). However, adult K. punctatus generally have a fixed migratory pattern, migrating annually only between spawning grounds along the shores of shallower waters and overwintering grounds in deeper waters, which may lead to the different genetic structures in different sea waters (Gwak et al. 2015; Liu et al. 2020; Myoung and Kim 2014; Ying 2011). For example, Myoung and Kim (2014) demonstrated that Korean populations are highly differentiated even in adjacent waters while Liu et al. (2020) found low genetic differentiation among the Chinese K. punctatus populations. These different structures may be caused by multiple factors, such as genetic differentiation and adaptation (Li et al. 2012). In addition, the resolution and number of markers are crucial for exploring genetic structure. For example, Ying (2011) detected the population structure and phylogeographic pattern of K. punctatus using four different markers [morphology, mitochondrial DNA (mtDNA) markers, amplified fragment length polymorphisms (AFLPs), and inter-simple sequence repeat (ISSR)]. These workers found morphology is less sensitive compared to the other three markers when detecting the degree of differentiation between the Japanese and Chinese populations. Meanwhile, different levels of differentiation within Chinese and Japanese populations were detected using mtDNA markers, AFLPs, and ISSR, respectively (Ying 2011). Most current markers have insufficient precision in detecting the population genetic structure of K. punctatus and limitations in reflecting selection or environmental influences due to quantitative limitations (Billington and Hebert 1991; Campbell et al. 2003). The advantage of restriction-site associated DNA sequencing (RAD-seq) technology is that it can efficiently detect genome-wide single-nucleotide polymorphisms (SNPs) even at low sequencing depth, which ultimately provides an efficient and accurate solution for studying the genetic structure of a species, even those with low genetic differentiation (Zhang et al. 2019). Simple library construction, short duration of the experiment, and well-developed data processing and analysis pipelines have made the RAD-seq technology popular in SNP identification and population genetics studies (Eaton and Ree 2013; McCormack et al. 2013). Furthermore, RAD-seq technology has been successfully applied to detect SNPs in the whole genome of numerous marine species, such as Mugil cephalus (Krück et al. 2013), Carcinus maenas (Jeffery et al. 2017) and Setipinna tenuifilis (Peng et al. 2021), to provide more reliable evidence for studying their genetic structure and adaptive evolution (Miller et al. 2007). However, no population genetics study has been conducted to detect genome-wide SNPs of K. punctatus based on RAD-seq. Therefore, a comprehensive population genetic analysis of K. punctatus based on SNPs via RAD-seq is needed.
In the present study, individuals of K. punctatus were collected from nine sampling locations along the coasts of China, Japan, and Korea. RAD-seq was first used to identify genome-wide SNPs in K. punctatus. Population genomic methods have been used to identify genetic variation and explore the adaptive evolution of K. punctatus populations. Finally, the results of this study will improve our understanding of the population structure and habitat-adaptive differentiation of K. punctatus.
Materials and methods
Collection and RAD-seq of K. punctatus samples
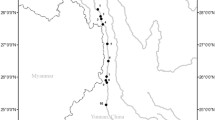
We collected samples of 146 K. punctatus from nine locations, including Qinhuangdao (QHD), Dandong (DD), Shouguang (SG), Lianyungang (LYG), Jeolla-Do (HQ), Saga (RZH), Nantong (NT), Zhoushan (ZS), and Wenzhou (YQ) along the coasts of China, Japan, and Korea (Fig. 1; Supplementary Table S1). Muscle tissue from each fish was preserved in 95% ethanol for DNA extraction. Total genomic DNA was extracted using a standard procedure (Dhaliwal 2013). DNA completeness and concentration were checked by gel electrophoresis and spectrophotometry. The standard for high-quality extracted DNA was that the range of OD260/280 was between 1.6 and 1.8. At the same time, the electrophoretic bands are well-defined and there is no significant dispersion.

Map of the studied area depicting sampled populations and currents. 1 Kuroshio Current, 2 Yellow Sea Warm Current, 3 Yellow Sea Cold Water Mass, 4 Bohai Circulation, 5 China Coastal Current, 6 South China Sea Warm Current. Chinese populations: Qinhuangdao (QHD), Dandong (DD), Shouguang (SG), Lianyungang (LYG), Nantong (NT), Zhoushan (ZS), and Wenzhou (YQ). Korean population: Jeolla-Do (HQ). Japanese population: Saga (RZH)
After that, high-quality genomic DNA was used to construct the RAD library (Etter et al. 2011). Firstly, enzyme digestion was performed after the restriction endonuclease was added to each genomic DNA sample. P1 adapters were added to the enzymatically digested fragments. Enzymatically digested fragments ligated to the P1 adapter were mixed and end-repaired. Then, dA and Illumina P2 adapters were added to the mixture fragments. PCR was employed to amplify the fragments with P1 and P2 adapters. Finally, paired-end sequencing was conducted on the reads of the library (150 bp) through the Illumina Novaseq 6000 sequencing platform.
SNP calling
Raw data was acquired and subjected to a quality control step. The Cutadapt program (version 2.6) (Martin 2011) was used to remove low-quality read pairs following the parameter listed below: -q 20 (trim low-quality reads), -e 0.1 (maximum allowed error rate by keeping the default value 0.1 = 10%), -n 1 (remove up to one adapter from each read) and -m 20 (discard trimmed reads that are shorter than 20 bp). Then the clean reads were aligned with the reference genome of K. punctatus at NCBI (Genome accession number: PRJNA664710, scaffold N50: 32.23 Mb, BUSCO completeness: 93.54%) (Liu et al. 2022a) using the algorithm of BWA-MEM in the BWA software (Li and Durbin 2010) with the default settings. After that, SAMtools (Li et al. 2009) was used to sort reads. Duplicates formed in PCR were cleared using the Picard tool (Zhao 2018) in the Java program. GATK (McKenna et al. 2010) was applied for SNP calling. SNPs were selected using VariantFiltration according to the following conditions: QD < 2.0, MQ < 40.0, FS > 60.0, SOR > 3.0, MQRankSum < − 12.5, ReadPosRankSum < − 8.0. Finally, VCFtools (Danecek et al. 2011) was performed to filter SNPs with the parameters: -maf 0.05; -max-missing 0.9; -minQ 30; -min-alleles 2 and -max-alleles 2. The filtered SNPs were used for subsequent analysis.
Population genetic analysis
The genetic parameters of nine sampling locations were statistically performed for K. punctatus, including polymorphic loci, the nucleotide diversity (π), observed heterozygosity (HO), expected heterozygosity (HE), the inbreeding coefficient (FIS), and pairwise FST values (Meirmans and Hedrick 2011) by the “populations” module of Stacks software (Catchen et al. 2013).
Population stratification and genetic structure
Linkage disequilibrium (LD) filter was performed using VCFtools (Danecek et al. 2011) before population structure analysis according to the criteria: -indep-pairwise 50 10 0.2. Briefly, principal component analysis (PCA) analysis was performed through the Plink program (Purcell et al. 2007) based on 259,449 SNPs. The contribution ratios for each principal component were calculated and the PCA diagram was drawn in the R software (Team RC 2013) environment. Furthermore, the admixture history and the cross-validation error for the hypothetical runs from K (the number of ancestral populations) = 1–4 were determined using the Admixture program (Alexander et al. 2009). The optimal K-value was determined through the cross-validation (CV) error plot using the received log data. Finally, R software (Team RC 2013) was used to draw admixture plots using the Q estimate files created by the Admixture programs. In addition, the phylogenetic tree was constructed based on the Neighbor-joining (NJ) method. First, a Perl script was used to convert the VCF file into a PHYLIP file (Retief 2000). A configuration file was then prepared for the dnadist program in the PHYLIP package to run the distance matrix. Finally, the NJ tree was formed through the Neighbor program in PHYLIP (Saitou and Nei 1987). The image of the phylogenetic tree was beautified using iTOL (Letunic and Bork 2021). To precisely resolve the genetic relationships among 146 K. punctatus individuals, the R package NETVIEW (Steinig et al. 2016) was used to construct a genetic relationship network map for them. Similarly, the number of mutual nearest neighbor graphs (mkNNGs) needs to be determined before running NETVIEW. It is essential to find a network topology within the possible range of mkNNGs that presents the genetic similarity among all individuals at an appropriate resolution.
Gene flow and population history
To find the genetic drift and infer gene flows of historical population splits and mixtures among the nine sampling locations of K. punctatus, Treemix software (Pickrell et al. 2012) was applied using Fitak (2021)’s methods. Population history was modeled from zero to ten migration events (1 ≤ m ≤ 10) and run for 10 replicates with the RZH as the root. To avoid converging on the same composite likelihood for each replicate, the number of SNPs per window was varied across runs from 100 to 1000 in 50 SNP increments. Once completed, the Treemix output files were analyzed with OptM v0.1.5 (Fitak 2021) using default parameters. Finally, the tree was visualized using the R script embedded in Treemix.
In addition to the above method of detecting population history dynamics by inferring gene flow, we also inferred the dynamic history of population contraction or expansion of K. punctatus based on the Site Frequency Spectrum (SFS). First, the SFS was calculated by the ANGSD 0.935 software (Korneliussen et al. 2014) based on the bam file and filtered with the parameter "-minMapQ 30 -minQ 20" for low-quality maps. Then, the likelihood of SFS was evaluated using -doSaf based on individual genotype likelihoods to obtain the.Saf file. Next, the realSFS was employed to generate the folded SFS based on the estimation of maximum likelihood. Finally, Stairway plot 2 (Liu and Fu 2020) was applied to infer the effective population size (Ne) of K. punctatus populations under a mutation rate of 1.29e−9 per generation and a generation time of one and a half years.
Detection of loci under putative selection and gene annotation
To clarify the mechanisms of local adaptation for K. punctatus populations, we combined the redundancy analysis (RDA) (Forester et al. 2018) and BayeScan methods (Foll et al. 2008). RDA as a genotype-environment association method was applied to evaluate the percentage of genomic variation interpreted by environmental variables and to detect SNPs subject to environmental selection (Lasky et al. 2012; Waits and Sork 2010). The principle of the method is to perform multiple linear regression analysis (MLR) on genetic and environmental data through the RDA function of the R package vegan 2.6 and generate a matrix of fitted values (Oksanen et al. 2007; Zulliger et al. 2013). Then PCA of the fitted values was performed to produce canonical axes, which were linear combinations of the factors (Legendre and Legendre 2012). The main steps were as follows: first, the allele frequencies of 259,449 SNPs were assessed through Plink v 1.9 (Purcell et al. 2007) software and the missing values in the plink. The raw file was replaced with the most common genotypic substitution at the same position in all individuals. Second, 19 marine environmental variables were collected through the Bio-ORACLE database (Assis et al. 2018), including mean seawater temperature (Tavg), mean warmest seawater temperature (Tmax), mean coldest seawater temperature (Tmin), mean seawater salinity (Sarg), mean highest seawater salinity (Smax), mean lowest seawater salinity (Smin), mean dissolved oxygen (Oarg), mean maximum dissolved oxygen (Omax), mean minimum dissolved oxygen (Omin), mean pH (pHarg), mean water flow rate (Carg), mean fastest water flow rate (Cmax), mean slowest water flow rate (Cmin), mean nitrate content (Narg), mean maximum nitrate content (Nmax), mean minimum nitrate content (Nmin), mean phosphate content (Phoarg), mean maximum phosphate content (Phomax), and mean minimum phosphate content (Phomin). Third, the ArcGIS software (Wampler et al. 2013) was then used to extract the marine environmental data of the K. punctatus sampling sites. Due to the correlation between 19 environmental variables, one of the two highly related (|r| > 0.7) environmental variables was removed using Pearson correlation analysis to avoid environmental redundancy in RDA. Five environmental variables were retained, including Tmax, pHavg, Cmax, Nmax, and Phomax (Supplementary Fig. S1; Supplementary Table S2). The significances of global RDA and constrained axes were assessed using 1000 permutations and a P-value threshold of 0.05. Finally, we screened out outlier SNPs loading ± 3 SD from the mean loading of significant RDA axes and classified SNPs associated with different environmental variables (Forester et al. 2018). In addition to RDA, a Bayesian method implemented in BayeScan version 2.1 (Foll et al. 2008) was used to identify outliers. BayeScan was run for 20 pilot runs with a length of 5000 iterations after a burn-in of 50,000 steps with prior odds of 1000. SNPs with a false discovery rate (FDR) lower than 0.01 were considered outliers.
To more precisely identify loci associated with the adaptive evolution of K. punctatus populations, we intersected the loci screened by BayeScan 2.1 and RDA analysis to statistically identify highly differentiated SNPs that were significantly related to different environmental variables. Then, the BEDTools (Quinlan and Hall 2010) was applied to extract genes in the 10 kb upstream and downstream range of these loci, and eggNOG-mapper (Cantalapiedra et al. 2021) was employed to annotate these genes. Finally, the screened environmentally associated genes were enriched for Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functions using ClusterProfiler (Yu et al. 2012). Only the top 25% of pathways with P < 0.05 were retained when GO enrichment was performed, and only the top 50% of pathways were retained when KEGG enrichment was conducted. In addition, we screened genes with multiple environmental associations from the above genes for subsequent discussion.
Results
RAD-seq and SNP identification
We collected 146 K. punctatus individuals from nine locations along the coasts of China, Japan, and Korea (Fig. 1; Supplementary Table S1). RAD-seq of 146 K. punctatus individuals generated 1468 million paired-end data, ranging from 6,431,148 to 15,838,216 (Supplementary Table S3). After the low-quality filtering step, 1,466,640,306 reads were retained (99.9%). Generally, the GC content of filtered clean reads was between 41.81% and 42.06%, which was relatively stable (Supplementary Table S3). Quality scores ranged from 97.68% to 97.99% for Q20, and 93.15% to 94.30% for Q30, indicating the high quality of RAD sequencing results. Most mapping rates of clean reads were higher than 97% (Supplementary Table S3). After individual sequence alignment and SNP extraction, a total of 6,510,125 pre-filtered SNPs were eventually detected. When the low-quality SNPs were excluded, a total of 632,090 SNPs were obtained over the whole genome of the K. punctatus populations. After linkage disequilibrium (LD) filtering, 259,449 SNPs were obtained for population genetic structure analysis.
Genomic diversity of K. punctatus
We calculated the diversity index of nine locations for K. punctatus based on 632,090 SNPs (Table 1). The whole-genome level of genetic diversity of nine K. punctatus locations was evaluated. The results indicated values of HE (0.17–0.25), HO (0.17–0.25), FIS (0.03–0.10), and π (0.18–0.26) of the nine locations, which showed the lowest value in the RZH (Table 1). Furthermore, percentages of polymorphic loci (80.35–95.60%) for other K. punctatus sampling locations were high except for RZH (59.53%) (Table 1).
Genetic differentiation levels and genetic structure of K. punctatus
The fixation index (FST) evaluation was performed to assess levels of genetic differentiation among nine sampling locations for K. punctatus (Meirmans and Hedrick 2011). In practice, low differentiation is indicated when FST ranges from 0.00 to 0.05 while 0.05–0.15 indicates moderate differentiation, and a high level of differentiation exists when FST > 0.15 (Govindaraju 1989). The FST among nine K. punctatus sampling locations ranged from 0.01 (between YQ and NT) to 0.17 (between ZS and RZH). Generally, pairwise FST values were low among different locations, especially between YQ and NT, indicating free interbreeding between them (Table 2). It should be noted that FST values between the RZH and other locations were all larger than 0.13, indicating a large genetic distance between them (Table 2).
Furthermore, the analysis of population structure among different locations via the Admixture program (Alexander et al. 2009) revealed that the best K-value was 2, which indicated that 146 K. punctatus individuals used in this study belonged to two clusters (Cluster A and Cluster B) (Supplementary Fig. S2). Individuals in Cluster B were highly differentiated from other individuals in Cluster A (Fig. 2A). The principal component analysis (PCA) results indicated the presence of two subgroups among 146 accessions in our study (Fig. 2B), which was consistent with the K = 2 analysis of the population structure analysis (Fig. 2A). The first and second coordinates explained 8.72% and 1.62% of the variation, respectively. The PCA plot showed that all samples were divided into two populations (Cluster A and Cluster B) along the first discriminant function (PC1) while individuals from Cluster A were evenly distributed along the second discriminant function (PC2) (Fig. 2B). What is more, the Neighbor-joining (NJ) tree also showed that Cluster B was highly differentiated from Cluster A (Fig. 2C), and individuals from Cluster A were mixed and lowly differentiated, which was consistent with the Admixture and PCA cluster results (Fig. 2A, B).

Population structure and genetic differentiation based on 259,449 SNPs. A Population structure. The length of each colored segment represents the proportion of the individual genome inferred from ancestral populations (K = 2). B Principal component analysis. C NJ tree showing the phylogenetic relationships. Chinese populations: Qinhuangdao (QHD), Dandong (DD), Shouguang (SG), Lianyungang (LYG), Nantong (NT), Zhoushan (ZS), and Wenzhou (YQ). Korean population: Jeolla-Do (HQ). Japanese population: Saga (RZH)
To achieve a finer resolution of genetic relationships among 146 K. punctatus, multiple algorithms of NETVIEW (Fast-Greedy, Infomap, Walktrap) were applied to pick the best K value in the range of K = 1 to 100 (Steinig et al. 2016). As shown in Supplementary Fig. S3, the results of all algorithms were generally consistent, which showed that the decline was flat at K = 35 and the mutual nearest neighbor graphs (mkNNGs) gradually emerged as a fine genetic structure (Fig. 3). Therefore, the genetic network structure among K. punctatus individuals was constructed at K = 35. A high degree of differentiation was also detected between Cluster A and Cluster B (Fig. 3), which agreed with plots of Admixture, PCA and NJ tree (Fig. 2). In addition, details of the clustering among individuals of Cluster A showed that most individuals sampled from the QHD, DD, SG, LYG, and HQ locations clustered on the left side of the genetic network graph while most individuals sampled from the NT, ZS and YQ locations clustered on the right side of the genetic network graph (Fig. 3). The results suggested that although the kinship of individuals in Cluster A was close, there was a potential differentiation in Cluster A and there may be a hidden genetic structure among these individuals.

Network of K. punctatus based on 259,449 SNPs, color shades represent sampling locations (K = 35, n = 146). Chinese populations: Qinhuangdao (QHD), Dandong (DD), Shouguang (SG), Lianyungang (LYG), Nantong (NT), Zhoushan (ZS), and Wenzhou (YQ). Korean population: Jeolla-Do (HQ). Japanese population: Saga (RZH)
Gene flow among K. punctatus sampling locations
We inferred the history of gene flow among nine sampling locations using Treemix software (Pickrell et al. 2012) to figure out the mechanism of differentiation for K. punctatus. The 99.8% variation cutoff inferred by OptM v0.1.5 (Fitak 2021) showed that m = 4 was the optimal value (Fig. 4A). Regarding gene flow, our results supported the migration signal from the RZH to the HQ, QHD, and LYG. In addition, there was a gene flow from the ZS to the YQ and NT. As for the Treemix phylogeny, individuals of K. punctatus were divided into two main branches, one was the RZH, and the second continued to split into the QHD, DD, SG, LYG, HQ, ZS, as well as the YQ and NT (Fig. 4B).

Gene flow and Spread history of K. punctatus. A The output produced by OptM for the simulated dataset with m = 4 migration edges. The mean and standard deviation (SD) across 10 iterations for the composite likelihood L(m) (left axis, black circles) and proportion of variance explained (right axis, red “x” s). The 99.8% threshold (horizontal dotted line) is that recommended by Pickrell and Pritchard. B A population-based maximum likelihood Treemix analysis. Branch lengths are equivalent to a genetic drift parameter. Chinese populations: Qinhuangdao (QHD), Dandong (DD), Shouguang (SG), Lianyungang (LYG), Nantong (NT), Zhoushan (ZS), and Wenzhou (YQ). Korean population: Jeolla-Do (HQ). Japanese population: Saga (RZH)
Population history dynamics of K. punctatus
We reconstructed the population history dynamics of Cluster A and Cluster B, respectively to further understand the reasons for differentiation between them. Stairway plot 2 results showed that the first population expansion of Cluster A began nearly 2 million years ago (Mya) (Fig. 5A). Meanwhile, we found that K. punctatus of Cluster A had experienced three bottleneck events since then, including 780–500 thousand years ago (Kya), 280–180 Kya and 95–35 Kya bottleneck (Fig. 5A). Then, Cluster A contracted once again around 20 Kya, and the effective population size (Ne) remained stable (3 × 106) from 5 Kya until recently (Fig. 5A). However, we detected only one bottleneck and one significant population contraction of Cluster B within 1 Mya (Fig. 5B). Cluster B experienced a bottleneck around 420–180 Kya, and contracted again at about 80 Kya. Since 15 Kya, the Ne of Cluster B was stable at around 2.2 × 106 (Fig. 5B). The above results indicated that the population of K. punctatus showed an overall expansion trend, and the population size fluctuated within the Pleistocene ice age (2.6–11.7 Kya).

Demographic history of K. punctatus. A Demographic history of Cluster A. B Demographic history of Cluster B. The stairway plot shows the historical effective population size Ne (y-axis) with a generation time of one and a half years. The blue shadows represent population bottlenecks. The red line represents the median of Ne. Dark gray and light gray lines represent 75 and 95% confidence intervals, respectively
Candidate loci and functional genes under putative selection
Five variables (Tmax, pHavg, Cmax, Nmax, and Phomax) were retained after removing highly correlated environmental factors based on the Pearson correlation analysis (Supplementary Fig. S1; Supplementary Table S2). Then, the results of the multiple linearity analysis of RDA explained approximately 4.4% of the variance (adjusted R2 = 0.04402). The RDA1 and RDA2 axes were significant (P = 0.001) while the RDA1 axis explained most of the variance, and five environmental factors retained in RDA were also significant as explanatory variables (P = 0.001) (Fig. 6A, B). The PCA of the fitted values after multiple linear analysis also showed that 146 K. punctatus individuals differentiated into two clusters (Cluster A and Cluster B), and Cluster A had a differentiation between the northern (QHD, DD, SG, LYG and HQ) and southern (NT, ZS and YQ) population (Fig. 6B), which was consistent with the result of NETVIEW (Fig. 3). As shown in Fig. 6B, pHarg and Phomax were significantly correlated with high differentiation between Cluster A and Cluster B, while Cmax, Nmax and Tmax were correlated with differentiation among northern and southern populations of K. punctatus in Cluster A (Fig. 6B), suggesting that the weak differentiation among individuals in the Cluster A shown in NETVIEW (Fig. 3) are existent and significantly related to the marine environment.

Genomic footprints of adaptation to environmental heterogeneity. A The significance of the RDA constraint axis. B Redundancy analysis (RDA) on axes 1 and 2 performed with 259,449 SNP loci. The dark grey points at the center of each plot represent the SNPs, and the colored points represent K. punctatus individuals of different sampling locations. Blue vectors represent environment variables. C Magnification of B to highlight SNP loadings on axes 1 and 2. Candidate SNPs are shown as colored points with coding by the most highly correlated environmental variables. SNPs not identified as candidates (neutral SNPs) are shown in light grey. Blue vectors represent environmental variables. D Bayesian test for selection performed with 259,449 SNPs using the program BayeScan. Red dots on the right side of the picture are above a 0.99 probability of being candidates of selection. Yellow dots on the left side are neutral SNPs. Chinese populations: Qinhuangdao (QHD), Dandong (DD), Shouguang (SG), Lianyungang (LYG), Nantong (NT), Zhoushan (ZS), and Wenzhou (YQ). Korean population: Jeolla-Do (HQ). Japanese population: Saga (RZH)
Among all SNPs, a total of 957 candidate loci subject to environmental selection were screened, of which 128 were on the RDA1 axis, 689 on the RDA 2 axis and 143 on the RDA 3 axis (Fig. 6C). Notably, the RDA detected the largest proportion of loci associated with pHarg (281; 29.36%), indicating that pHarg explained more allelic variation than any other environmental variable tested, followed by Tmax (244; 25.50%), then Phomax (240; 25.07%), then was Nmax (183; 19.12%), and finally, Cmax (9; 0.94%) (Fig. 6C). In addition, we screened 309 significant highly differentiated SNPs using BayeScan 2.1 (Fig. 6D). Therefore, we intersected the candidate loci detected by RDA and BayeScan 2.1 and obtained 244 SNP loci associated with local adaptation of K. punctatus, of which 78 loci were associated with Nmax, 64 loci with pHarg, 63 loci with Tmax, 36 loci with Phomax and 3 loci with Cmax (Supplementary Table S4).
Functional annotation of adaptive genes
BEDTools was used to find genes in the 10 kb range upstream and downstream of the adaptive loci, and 56, 53, 45 and 24 genes were detected near the Nmax, pHarg, Tmax and Phomax association loci, respectively, while none were found near the Cmax association loci (Supplementary Table S5). As shown in Fig. 6C, pHarg association genes were significantly correlated with the differentiation between Cluster A and Cluster B while Tmax association genes correlated with the differentiation between the northern and southern populations of K. punctatus in Cluster A. So, Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment were only performed for genes associated with pHarg and Tmax, respectively. When pHarg association genes were enriched in GO terms, most of them were engaged in molecular functions (MF) such as the epidermal growth factor receptor binding (GO:0005154), the regulation of intramolecular phosphotransferase class activity (GO:0016868) and the regulation of peptidase activity (GO:0061134) (Supplementary Table S6). In addition, there were some genes engaged in a few biological processes (BP) such as the positive regulation of the intrinsic apoptotic signaling pathway (GO:2001244), the negative regulation of centrosome replication (GO:0010826) and the regulation of cytoplasmic mRNA assembly (GO:0010603) (Supplementary Table S6). The most significant 39 GO pathways enriched for Tmax association genes contained 17 cellular components (CC) such as extracellular matrix components (GO:0044420), collagen trimer (GO:0005581) and endoplasmic reticulum lumen (GO:0005788); 22 molecular functions (MF) mainly included binding to extracellular matrix (GO:0050840), protein self-association (GO:0043621), vascular endothelial growth factor receptor activity (GO:0005021), and oxidoreductase activity (GO:0016722), etc. (Supplementary Table S7).
KEGG enrichment results showed that these adaptive genes were mainly engaged in five physiological processes including metabolism, genetic information processing, environmental information processing, cellular processes and organismal systems (Fig. 7). The genes associated with pHarg were enriched to 21 pathways, five of which were related to cellular processes, namely apoptosis, cellular senescence, p53 signaling pathway, cell cycle and autophagy (Fig. 7A). Seven pathways were related to sugar metabolism, including biosynthesis of nucleotide sugars, amino and nucleotide sugar metabolism, pentose phosphate pathway, starch and sucrose metabolism (Fig. 7A). In addition, some pathways related to environmental information processing such as the VEGF signaling pathway, ErbB signaling pathway and FoxO signaling pathway were found as well (Fig. 7A). Among the 30 KEGG pathways enriched for Tmax association genes, there were many pathways related to sugar metabolism as well, but the pathways enriched for the most genes are the cellular focal adhesion and NOD-like receptor signaling pathways (Fig. 7B). In addition, some signaling pathways widely present in vertebrates and invertebrates were identified, such as the Wnt signaling pathway, Hedgehog signaling pathway, and GnRH signaling pathway (Fig. 7B).

The results of Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment. A KEGG pathway of genes associated with pHarg. B KEGG pathway of genes associated with Tmax
Furthermore, we screened eight genes associated with multiple environmental variables from the above genes. Among them, the genes associated with Nmax and pHarg were KIDINS220 and SCRIB; the genes associated with Nmax and Tmax were VAV3 and EPB41L3; the genes associated with pHarg and Tmax were PAN3 and PGM5; the genes associated with Phomax and Tmax were CCT7 and HSPA12B (Table 3). These genes were enriched for GO biological process pathways (P < 0.050). Except for HSPA12B, all genes were enriched in a variety of GO biological processes, with SCRIB enriched in the most pathways (73), followed by VAV3 (37), then EPB41L3 (30), CCT7 and PGM5 enriched in 18 and 14 pathways, respectively, and PAN3 and KIDINS220 enriched in the least pathways, as detailed in Supplementary Table S8.
Discussion
In this study, we successfully detected 632,090 SNPs across the genome of K. punctatus populations based on RAD-seq for the first time, and analyzed population genetic diversity, population genetic structure and adaptation evolution of K. punctatus off the coast of China, Japan, and Korea. The following are the main findings of our study: (1) the population genetic diversity results indicated that all K. punctatus sampling locations had high genetic diversity whereas RZH had lower genetic diversity than other locations. (2) The results of pairwise FST, Admixture, PCA and NJ tree indicated that 146 K. punctatus from nine different locations differentiated into two Clusters (Cluster A and Cluster B), but the NETYIEW plot presented that a small but remarkable genetic differentiation existed between northern and southern population in the Cluster A. (3) Interpopulation gene flow analysis revealed weak gene flow from Cluster B (RZH) to some locations in Cluster A (QHD, HQ and LYG). (4) The results of historical population dynamics showed that Cluster A and Cluster B of K. punctatus experienced different times of population contraction and expansion, but generally showed an expansion trend. (5) The RDA plots proved that Cluster A had a differentiation between the northern (QHD, DD, SG, LYG and HQ) and southern (NT, ZS and YQ) population. In addition, the research on the local adaptation of K. punctatus found some genes which mediated physiological processes such as growth and development, substance metabolism and immune response of K. punctatus. The following is a discussion of the results of this study.
Population structure and spread history
In this study, we successfully identified genome-wide SNPs using RAD-seq to explore population genetic diversity and population genetic structure of nine sampling locations for K. punctatus in China, Japan, and Korea firstly. In terms of the population genetic diversity, the mean values of HE, HO and π of nine K. punctatus locations based on SNPs were 0.24, 0.23 and 0.25, respectively (Table 1). The RZH exhibited a lower nucleotide diversity than the other eight locations, which was consistent with the results of the proportion of polymorphic loci (Table 1). Since the proportion of polymorphic loci of RZH (59.53%) was significantly lower than the mean value (86.57%) (Table 1), resulting in the low level of nucleotide diversity of RZH, suggesting that the other eight K. punctatus locations all generated more genetic variation and had more population resilience during adaptive evolution, showing a more capable of adapting to the changing marine environment than RZH. Furthermore, the level of population genetic diversity for K. punctatus in this study was similar to that of some other fishes. For example, Zhang et al. (2016) investigated the population genetic structure and local adaptation of small yellow croaker (Larimichthys polyactis) off the coasts of China using RAD-seq, and the population genetic diversity study based on 27,556 SNPs revealed that the mean value of HE, HO and π for L. polyactis populations were 0.22, 0.23 and 0.22, respectively. In addition, the level of population genetic diversity of some endangered fishes was lower than that in this study. For example, the mean value of HE, HO and π for Anguilla japonica populations along the Chinese and Japanese coasts were 0.15, 0.16 and 0.15, respectively (Liu 2017), all lower than the values of K. punctatus populations in this study. In a word, population genetic diversity results revealed that all K. punctatus locations had relatively high genetic diversity except for RZH.
In terms of the population genetic structure, the mean value of FST (0.05) showed a moderate genetic differentiation of K. punctatus populations (Meirmans and Hedrick 2011), indicating little variation happened among the K. punctatus populations. Overall, only the RZH revealed a high level (FST > 0.15) and significant differentiation in the case of SNPs while other locations of K. punctatus showed a low-level differentiation (Table 2), which was like the results of Liu et al. (2020)’s study on K. punctatus populations distributed along the coast of China. Furthermore, the results of Admixture, PCA and NJ tree all showed significant differentiation between Cluster A and Cluster B (Fig. 2), which was consistent with previous population genetic studies based on the mtDNA control region (Gwak et al. 2015; Song et al. 2016; Ying 2011). However, the NETVIEW and RDA plot in this study showed a small but significant genetic differentiation between northern and southern population in Cluster A, which was never observed in previous studies based on other genetic markers (Gwak et al. 2015; Song et al. 2016; Ying 2011).
There were three reasons accounting for the population genetic differentiation of K. punctatus. (1) The ecological habits of K. punctatus. K. punctatus has a fixed migratory pattern, as an offshore fish, spawning and nursing almost exclusively in the nearshore and estuaries. However, populations distributed in the Yellow Sea and Bohai Bay have a common overwintering site in the southeastern Yellow Sea. After overwintering, they migrate to shallower waters off the coast of Bohai Bay and Shandong, China for spawning respectively, while populations in other waters do not migrate long distances (Zhang 2001). The relatively independent migratory routes limit the dispersal of southern East China Sea populations of K. punctatus, which may lead to the potential differentiation between the northern (QHD, DD, SG, LYG and HQ) and southern (NT, ZS and YQ) population in the Cluster A (Song et al. 2016) (Figs. 3, 6B). However, although the existence of common overwintering sites and the overlap of a few migratory routes may cause the mixing of adjacent K. punctatus individuals in Cluster A. The K. punctatus in Cluster B only make inshore spawning and overwintering migrations, which may lead to the genetic differentiation between Cluster B and the geographically distant Cluster A due to the long absence of gene exchange (Ying 2011). (2) The influence of ocean currents. Like other fishes, the spawning and hatching grounds of K. punctatus tend to be in coastal waters with higher nutrient and productivity levels. But ocean currents may have an impact on the content and distribution of nutrients and productivity in the ocean. For example, there is a branch of Kuroshio warm current between Cluster A and Cluster B (Fig. 1), which makes the chlorophyll A content of the flowing sea area significantly lower than that of the East China Sea shelf and the Japanese coast in spring. The gradual strengthening of the warm current in the summer makes the high productivity only in a small land area along the East China Sea (Fig. 1) (Hamilton et al. 2008). Therefore, the dispersion of spawning and rearing grounds farther offshore is not conducive to the post-migration survival of K. punctatus, which may have indirectly led to the gradual differentiation between Cluster A and Cluster B (Ying 2011). (3) Historical climate changes. When Ying (2011) detected the population genetic structure of K. punctatus off the coasts of China and Japan based on mtDNA markers, he concluded that the genetic differentiation among them was mainly due to the East China Sea being closed to an inland sea as the drop of sea level during the Last Glacial Maximum (LGM). This has resulted in K. punctatus populations distributed in the East China Sea and other seas being isolated in different refuges, promoting differentiation among populations (Wang 1999; Ying 2011). Some marine organisms with similar geographic distribution patterns to K. punctatus have also formed similar population genetic structures, such as CluPea pallasii (Liu et al. 2007b), Pennahia argentata (Han et al. 2008), and Scapharca broughtonii (Yokogawa 1997). All population structures of them were affected by the LGM, generating a differentiation among populations along the East China Sea coast and the Japan Sea coast. In addition, Gwak et al. (2015) surveyed the population genetic structure of K. punctatus offshore Korea and Japan using fragments of mtDNA and found that two genetic lineages of K. punctatus diverged during the Pleistocene glacial cycles. This study hypothesized that the differentiation between the Korean and Japanese populations of K. punctatus was caused by the sea level drop during the LGM. At that time, the Yellow Sea almost entirely disappeared and land masses appeared on the seabed of both the Yellow Sea and the southern coast of Korea, leaving the Korean and Japanese coasts separated by a strait-like seaway linking the Northwest Pacific Ocean and the Japan Sea (Park et al. 2006). In our study, Cluster A experienced a population contraction at around 20 Kya, close to the onset of the LGM (~ 22 Kya) (Fig. 5A). It is therefore hypothesized that the shrunk population size of Cluster A was affected by the drop in sea levels during the LGM. Meanwhile, populations in Cluster A were isolated due to the reduced area of the marginal sea (Han and Huang 2008; Yoo et al. 2016), which eventually led to the differentiation between Cluster A and Cluster B.
Population historical dynamics of K. punctatus
The results of the population genetic structure study revealed that there was a high and significant genetic differentiation between Cluster A and Cluster B, so we investigated whether there was gene flow between them using Treemix. The results showed that there was weak gene flow from the RZH to HQ, QHD and LYG, respectively (Fig. 4B), indicating that even though the genetic differentiation among RZH and other locations was significant, potential gene flow existed among them, which was not found in previous studies. In addition, we reconstructed the population history dynamics of Cluster A and Cluster B using Stairway plot 2 and found that Cluster A experienced three distinct bottlenecks in the last two million years (Fig. 5A), while Cluster B experienced only one (Fig. 5B). After the start of the Last Glacial Period (LGP, ~ 110–12 Kya) (Gibbard and Kolfschoten 2005), the population size of both Cluster A and Cluster B changed, the difference being that Cluster A experienced a bottleneck before the population contracted again during the LGM (~ 22 Kya) (Gibbard and Kolfschoten 2005), and then a slight population expansion at the end of the LGP (~ 12 Kya). In contrast, Cluster B experienced only one population contraction at the beginning of the LGP, and the population size gradually stabilized without being greatly affected by the LGM. Overall, there was an expansion trend of K. punctatus populations during the Pleistocene ice age. Furthermore, Ying (2011) also detected the expansion trend of the K. punctatus population when performing the population genetic analysis based on mitochondrial molecular markers. Combined with historical geographic factors, a series of dramatic climatic fluctuations during the Pleistocene ice age affected various factors of the marine environment, such as seawater temperature and sea level, which can cause changes in the ecological habits and habitat populations of many marine organisms (Liu et al. 2007a). The overall expansion trend of the K. punctatus population may be ascribed to the recovery of the marine environment and marine geological changes originally affected by the LGP. As the sea level rise, the isolated sea areas were reconnected, which led to the expansion of the habitat of K. punctatus, the increase in population size, and the re-establishment of genetic exchange between populations (Ying 2011). Analogous phenomena have occurred in other fish populations with similar geographic distribution patterns, such as Lateolabrax japonicus, Chelon haematocheilus and Lateolabrax maculatus, all of which have been detected as apparent historical population expansion events after the LGP (Liu et al. 2006a, 2007a). Other marine fishes such as Engraulis mordax and Sardinops sagax in the Northeast Pacific have also undergone population expansion after the Pleistocene ice age (Lecomte et al. 2004; Liu et al. 2006b).
The habitat adaptability of K. punctatus
In this study, we used RDA analysis to investigate the correlation between local adaptations of K. punctatus and marine environmental factors. After combining population genetic data and environmental data for PCA, we found a north–south differentiation among locations of Cluster A, which verified the NETVIEEW plot. However, both Ying (2011) and Liu et al. (2020) failed to detect the genetic structure in the Chinese coastal K. punctatus populations based on mitochondrial molecular markers. Therefore, we need to investigate the local adaptive mechanisms of K. punctatus populations. Based on RDA, we found that five environmental factors (Tmax, pHarg, Cmax, Nmax and Phomax) had a strong impact on the adaptive evolutionary process of K. punctatus populations.
As shown in Fig. 6B, pHarg was mainly associated with the genetic differentiation between Cluster A and Cluster B. It has been shown that seawater pH affects behavioral, cellular metabolism, immune response and other processes in marine fishes. For example, the decrease of seawater pH can significantly affect the predation, habitat selection and other behaviors of fishes such as Amphiprion percula, Pomacentrus amboinensis and Pomacentrus moluccensis, reducing their ability to recognize habitats and even changing their temporal rhythm of settlement (Devine et al. 2012; Munday et al. 2009). In addition, the clustering intensity, phototropic behavior, courage level, and the dispersal of marine fishes are all limited by the pH of seawater (Forsgren et al. 2013; Lopes et al. 2016; Maulvault et al. 2018). Meanwhile, we identified several pathways related to glucose metabolism, cellular processes and immune regulation during the KEGG enrichment for pHarg association genes (Fig. 7A). Among them, the p53 signaling pathway is involved in apoptosis, cell growth and differentiation, and DNA damage repair processes (Vousden and Lu 2002). It has been found to be able to mediate the resistance of L. crocea bodies to their own visceral nodular disease, suggesting that the p53 signaling pathway also exerts a significant role in the immune regulation of marine fishes (Bai et al. 2022). The p53 signaling pathway is likely to contribute to the adaptation of K. punctatus to environmental pH by repairing damage and altering cellular metabolism levels, which indicates that the mean seawater pH can influence various physiological activities in K. punctatus, thus affecting the process of adaptive evolution.
Tmax was positively correlated with the differentiation among populations in Cluster A, indicating that temperature was also a major factor influencing the adaptive evolution of K. punctatus (Fig. 6B). Previous studies have also shown that seawater temperature can influence the adaptive differentiation of K. punctatus. For example, Myoung and Kim (2014) analyzed the population genetic structure of K. punctatus along the Korean coasts using mtDNA markers and found that K. punctatus populations diverged into two lineages with different sensitivity to temperature, which influenced the local adaptation of K. punctatus. Furthermore, it has been shown that temperature may affect the growth, reproduction, and migration of marine fishes, and may drive their adaptive evolution during different life history stages (Johansen and Jones 2011; Pankhurst and Munday 2011). The presence of the GnRH signaling pathway in the KEGG enrichment also supports this conclusion (Fig. 7B), as this signaling pathway can take a vital role in the control of reproductive function, growth and development of fishes (Okubo and Nagahama 2008). Also, Liu et al. (2022a, b) have found that this signaling pathway in male Opsariichthys bidens was able to induce the action of growth hormone in males by regulating the secretion of luteinizing hormone and glycoprotein hormone, making males grow faster than females (Liu et al. 2022b). In addition, we identified the Notch signaling pathway that may be directly related to the adaptive evolution of K. punctatus (Fig. 7B), which participates in several processes of cell development, including cell morphogenesis and proliferation and differentiation (Skoda et al. 2018; Weng et al. 2004). When Notch receptors bind to ligands, neighboring cells can expand molecular differences between cells by transmitting Notch signals, determining cell fates and ultimately affecting organ formation and morphogenesis (Metz and Bridges 1917). Mutations of loci in the Notch signaling pathway may also cause alterations in biological phenotypes (Mohr 1919), suggesting that it may increase differences among K. punctatus populations at both molecular and morphological levels during their adaptation to seawater temperature. Furthermore, even if temperature was not directly determining the local adaptation of K. punctatus, other factors closely related to temperature may contribute to the evolution of adaptation. For example, increased water temperatures may reduce the overall oxygen content of the living environment for marine fishes, and less oxygen means less energy to support the physiological performance and movement of marine fishes (Pörtner and Knust 2007), all of which have the potential to affect the environmental adaptation of K. punctatus.
Furthermore, we combined RDA and BayeScan 2.1 to screen for highly differentiated environmentally associated genes in K. punctatus populations, based on which we identified eight genes associated with multiple environmental variables (Table 3) and enriched them for GO functions (Supplementary Table S8). Among them, the KIDINS220 gene was mainly engaged in the GO pathways such as the nerve growth factor signaling pathway, the obsolete activation of MAPKK activity, and the cellular response to nerve growth factor stimulus (Supplementary Table S8). The mitogen-activated protein kinase kinase (MAPKK) is engaged in the MAPK signaling pathway, which is essential in regulating cell growth, differentiation, stress adaptation to the environment, inflammatory response and other physiological processes. It had been shown that KIDINS220 gene expression in zebrafish activated the MAPK signaling pathway (Howe et al. 2013), which had the functions of promoting neuronal cell development and regulating neuronal cell survival in animals (Sutachan et al. 2010; Zhou 2011). Thus, the KIDINS220 gene may regulate processes, such as cellular responses to environmental stimuli, neurotransmitter receptor transport, or regulate metabolic and immune processes in K. punctatus through the MAPK signaling pathway, thus promoting the adaptation of cells or organs to the nitrate content and pH of the marine environment. The protein encoded by the VAV3 gene can catalyze the interconversion of GDP and GTP and affect the process of intracellular gene transcription (Ulc et al. 2017). Meanwhile, pathways associated with the activation of GTPase activity and the positive regulation of GTPase activity were also identified in GO enrichment (Supplementary Table S8), indicating that the VAV3 gene can play a role in signaling, material transport, and many cellular activities in which G proteins are involved (Dong and Wen 2005). In addition, GO enrichment revealed that the VAV3 gene can be involved in 37 biological processes, among which the more important ones for the adaptive evolution of K. punctatus were the positive regulation of B cell proliferation, the positive regulation of B cell activation and the involvement of cell surface receptor signaling pathways regulating the immune response in phagocytosis (Supplementary Table S8). B cells are known to produce specific antibodies against invading antigens in vertebrates and have a major role in adaptive immunity (Kincade and Cooper 1971). In addition, B cells can perform phagocytic and bactericidal functions by forming phagocytic lysosomes, which perform a key role in bridging the gap between intrinsic and adaptive immunity in teleost fishes (Wu et al. 2020). The presence of a pathway regulating the involvement of immune response cells in phagocytosis could also corroborate the importance of the VAV3 gene in the adaptive immunity of K. punctatus. Thus, the multiple functions of the protein encoded by the VAV3 gene are beneficial to the local adaptation of K. punctatus to the variable marine environment. The genes associated with pHarg and Tmax were PAN3 and PGM5 (Table 3). The protein encoded by the PAN3 gene is conserved and belonged to the RAS protein family, which may indirectly inhibit excessive apoptosis by suppressing apoptosis protein activity, and inhibit tumor invasion and migration (Yang et al. 2017). Therefore, the PAN3 gene may enable K. punctatus to survive in different seawater pH and water temperatures by inhibiting excessive apoptosis or disease development process. Although the protein encoded by the PGM5 gene is a muscle structural protein with no enzymatic activity, it has been shown that the PGM5 gene has a facilitative role in adaptive differentiation between Atlantic herring (Clupea harengus harengus) and Baltic herring (C. harengus membras) (Gustafsson et al. 2020). In addition, zebrafish embryos have been found to develop severe cardiac and skeletal myopathies with signs of paralysis after knocking out the gene for PGM5 (Molt et al. 2014). These researches are sufficient to argue that the PGM5 gene has an important effect on the adaptive evolution and embryonic development of fish. The PGM5 gene was mainly enriched in the pathways of glucose metabolism and energy metabolism (Supplementary Table S8), suggesting that it also has an impact on physiological processes such as cell metabolism, which may help K. punctatus adapt to the marine environment. Furthermore, genes associated with Phomax and Tmax were found (Table 3). The CCT7 gene encodes the protein which can involve in the regulation of protein localization to chromosome, positive regulation of telomere maintenance, binding of sperm to zona pellucida and positive regulation of DNA biosynthesis (Supplementary Table S8). Surprisingly, ten GO pathways associated with telomeres were enriched. As we know, telomeres have functions in maintaining the lifespan of plant and animal cells, protecting chromosomes, mediating chromosome replication, and regulating cellular senescence, and are closely related to the development of many disease processes. Meanwhile, telomeres have also been found in some long-lived fish to play an active role in the control of the cell cycle, cellular self-phagocytosis and growth regulation, as well as in the regulation of cellular senescence and lifespan (Sauer et al. 2021). Therefore, the CCT7 gene may be involved in regulating DNA biosynthesis, protein folding and cellular life processes in K. punctatus, promoting its adaptation to environmental Phomax and Tmax. In addition, we detected a heat shock protein-encoding gene, HSPA12B, which is the latest member of the HSP70 protein family (Zhou et al. 2010). The HSPA12B gene can participate in the synergistic immunity of animals, which is closely related to angiogenesis and the functional performance of cardiac organs (Zhou et al. 2010). Heat shock proteins are widely present in organisms and play a protective role in the adaptation of organisms to high temperatures. The mean warmest water temperature at the sampling sites of K. punctatus populations in this study reached 25–28 °C (Supplementary Table S2). The HSPA12B gene may have mobilized the process of synergistic immunity in the body of K. punctatus to play a protective role under temperature stress. All these functionally enriched genes detected in highly divergent genomic regions of K. punctatus populations may be essential in regulating molecular mechanisms that allow habitat adaptation in K. punctatus. Further functional verification of these adaptive genes is needed. In general, the selection signals based on RAD-seq in this study provided a powerful step to understand the potentially adaptive evolution of K. punctatus. Furthermore, these findings will help us better protect and exploit the genetic resources of K. punctatus and provide a valuable resource for association analysis.
Conclusions
We successfully employed RAD-seq to explore the population genetic diversity and genetic structure of nine sampling locations for K. punctatus in China, Japan, and Korea. The results based on all SNPs revealed significant genetic differentiation between the RZH and other locations. Furthermore, genetic differentiation between northern (QHD, DD, SG, LYG and HQ) and southern (NT, ZS and YQ) population was found. At the same time, we confirmed that the direction of gene flow was from the RZH to HQ, QHD and LYG, and reconstructed the population history dynamics of K. punctatus. In addition, we identified outlier SNPs under putative selection and found genes associated with multiple environmental variables that may be crucial in K. punctatus adaptation to local habitat heterogeneity. In conclusion, population genetic structure and local habitat adaptation research of K. punctatus in this study will provide a strong scientific basis for the conservation and rational use of K. punctatus resources. In the future, functional verification of these adaptive genes should be performed to clarify the physiological mechanism of the local adaptation of K. punctatus to the marine environment.
Data availability
The whole genome of K. punctatus at NCBI is under Bioproject accession number PRJNA664710. RAD -seq data of K. punctatus is publicly accessible at GenBank under Bioproject accession number PRJNA746100 and Biosample accession numbers SAMN20181051–SAMN20181086, SAMN20181096, SAMN20181098-SAMN20181102, SAMN20181104-SAMN20181168, SAMN20181215- SAMN20181233 and SAMN20181234-SAMN20181254.
References
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Assis J, Tyberghein L, Bosch S, Verbruggen H, Serrão EA, De Clerck O (2018) Bio-ORACLE v2. 0: extending marine data layers for bioclimatic modelling. Glob Ecol Biogeogr 27:277–284
Bai Y, Qu A, Liu Y, Chen X, Wang J, Zhao J, Ke Q, Chen L, Chi H, Gong H (2022) Integrative analysis of GWAS and transcriptome reveals p53 signaling pathway mediates resistance to visceral white-nodules disease in large yellow croaker. Fish Shellfish Immunol 130:350–358
Billington N, Hebert PD (1991) Mitochondrial DNA diversity in fishes and its implications for introductions. Can J Fish Aquat Sci 48:80–94
Campbell D, Duchesne P, Bernatchez L (2003) AFLP utility for population assignment studies: analytical investigation and empirical comparison with microsatellites. Mol Ecol 12:1979–1991
Cano JM, Shikano T, Kuparinen A, Merilä J (2008) Genetic differentiation, effective population size and gene flow in marine fishes: implications for stock management. J Integr Field Biol 5:1–10
Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J (2021) eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 38:5825–5829
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Conover D, Clarke L, Munch S, Wagner G (2006) Spatial and temporal scales of adaptive differentiation in marine fishes and the implications for conservation. J Fish Biol 69:21–47
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Devine B, Munday P, Jones G (2012) Rising CO2 concentrations affect settlement behaviour of larval damselfishes. Coral Reefs 31:229–238
Dhaliwal A (2013) DNA extraction and purification. Mater Methods 3:191
Dong J, Wen J (2005) Structure and function of small molecule GTPases. Chem Life 25:422–424
Eaton DA, Ree RH (2013) Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst Biol 62:689–706
Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA (2011) SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol Biol 772:157–178
Fernández MV, Heras S, Maltagliati F, Turco A, Roldán MI (2011) Genetic structure in the blue and red shrimp Aristeus antennatus and the role played by hydrographical and oceanographical barriers. Mar Ecol Prog Ser 421:163–171
Fitak RR (2021) OptM: estimating the optimal number of migration edges on population trees using Treemix. Biol Methods Protoc 6:bpab017
Foll M, Beaumont MA, Gaggiotti O (2008) An approximate Bayesian computation approach to overcome biases that arise when using amplified fragment length polymorphism markers to study population structure. Genetics 179:927–939
Forester BR, Lasky JR, Wagner HH, Urban DL (2018) Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Mol Ecol 27:2215–2233
Forsgren E, Dupont S, Jutfelt F, Amundsen T (2013) Elevated CO2 affects embryonic development and larval phototaxis in a temperate marine fish. Ecol Evol 3:3637–3646
Gibbard P, Kolfschoten T (2005) The pleistocene and holocene epochs. In: Gradstein FM, Ogg JG, Smith AG (eds) A geologic time scale 2004. Cambridge University Press, Cambridge, pp 441–452
Govindaraju DR (1989) Variation in gene flow levels among predominantly self-pollinated plants. J Evol Biol 2:173–181
Gustafsson R, Eckhard U, Ye W, Enbody ED, Pettersson M, Jemth P, Andersson L, Selmer M (2020) Structure and characterization of phosphoglucomutase 5 from Atlantic and Baltic herring—an inactive enzyme with intact substrate binding. Biomolecules 10:1631
Gwak WS, Lee YD, Nakayama K (2015) Population structure and sequence differentiation in the mitochondrial DNA control region of gizzard shad Konosirus punctatus in Korea and Japan. Ichthyol Res 62:379–385
Hamilton SL, Regetz J, Warner RR (2008) Postsettlement survival linked to larval life in a marine fish. Proc Natl Acad Sci 105:1561–1566
Han G, Huang W (2008) Pacific decadal oscillation and sea level variability in the Bohai, Yellow, and East China seas. J Phys Oceanogr 38:2772–2783
Han ZQ, Gao TX, Yanagimoto T, Sakurai Y (2008) Deep phylogeographic break among white croaker Pennahia argentata (Sciaenidae, Perciformes) populations in North-western Pacific. Fish Sci 74:770–780
Hellberg ME, Burton RS, Neigel JE, Palumbi SR (2002) Genetic assessment of connectivity among marine populations. Bull Mar Sci 70:273–290
Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L (2013) The zebrafish reference genome sequence and its relationship to the human genome. Nature 496:498–503
Jeffery NW, DiBacco C, Van Wyngaarden M, Hamilton LC, Stanley RRE, Bernier R, FitzGerald J, Matheson K, McKenzie CH, Ravindran PN, Beiko R, Bradbury IR (2017) RAD sequencing reveals genomewide differentiation between independent invasions of the European green crab (Carcinus maenas) in the Northwest Atlantic. Ecol Evol 7:2513–2524
Johansen J, Jones G (2011) Increasing ocean temperature reduces the metabolic performance and swimming ability of coral reef damselfishes. Glob Change Biol 17:2971–2979
Kincade PW, Cooper MD (1971) Development and distribution of immunoglobulin-containing cells in the chicken: an immunofluorescent analysis using purified antibodies to µ, γ and light chains. J Immunol 106:371–382
Kong L, Kawasaki M, Kuroda K, Kohno H, Fujita K (2004) Spawning characteristics of the konoshiro gizzard shad in Tokyo and Sagami Bays, central Japan. Fish Sci 70:116–122
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinform 15:1–13
Krück NC, Innes DI, Ovenden JR (2013) New SNP s for population genetic analysis reveal possible cryptic speciation of eastern Australian sea mullet (Mugil cephalus). Mol Ecol Resour 13:715–725
Lasky JR, Des Marais DL, McKAY JK, Richards JH, Juenger TE, Keitt TH (2012) Characterizing genomic variation of Arabidopsis thaliana: the roles of geography and climate. Mol Ecol 21:5512–5529
Lecomte F, Grant WS, Dodson J, Rodríguez-Sánchez R, Bowen B (2004) Living with uncertainty: genetic imprints of climate shifts in East Pacific anchovy (Engraulis mordax) and sardine (Sardinops sagax). Mol Ecol 13:2169–2182
Lee T (1983) Age composition and reproductive period of the shad, Konoserus punctztus, in Cheonsu Bay. J Korean Oceanogr Soc 18:161–168 (in Korean)
Legendre P, Legendre L (2012) Ordination in reduced space. Dev Environ Model 24:425–520
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Li M, Shi S, Wang M, Lun Z, Yang T (2012) Complete mitochondrial genome of the dotted gizzard shad Konosirus punctatus (Teleostei, Clupeidae). Mitochondrial DNA 23:295–297
Liu B (2017) Population genetic structure and local adaptation of the small yellow croaker (Larimichthys polyactis) and Japanese eel (Anguilla japonica). Dissertation, Institute of Oceanology, Chinese Academy of Sciences
Liu X, Fu YX (2020) Stairway Plot 2: demographic history inference with folded SNP frequency spectra. Genome Biol 21:1–9
Liu JX, Gao TX, Yokogawa K, Zhang YP (2006a) Differential population structuring and demographic history of two closely related fish species, Japanese sea bass (Lateolabrax japonicus) and spotted sea bass (Lateolabrax maculatus) in Northwestern Pacific. Mol Phylogenet Evol 39:799–811
Liu JX, Gao TX, Zhuang ZM, Jin XS, Yokogawa K, Zhang YP (2006b) Late Pleistocene differentiation and subsequent population expansion of two closely related fish species, Japanese anchovy (Engraulis japonicus) and Australian anchovy (Engraulis australis). Mol Phylogenet Evol 40:712–723
Liu JX, Gao TX, Wu SF, Zhang YP (2007a) Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol Ecol 16:275–288
Liu M, Wang Y, Gao T, Yanagimoto T, Zhuang Z, Sakurai Y (2007b) Morphological comparison of Pacific herring Clupea pallasii in China and Japan. J Ocean Univ China 37:131–136
Liu B, Zhang K, Zhu K, Shafi M, Gong L, Jiang L, Liu L, Muhammad F, Lü Z (2020) Population genetics of Konosirus punctatus in Chinese coastal waters inferred from two mtDNA genes (COI and Cytb). Front Mar Sci 7:534
Liu BJ, Zhang K, Zhang SF, Liu YF, Li JS, Peng Y, Jin X, Wang YP, Zheng SX, Gong L, Liu LQ, Lyu ZM (2022a) Chromosome-level genome assembly of the dotted gizzard shad (Konosirus punctatus) provides insights into its adaptive evolution. Zool Res 43:217
Liu D, Gui L, Zhu Y, Xu C, Zhou W, Li M (2022b) Chromosome-level assembly of male Opsariichthys bidens genome provides insights into the regulation of the gnrh signaling pathway and genome evolution. Biology 11:1500
Lopes AF, Morais P, Pimentel M, Rosa R, Munday PL, Gonçalves EJ, Faria AM (2016) Behavioural lateralization and shoaling cohesion of fish larvae altered under ocean acidification. Mar Biol 163:1–10
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12
Maulvault AL, Santos LH, Paula JR, Camacho C, Pissarra V, Fogaça F, Barbosa V, Alves R, Ferreira PP, Barceló D (2018) Differential behavioural responses to venlafaxine exposure route, warming and acidification in juvenile fish (Argyrosomus regius). Sci Total Environ 634:1136–1147
McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT (2013) Applications of next-generation sequencing to phylogeography and phylogenetics. Mol Phylogenet Evol 66:526–538
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Meirmans PG, Hedrick PW (2011) Assessing population structure: FST and related measures. Mol Ecol Resour 11:5–18
Metz CW, Bridges CB (1917) Incompatibility of mutant races in Drosophila. Proc Natl Acad Sci 3:673–678
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17:240–248
Mohr OL (1919) Character changes caused by mutation of an entire region of a chromosome in Drosophila. Genetics 4:275
Molt S, Bührdel JB, Yakovlev S, Schein P, Orfanos Z, Kirfel G, Winter L, Wiche G, van der Ven PF, Rottbauer W (2014) Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J Cell Sci 127:3578–3592
Munday PL, Dixson DL, Donelson JM, Jones GP, Pratchett MS, Devitsina GV, Døving KB (2009) Ocean acidification impairs olfactory discrimination and homing ability of a marine fish. Proc Natl Acad Sci 106:1848–1852
Myoung SH, Kim JK (2014) Genetic diversity and population structure of the gizzard shad, Konosirus punctatus (Clupeidae, Pisces), in Korean waters based on mitochondrial DNA control region sequences. Genes Genomics 36:591–598
Nielsen EE, Hemmer-Hansen J, Poulsen NA, Loeschcke V, Moen T, Johansen T, Mittelholzer C, Taranger G-L, Ogden R, Carvalho GR (2009) Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua). BMC Evol Biol 9:1–11
Okamura O, Yamada U (1986) Fishes of the East China Sea and the Yellow Sea. Contrib Seikai Reg Fish Res Lab 422
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’hara R, Simpson G, Solymos P (2007) The vegan package. Community Ecol Package 10:719
Okubo K, Nagahama Y (2008) Structural and functional evolution of gonadotropin-releasing hormone in vertebrates. Acta Physiol 193:3–15
Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma RK, Hedrick PW (2010) Conservation genetics in transition to conservation genomics. Trends Genet 26:177–187
Pankhurst NW, Munday PL (2011) Effects of climate change on fish reproduction and early life history stages. Mar Freshw Res 62:1015–1026
Park SY, Kim JY, Yoon JM (2006) Geographic variations and DNA polymorphisms in gizzard-shad (Konosirus punctatus). Korean J Ichthyol 18:300–310
Peng Y, Li J, Zhang K, Liu Y, Li X, Zhang H, Gong L, Liu L, Lü Z, Liu B (2021) Identification of a large dataset of SNPs in hair-fin anchovy (Setipinna tenuifilis) based on RAD-seq. Anim Genet 52:371–374
Pickrell J, Pritchard J, Tang H (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8:e1002967
Pörtner HO, Knust R (2007) Climate change affects marine fishes through the oxygen limitation of thermal tolerance. Science 315:95–97
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Retief JD (2000) Phylogenetic analysis using PHYLIP. Bioinformatics methods and protocols. Springer, Berlin, pp 243–258
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sauer DJ, Heidinger BJ, Kittilson JD, Lackmann AR, Clark ME (2021) No evidence of physiological declines with age in an extremely long-lived fish. Sci Rep 11:1–10
Shanks AL, Grantham BA, Carr MH (2003) Propagule dispersal distance and the size and spacing of marine reserves. Ecol Appl 13:159–169
Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L (2018) The role of the hedgehog signaling pathway in cancer: a comprehensive review. Bosn J Basic Med Sci 18:8
Song N, Gao T, Ying Y, Yanagimoto T, Han Z (2016) Is the Kuroshio Current a strong barrier for the dispersal of the gizzard shad (Konosirus punctatus) in the East China Sea? Mar Freshw Res 68:810–820
Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR (2016) netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Resour 16:216–227
Sutachan JJ, Chao MV, Ninan I (2010) Regulation of inhibitory neurotransmission by the scaffolding protein ankyrin repeat-rich membrane spanning/kinase D-interacting substrate of 220 kDa. J Neurosci Res 88:3447–3456
Team RC (2013) R: a language and environment for statistical computing. Computing 1:12–21
Ulc A, Gottschling C, Schäfer I, Wegrzyn D, Sv L, Luft V, Reinhard J, Faissner A (2017) Involvement of the guanine nucleotide exchange factor Vav3 in central nervous system development and plasticity. Biol Chem 398:663–675
Vousden KH, Lu X (2002) Live or let die: the cell’s response to p53. Nat Rev Cancer 2:594–604
Waits L, Sork V (2010) Landscape genetics. Mol Ecol 19:3489–3835
Wampler PJ, Rediske RR, Molla AR (2013) Using ArcMap, Google Earth, and global positioning systems to select and locate random households in rural Haiti. Int J Health Geogr 12:1–8
Wang P (1999) Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Mar Geol 156:5–39
Weng AP, Ferrando AA, Lee W, Morris JP IV, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306:269–271
Wu L, Qin Z, Liu H, Lin L, Ye J, Li J (2020) Recent advances on phagocytic B cells in teleost fish. Front Immunol 11:824
Yang M, Wu J, Zhang L, Huang Y, Zi Y, Zhang Y (2017) Effects of miR-143 on invasion and migration of B cell lymphoma cells through regulating expression of PAN3 protein. Chin J Immunol 33:1774
Yao S, Bo Z, Xuewu G, Jun W, Qisheng T (1999) Feeding, growth and ecological conversion efficiency of Konosirus punctatus—field gastric content method in vivo. Mar Fish Res 20:12–16 (in Chinese)
Ying Y (2011) Population genetic structure and molecular phylogeography of Konosirus punctatus and Sardinella zunasi. Dissertation, Ocean University of China
Yokogawa K (1997) Morphological and genetic differences between Japanese and Chinese Red Ark Shell (Scapharca broughtonii). Fish Sci 63:332–337
Yoo DG, Lee GS, Kim GY, Kang NK, Yi BY, Kim YJ, Chun JH, Kong GS (2016) Seismic stratigraphy and depositional history of late Quaternary deposits in a tide-dominated setting: an example from the eastern Yellow Sea. Mar Pet Geol 73:212–227
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287
Zhang S (2001) Fauna sinica: Osteichthyes, Acipenseriformes, Elopiformes, Clupeiformes, Gonorhynchiformes. Science Press
Zhang BD, Xue DX, Wang J, Li YL, Liu BJ, Liu JX (2016) Development and preliminary evaluation of a genomewide single nucleotide polymorphisms resource generated by RAD-seq for the small yellow croaker (Larimichthys polyactis). Mol Ecol Resour 16:755–768
Zhang BD, Xue DX, Li YL, Liu JX (2019) RAD genotyping reveals fine-scale population structure and provides evidence for adaptive differentiation in a commercially important fish from the northwestern Pacific Ocean. PeerJ 7:e7242
Zhao Q (2018) A study on optimizing markduplicate in genome sequencing pipeline. In: Proceedings of the 2018 5th international conference on bioinformatics research and applications, pp 8–15
Zhou Y (2011) Function research of Kidins220/ARMS on the mouse embrvonic stem cells. Dissertation, Zhejiang University
Zhou H, Qian J, Li C, Li J, Zhang X, Ding Z, Gao X, Han Z, Cheng Y, Liu L (2010) Attenuation of cardiac dysfunction by HSPA12B in endotoxin-induced sepsis in mice through a PI3K-dependent mechanism. Cardiovasc Res 89:109–118
Zulliger D, Schnyder E, Gugerli F (2013) Are adaptive loci transferable across genomes of related species? Outlier and environmental association analyses in Alpine Brassicaceae species. Mol Ecol 22:1626–1639
Acknowledgements
The study was supported by the National Natural Science Foundation of China (NSFC) (No. 41806156); Zhejiang Provincial Natural Science Foundation of China (LY22D060001&LY20C190008); Science and Technology Project of Zhoushan (2020C21016). Fundamental Research Funds for Zhejiang Provincial Universities and Research Institutes (No. 2021J008). Fund of Guangdong Provincial Key Laboratory of Fishery Ecology and Environment (FEEL-2021-8).
Author information
Authors and Affiliations
Contributions
YP, YFL, and BJL conceived and designed the research. YP, YFL, BJL, JSL, KZ, XJ, SXZ, YPW, ZML, LQL, and LG conducted experiments, analyzed data, and wrote the manuscript. The authors critically reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors do not have any conflicts of interest to declare.
Animal and human rights statement
All international, national, and institutional guidelines for the care and use of animals were followed.
Additional information
Edited by Jiamei Li.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, Y., Liu, Y., Li, J. et al. New perspectives on the genetic structure of dotted gizzard shad (Konosirus punctatus) based on RAD-seq. Mar Life Sci Technol 6, 50–67 (2024). https://doi.org/10.1007/s42995-024-00216-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-024-00216-2