
Abstract
Hydroxyl-terminated polybutadiene (HTPB) has long been used as a binder in propellants and explosives. However, cured HTPB polyurethanes have not been characterized in a systematic fashion as a function of plasticizer content. In this study, three isocyanate-cured HTPB variants with different amounts of plasticizer were formulated. The materials were characterized across a range of strain rates from 10−3 to 106 s−1. Group interaction modeling (GIM) was used to predict the material behavior based on the underlying structure of the polymer. Increasing the amount of plasticizer was found to reduce the strength of the material across all strain rates. GIM was found to overpredict the modulus but predicted the shock response very well.
Similar content being viewed by others

Introduction
Hydroxyl-terminated polybutadiene (HTPB) resin is used as the basis of a family of rubbery polyurethanes with varying amounts of plasticizer and curing agent, which results in materials with varying glass transition temperature (Tg). This variation in Tg allows for materials to vary from rigid to highly flexible at room temperature. HTPB is a crosslinked elastomer, containing the main, crosslinked network (gel) and the material not attached to the network (sol). The addition of plasticizer to the system not only changes the Tg, but also results in a change in the way the network forms, not just the density of crosslinks. Therefore most mechanical properties are expected to be affected. This wide range of available properties results in HTPB being used in a wide range of applications, such as adhesives, elastomeric wheels and tires, and binders for rocket propellants and explosives.
There have been limited studies on the mechanical response of HTPB in the literature. Understanding of the mechanical response across strain rates and temperatures is critical for developing constitutive models for this material, which allows for mesoscale mechanical modeling of composites, such as polymer bonded explosives (PBXs). Studies investigating the quasi-static mechanical properties as a function of composition [1–7] have demonstrated that the mechanical properties of HTPB can be modified by the choice of isocyanate and chain extenders [2, 6] and also by the degree of crosslinking strongly correlates with the strength [4, 5].
Studies of the high rate compressive response, critical for energetic materials, are much more limited [8, 9]. For the split Hopkinson pressure bar technique, the low impedance of HTPB results in a testing challenge, particularly in ensuring the sample is in equilibrium. However, a few studies have been performed using cylindrical samples, and the strength of HTPB was found to increase with increasing strain rate and decreasing temperature [8, 9]. For the most part, these studies have focused on a particular HTPB composition. The shock response of HTPB has also been investigated in a limited way. A previous study indicated that the Hugoniot of HTPB is dependent on formulation, although in the work cited the complete formulation data for only one HTPB variant is given [10]. In contrast, in epoxy, another crosslinked polymer, it has been shown that the Hugoniot is not dependent on the degree of crosslinking or the specifics of the polymer formulation [11].
Group Interaction Modeling (GIM) is a mean-field group contribution method that has been demonstrated to predict the properties of homopolymers well [12, 13]. The method takes the groups, usually mer units, involved in interactions within the polymer and sums their contributions to physical parameters such as cohesive energy, vibrational degrees of freedom, and van der Waals volume in order to calculate energy stored and dissipated through thermomechanical loading. The ability to deal with relaxation allows predictions to be made both above and below the glass transition. The developments needed for plasticized, networked polymers center around predicting the interactions that occur within the material and how they contribute to the overall loss tangent.
In this study, three HTPB variants were prepared with different amounts of plasticizer ranging from 0 to 64 wt%. The compressive mechanical properties were investigated across a range of strain rates (10−3 to 106 s−1). Shock experiments on two of the materials were conducted in order to determine whether the crosslinking and formulation details affect the Hugoniot. GIM is used to predict these properties of the three HTPB variants based on the underlying polymer structure.
Experimental Approach
Materials
Three HTPB-based polymers were prepared using varying amounts of plasticizer, Dioctyl Adipate, as described in Table 1, in comparison with other HTPB-based materials from the literature [8–10]. This was done to systematically vary the mechanical properties of the polymers to such a degree that the inherent properties changed dramatically between materials. For the dynamic mechanical analysis (DMA) and quasi-static and high strain rate compression experiments, the polymer was cast into greased molds of the same size as the desired sample, so that cutting was not necessary. The density of each sample was measured using helium pyncnometry and is included in Table 1. The measured density increased with increasing percent total plasticizer content.
Mechanical Characterization
DMA was performed using a thin film configuration at 1, 10, and 100 Hz over temperatures from 150 to 300 K. The analysis was performed using a TA Instruments Q800 Dynamic Mechanical Analyzer with nominal sample geometries of 30 mm (L), 8 mm (W), and 2 mm (T). During a DMA test, a small polymer sample is subjected to a sinusoidal stress of a certain frequency at a certain temperature and the resultant sinusoidal displacement is measured. For viscoelastic materials, there is a phase difference between the applied stress and measured strain, which is used to determine the storage and loss moduli and the loss tangent, tan δ, of the polymeric material. The loss tangent is the ratio of the storage modulus to the loss modulus, and peaks in the loss tangent can correspond to phase changes in the material, e.g. the glass transition [14].
Compression experiments were performed across a range of strain rates from 10−4 to 103 s−1 at room temperature. For the low strain rate experiments, an Instron model 55R4201 with a 100 N load cell was used for quasi-static loading, in which the cylindrical samples were nominally 12.7 mm diameter by 12.7 mm long. The strain in the sample was determined by tracking the relative displacement of two dots nominally 3 mm above and below the sample centerline with Instron’s Advanced Video Extensometer (model 2663-821), and the stress was determined from the load cell output. The strain rates tested were 3 × 10−2, 3 × 10−3, and 3 × 10−4 s−1. All data was acquired using Instron’s Bluehill 2.15 software.
The high strain rate compression experiments (~2 × 103 s−1) were conducted using a split Hopkinson pressure bar (SHPB) system located at AFRL/RWME, Eglin AFB, FL [15]. The 6061-T6 aluminum incident and transmitted bars were 1524 mm long and 19.05 mm diameter. The striker is 610 mm long and made of the same material as the other bars. The samples were nominally 12.3 mm diameter and 7 mm long, where the sample size was chosen to minimize the axial and radial inertial effects [16, 17] and to ensure the samples could be manufactured. The length to diameter ratios for our samples is 0.55, which is close to the ideal ratio of \(\frac{\sqrt 3 }{4}\) as given by Ref. [17]. However, it is recognized that, due to the ability to manufacture the samples, these samples are fairly thick for testing these types of materials [18]. They are positioned between the incident and transmitted bars. The bar faces are lightly lubricated with grease to reduce friction. A thin copper pulse shaper was used to control the input pulse to the sample. The bar system uses Kulite semiconductor strain gages (Kulite AFP-500-90) in a voltage divider to acquire the incident and transmitted signals. The voltage pulses are converted to force using a measured calibration [15]. The forces calculated from the input and reflected waves (two wave analysis) were compared with the results from the transmitted wave (one wave analysis) during each experiment to ensure that equilibrium was reached in the samples. It was observed that the two-wave analysis oscillated about the one-wave analysis, an indication of equilibrium in the sample, by approximately 5 % strain. All SHPB data presented in this paper were corrected for dispersion using a post-processing dispersion correction algorithm [19, 20].
Shock Characterization
Planar impact experiments were conducted to determine if the relative amount of plasticizer affected the Hugoniot of the cured HTPB. The materials chosen were the HTPB0, containing no plasticizer, and HTPB2, containing the most plasticizer. A 60 mm powder gun at AFRL/RWMW, Eglin AFB, FL was used to impact the HTPB samples in a rough vacuum (~100 mtorr) at impact velocities ranging from 300 to 1400 m/s. In all cases, the impactor was a 6061 aluminum disc 4 mm thick and 55 mm diameter. As shown in the schematic in Fig. 1, the target was composed of several layers. The first layer was a 2 mm thick 6061 Al disc, 46 mm diameter. The second layer was the HTPB sample, cut from a ~ 4 mm thick cast sheet into samples roughly 37 mm diameter. The last layer was a PMMA disc, 12.7 mm thick, which was coated with a thin (<1 μm) layer of aluminum on one side to serve as a reflector for the laser-based velocity measurement diagnostics.

Schematic of the experimental configuration for the impact experiments. On the left is the front of the projectile, which is moving at the impact velocity. On the right is the layered target, which is stationary prior to impact. The various velocity probe locations are shown. Note that the spacer ring was only used for dimensional stability of the soft samples and was not used for the hard samples
Due to the extreme compliance of the softer HTPB2 relative to that of the harder HTPB0, two different sample setups were used. For the soft material, a confining cell was necessary to ensure dimensional accuracy, and so an aluminum spacer, just thinner than the HTPB2 sample, was used to “force” the sample to the thickness of the spacer. This spacer was glued to both the 2 mm Al disc and the PMMA disc. For the hard HTPB0 samples, no spacer was needed, and so the HTPB0 was glued directly to the 2 mm Al disc and the PMMA disc. The experimental setup for the softer material (including the spacer) is illustrated in Fig. 1. As shown, the inner diameter of the spacer was larger than the sample to allow for slight deformation of the sample, and the spacer was vented to allow for evacuation of the sample chamber. The compression of the soft samples when enclosed in the cell was approximately 100 μm.
The incoming projectile velocity was measured using PDV 1 and 2 [21]. The motion of the aluminum plate (the first layer of the target) was also monitored using PDV at location PDV 3. Similarly, the mirror at the interface between the HTPB and the PMMA was monitored using both PDV at location PDV 4 and VISAR [22]. The tilt of the impactor relative to the target was also measured using four shorting pins (not shown in Fig. 1).
Group Interaction Modeling of HTPB
GIM for predicting properties of polymers based upon their chemical composition and microstructure has been given by Porter [12], and this reference also contains estimates of the parameters for butadiene mer units. The initial prediction of glass transition temperature for trans 1,4-poly(butadiene) was not adequate, so a modification was made to the value of cohesive energy E coh . The parameters used for the predictions are given in Table 2. For IPDI, the parameters relate to the networked molecule. The glass transition temperature, T g , given in Table 2 was calculated from the parameters according to Eq. 1 for a frequency of 1 Hz [12]:
where θ 1 is the one-dimensional Debye temperature for skeletal modes in the Tarasov model, E coh is the cohesive energy, and N is the number of skeletal modes associated with the mer or molecule. The other parameters in Table 2 are: the mass of the mer or molecule, the van der Waals volume, V w , and the number of vibrations normal to the chain axis, N c . All contributions to loss tangent are presumed to vary with rate according to a Vogel-Fulcher response as set out in [13] apart from the plasticizer, which appears not to have a rate dependence for its T g [23].
The average HTPB parameters were calculated by summing the contributions from a typical HTPB chain and dividing by the number of mer units expected in that chain. Details of the composition of a typical HTPB chain will vary depending on source and can be inferred from information provided by the manufacturer. In order to understand interactions within the material, these components need to interact as units of equivalent size. One DOA molecule is approximately the same volume as two IPDI molecules and seven average HTPB mers, which will inform the units chosen. It is presumed that there are sufficient molecules in the samples chosen for the probabilities of interaction to be interpreted as number and volume fractions.
Taking a simple approach to loss, each unit within the material is presumed to interact on average with six other units and they share a glass transition which is represented by a Gaussian with width 6 K. The Gaussians from all the interactions are summed to calculate the final loss tangent spectrum as a function of temperature. The composition and curing strategy needs to inform the probabilities of interaction. Plasticizer molecules are presumed to be ubiquitous and homogeneously distributed so will take part in all interactions where plasticizer is present. The network will have three types of interaction volume: volumes around IPDI molecules that link HTPB chains together; volumes far from chain ends where there is no IPDI involvement; and volumes associated with the disconnected chain ends that characterize the sol fraction. The contribution from disconnected chain ends needs to be understood as it will be different in two respects: the chain end has an extra six degrees of freedom relative to the network and the chain will terminate either in a hydroxyl group or in an IPDI group. Table 3 gives the group parameters for each of these units. It can be seen that the T g of an HTPB-OH unit is not significantly different than the average networked HTPB and so this contribution will be neglected. The model requires the number of unconnected ends in order to determine the volume fraction of interactions that include these ends. This should be able to be measured by solid state NMR but, for this study, needs to be inferred from the likely sol fraction.
In order to understand how the loss tangent spectrum as a function of temperature is built up, the range over which particular molecule shares energy and, thus, the volume of interaction must be understood. In the absence of measurements or values derived from molecular mechanics, this is essentially a fitting parameter for a particular molecule. If the molecule only shared energy with its six nearest neighbors, then the interaction volume would be a factor four larger than the volume of the molecule. When predicting the properties of the unplasticized HTPB, it was found that a factor seven was the most appropriate interaction volume, which was used consistently for all interaction volumes.
For HTPB0, the sol fraction will be around 5 % by mass [23]; if it is presumed that this is made from isolated HTPB molecules with all reaction sites unconnected then, for every 100 HTPB molecules containing 1460 HTPB units, there will be approximately 15 free IPDIs and 110 network IPDIs. This splits into three volumes: a volume that includes a network IPDI in the set of seven interacting units; a volume that includes a free IPDI unit in the set of interacting units and a volume in which all interacting units are HTPB alone. If the interaction volume around a network IPDI is a factor seven times the volume of the IPDI unit then these three volumes will split into:
-
110 network IPDIs + 8 free IPDIs + 740 HTPB units.
-
7 free IPDIs + 50 HTPB units.
-
670 HTPB units.
In the first of these volumes, the central network IPDI is presumed to be surrounded by six random neighboring units so there are: n1 other network IPDIs, n2 free IPDIs and n3 HTPB units. The probability of site occupancy is the volume fraction. Each combination of seven interacting units from the set that makes up the interaction volume has a loss profile which is presumed to be Gaussian in temperature space centered on its own T g,i with width, i.e. standard deviation, s = 6 K and area tanΔ g,i . These are calculated via:
All of the contributions from all of the combinations of seven interacting units are summed to give the final prediction of loss tangent. This process is then carried out for the combinations that make up the other independent volumes to generate a final loss tangent spectrum as a function of temperature. For the compositions that contain plasticizer the same process is followed but there will be a different sol fraction. As well as shifting loss peaks to lower temperature the other main effect of plasticizers on loss tangent is to increase the area under the spectrum. This is interpreted as extra loss caused by liquid-like interactions between plasticizer molecules. This is represented in the GIM model by neglecting the plasticizer contribution to the degrees of freedom in Eq. 3.
Having predicted the loss tangent spectrum as a function of temperature and rate, the other mechanical properties are calculated from that loss, the thermal and mechanical energies and moduli as set out in [13]. The response is presumed to be dominated by the HTPB interactions. The instantaneous elastic bulk modulus, B e , due to the thermal energy density is subjected to dissipation via the development of glass transition degrees of freedom, ΔN g , and Young’s modulus, E, then develops via tan Δ g,i as in:
Results and Discussion
Dynamic Mechanical Analysis
The three materials were investigated using DMA to understand the effect of plasticizer on the glass transition temperature. The storage modulus as a function of temperature for all three materials is shown in Fig. 2a. Each material was tested three times, and it can be seen in Fig. 2a that there is considerable sample-to-sample variation for a given material. However, there is a trend above the glass transition temperature that increasing the plasticizer results in decreased storage modulus, which is expected since increasing the plasticizer results in a softer, more flexible material. Additionally, as shown in Fig. 2b, increasing the plasticizer, or decreasing the resin, results in reduced glass transition temperature.

a Dynamic Mechanical Analysis comparing the storage modulus for the three HTPB materials, where each material was replicated 2–3 times; b glass transition as a function of temperature for the experimental data in comparison to GIM calculations; and c comparison of tan δ between experiment and GIM
GIM predictions of the temperature of the main peak are outside the measurements by about 3 % for all three resins, as shown in Fig. 2c, underpredicting the T g for all three materials. The temperature of this peak is easily affected at this accuracy by the averaging process that led to the HTPB unit T g : slightly fewer or more cis-butadiene contributions to the chain can lead to such variation. The area under the main peak is significantly overestimated for HTPB2 and slightly overestimated for HTPB1 which suggests that the method proposed for including loss effects from plasticizers needs more attention and would benefit from a more systematic study. The loss spectrum for HTPB0 is in better agreement than the other two materials, corroborating the argument that the plasticizer treatment may be the largest source of error. It is also worth examining the loss at temperatures above the main peak which GIM ascribes to interactions with the IPDI molecules; for HTPB0 this part of the loss tangent is well predicted. The plasticized resins are less well predicted with the fraction of interactions that include the IPDI groups being underestimated. This aspect of GIM: the relative contributions of the various interactions and the combinations of interactions that lead to predictions, should be able to be validated using techniques such as solid-state NMR that directly interrogate the chemical environment that the groups find themselves within.
Compressive Response
The three HTPB polymers were tested at three quasi-static strain rates using an Instron load frame (3 × 10−2, 3 × 10−3, and 3 × 10−4 s−1) and one high strain rate using a split Hopkinson pressure bar (2.5 × 103 s−1). HTPB 1 was not tested at 3 × 10−3 s−1. Figure 3a shows the response of the materials at the highest quasi-static rate, and Fig. 3b shows the response of the three polymers at the highest strain rate tested. Increasing the amount of plasticizer resulted in decreased strength in the material. There is considerable scatter in the data, as seen from Fig. 3b, which may be due to both variations in given specimens tested as well as error associated with testing such soft materials. The scatter is not likely due to a lack of equilibrium in the sample, as the forces on the surfaces of the sample were in equilibrium over the vast majority of the experiment.

a Quasi-static (~5 × 10−1 s−1) and b dynamic (~2.5 × 103 s−1) compressive stress strain response of three HTPB materials with varying amounts of plasticizer
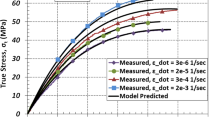
It can be seen from Fig. 4 that increasing the strain rate resulted in an increased strength in the material, and, from Fig. 3, an increase in the slope of the stress–strain curve. At low strain rates, there is little strain rate dependence. However, at dynamic strain rates, the measured stress increases greatly over what would be expected from an extrapolation from the quasi-static data. The experimental data from this study is in general agreement with that from Cady et al. [8] and Siviour et al. [9], in that there is increased stress with increased strain rate. However, although the Cady material has a similar amount of plasticizer to HTPB1 and the Siviour material is between HTPB0 and HTPB1, there is not agreement with the measured stress values. This discrepancy likely results from the differences in the types plasticizers and isocyanates used in those materials versus the HTPB in this study. Additionally, as the DMA experiments demonstrated, there can be considerable sample-to-sample variation even within a given material.

From Fig. 4, it can be seen that the GIM predictions for all resins overestimate the strength apart from at the highest rates which, given the significant overprediction of the loss tangent for HTPB2, is surprising. The predictions are derived for loading in tension, and thus the Poisson’s ratio is used to convert to compressive stress state, which may be a cause of the overprediction. Another factor that may be important is a potential underestimate of the presence of liquid-like interactions in the plasticizer and in the sol fraction. The predictions also represent ideal behavior in the polymer and take no account here of effects of distance between crosslinks and lower molecular weight fractions.
It is also obvious that the increase in stress at high strain rates is not captured by GIM for HTPB. The only method within GIM for an increase in modulus, following classical polymer physics, is a reduction in total loss caused by movement of the loss tangent spectrum. Studies on PMMA and PC [13] did capture an increase in stress at high rates; however, this was due to the shift in the beta transition peaks, which are not present in HTPB causing significant change in loss tangent to affect the mechanical properties. Shifting the stress by the difference seen in Fig. 4 would require an unphysical peak shifting of 20 K/decade, since most loss occurs at such low temperatures in these materials. It would be useful to be able to simulate the gauge traces directly using numerical simulation to better understand the effect of different loading conditions on the material, which adds additional complexity because a satisfactory constitutive model would need to be employed in finite element codes.
Shock Response
A total of five shots were performed to determine the shock response of the HTPB using the experimental setup previously described. PDV and VISAR traces were obtained for most experiments. On those experiments where traces were obtained, the VISAR and PDV probe on the HTPB/PMMA interface showed sharp rises to a well-defined constant velocity, indicating little to no viscous stress relaxation. On FY13-40, the data was truncated and so only arrival times could be determined. Representative traces are shown for both of the shots on soft HTPB2, FY13-43 and FY13-39, in Fig. 5.

Representative interferometry traces for two experiments conducted on HTPB2. The incoming velocity is clearly seen in PDV 1 and 2, and the arrival times and steady-state velocities are clearly observed in the PDV 3, 4, and VISAR. The slight structure observed in the beginning FY13-43 PDV 3 is due to the elastic precursor in 6061 Al; in FY13-39 it is overdriven. The velocities shown for VISAR and PDV are uncorrected for index of refraction effects in PMMA
The speed with which the pressure front propagates through the material is commonly referred to as the shock velocity, US. Shock velocity was determined from the transit time across the sample, as found from the arrival of the compressive wave at PDV3 and PDV4, with slight corrections to account for the tilt at impact. Then, using the measured initial density and impact velocity, the known Hugoniot for 6061 Al, and standard impedance matching techniques [24], the material velocity u p and stress σ were calculated. The values used for the Hugoniot of 6061 Al in the impedance matching were density of 2.70 g/cc, C o = 5.35 km/s, S = 1.3 [25]. Relevant measured and calculated values are shown in Table 4.
The U S -u P values are plotted in Fig. 6 below, along with linear fits to the data. The measured longitudinal acoustic wave speeds are also shown (shear wave speeds could not be determined) and are included in the linear fit. The seemingly reasonable agreement between the U S intercept and the measured longitudinal sound speed, the inability to obtain a shear wave speed, and the obvious compliance of the materials suggests that a the material might be adequately described as a liquid. A universal liquid Hugoniot has been previously reported [26] but underpredicts the data, as shown in the figure.

US-uP data for both the soft and hard HTPB formulations. The measured longitudinal acoustic sound speed is also shown and is incorporated into the linear fits shown in the figure as dashed lines. The Universal Liquid Hugoniot is also shown as solid lines for comparison, and slightly underpredicts the data in both cases. Comparison with GIM results (dashed grey line) is included and shows good agreement with HTPB 0. Data from the literature [10] is also included
It can be seen from Fig. 6 that the measured difference in the acoustic sound speed is maintained as an offset in the U S -u P Hugoniot across the range investigated in this work. Experimental data from the literature [10] is also shown for comparison. It is apparent in Fig. 6 than the formulation described in the literature as “Millett HTPB 2”{[10] agrees very well with the HTPB0 formulation containing no plasticizer, which is to be expected as the formulations are very similar as shown in Table 1. The “Millett HTPB 1” data [10] is a proprietary formulation, and so it is not clear how it differs from “Millett HTPB 2” or the formulations described in the present work. However, it is clear that the two formulations from Ref. [10] differ in ways that are distinct from the differences in the formulations described in the present work. In the present work, the aforementioned offset is maintained, whereas in the two formulations from the literature, no offset is present, but instead the slope of the U S -u P data differs significantly.
The GIM prediction of the data is excellent particularly given that it did not require knowledge of the mechanical properties to generate the prediction. This ties in well with other predictions of equation of state parameters for polymers previously predicted by GIM [27]. The wave speed at u p = 0 m/s is also predicted well and the curvature of the locus is a natural part of the interaction physics embodied in GIM. The lack of substantial difference in the data for plasticized and unplasticized HTPB can also be understood from the GIM predictions: the response is dominated by molecular interactions between trans-butadiene mer units far from the glass transition and these predominate not only in the main HTPB chains but also in the plasticizer. Dissipation on a macromolecular level is less important. It would be interesting to repeat these shock experiments at temperatures around the glass transition to see if there is better discrimination between the plasticized and unplasticized resins.
Conclusions
Although commonly used in a variety of defense and civilian applications, the mechanical response of HTPB has not been systematically investigated. In this study, three materials based on HTPB resin with varying amounts of plasticizer were characterized in compression across a range of strain rates (10−3 to 106 s−1). As expected, increasing the plasticizer resulted in decreased strength and decreased glass transition temperature. Increasing the plasticizer also resulted in decreased shock velocity for a given particle velocity. However, the difference appeared to be constant over the velocity regime tested indicating that this difference may be due to the difference in density between the two materials tested. GIM was employed to model the variation in material properties with varying amounts of plasticizer, and this study indicates the level of confidence that may be ascribed for different HTPB resins with differing amounts of plasticizer. Overall, Young’s modulus, and subsequently stress, and loss tangent are overpredicted but the shock response is very well predicted. Additional work on understanding these complex polymer materials using GIM is warranted to provide predictive capability for these materials where manufacturing and testing many sample variations is prohibitively expensive.
References
Chen TK, Hwung CJ, Hou CC (1992) Effects of number-average molecular weight of network chain on physical properties of cis-polybutadiene-containing polyurethane. Polym Eng Sci 32(2):115–121
Vilar W, Akcelrud L (1995) Effect of HTPB structure on prepolymer characteristics and on mechanical properties of polybutadiene-based polyurethanes. Polym Bull 35(5):635–639
Panicker SS, Ninan K (1997) Effect of reactivity of different types of hydroxyl groups of HTPB on mechanical properties of the cured product. J Appl Polym Sci 63(10):1313–1320
Haska SB et al (1997) Mechanical properties of HTPB-IPDI-based elastomers. J Appl Polym Sci 64(12):2347–2354
Sekkar V et al (2000) Polyurethanes based on hydroxyl terminated polybutadiene: modelling of network parameters and correlation with mechanical properties. Polymer 41(18):6773–6786
Wingborg N (2002) Increasing the tensile strength of HTPB with different isocyanates and chain extenders. Polym Testing 21(3):283–287
De La Fuente JL, Fernández-García M, Cerrada ML (2003) Viscoelastic behavior in a hydroxyl-terminated polybutadiene gum and its highly filled composites: effect of the type of filler on the relaxation processes. J Appl Polym Sci 88(7):1705–1712
Cady C et al (2006) Mechanical properties of plastic-bonded explosive binder materials as a function of strain-rate and temperature. Polym Eng Sci 46(6):812–819
Siviour CR et al (2008) High strain rate properties of a polymer-bonded sugar: their dependence on applied and internal constraints. Proc R Soc 464(2093):1229–1255
Millett J, Bourne N, Akhavan J (2004) The response of hydroxy-terminated polybutadiene to one-dimensional shock loading. J Appl Phys 95(9):4722–4727
Munson DE, May RP (1972) Dynamically Determined High-Pressure Compressibilities of Three Epoxy Resin Systems. J Appl Phys 43(3):962–971
Porter D (1995) Group Interaction Modelling of Polymer Properties. Marcel Dekker, New York
Porter D, Gould PJ (2009) Predictive nonlinear constitutive relations in polymers through loss history. Int J Solids Struct 46(9):1981–1993
Foreman J (1997) Dynamic mechanical analysis of polymers. TA Instruments, New Castle
Jordan JL, Foley JR, Siviour CR (2008) Mechanical properties of Epon 826/DEA epoxy. Mech Time Depend Mater 12(3):249–272
Gorham D (1989) Specimen inertia in high strain-rate compression. J Phys D Appl Phys 22(12):1888
Chen WW, Song B (2010) Dynamic characterization of soft materials. In: Shukla A, Ravichandran G, Rajapakse YDS (eds) Dynamic failure of materials and structures. Springer, New York, pp 1–28
Song B, Chen W (2004) Dynamic stress equilibration in split Hopkinson pressure bar tests on soft materials. Exp Mech 44(3):300–312
Gorham D (1983) A numerical method for the correction of dispersion in pressure bar signals. J Phys E 16(6):477
Bacon C (1998) An experimental method for considering dispersion and attenuation in a viscoelastic Hopkinson bar. Exp Mech 38(4):242–249
Strand OT et al (2006) Compact system for high-speed velocimetry using heterodyne techniques. Rev Sci Instrum 77:083108
Barker LM, Hollenbach RE (1972) Laser interferometer for measuring high velocities of any reflecting surface. J Appl Phys 43(11):4669–4675
Lawrence A (2014) Sol fraction, P. Gould, Editor
Forbes JW (2012) Shock wave compression of condensed matter. Springer, Berlin
Meyers MA (1994) Dynamic behavior of materials. Wiley, New York, p 668
Woolfolk RW, Cowperthwaite M, Shaw R (1973) A Universal Hugoniot for Liquids. Thermochim Acta 5:409–414
Porter D, Gould P (2006) A general equation of state for polymeric materials. J Phys IV 134:373–378
Acknowledgments
This work was funded by the Munitions Directorate, Air Force Research Laboratory. The authors would like to thank Mr. Tomislav Kosta for his assistance with testing. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the United States Air Force.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jordan, J.L., Montaigne, D., Gould, P. et al. High Strain Rate and Shock Properties of Hydroxyl-Terminated Polybutadiene (HTPB) with Varying Amounts of Plasticizer. J. dynamic behavior mater. 2, 91–100 (2016). https://doi.org/10.1007/s40870-015-0044-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40870-015-0044-0