
Abstract
Introduction
Upadacitinib (UPA) is a Janus kinase inhibitor that has demonstrated efficacy in moderate-to-severe rheumatoid arthritis (RA) with an acceptable safety profile. We investigated laboratory parameter changes in UPA RA clinical trials.
Methods
Pooled data from six randomized trials in the SELECT phase 3 program were included. Key laboratory parameters and safety data were measured for UPA 15 and 30 mg once daily (QD), adalimumab (ADA) 40 mg every other week + methotrexate (MTX), and MTX monotherapy. Exposure-adjusted event rates (EAERs) of adverse events were calculated.
Results
A total of 3209 patients receiving UPA 15 mg QD (10 782.7 patient-years [PY]), 1204 patients receiving UPA 30 mg QD (3162.5 PY), 579 patients receiving ADA + MTX (1573.2 PY), and 314 patients receiving MTX monotherapy (865.1 PY) were included, representing up to 6.5 years of total exposure. Decreases in mean levels of hemoglobin, neutrophils, and lymphocytes, and increases in mean levels of liver enzymes and creatinine phosphokinase were observed with UPA, with grade 3 or 4 changes observed in some patients. Mean low- and high-density lipoprotein cholesterol ratios remained stable for patients receiving UPA 15 mg QD. EAERs of anemia and neutropenia occurred at generally consistent rates between UPA and active comparators (3.1–4.3 and 1.7–5.0 events [E]/100 PY across treatment groups, respectively). Rates of hepatic disorder were higher with MTX monotherapy, UPA 15 mg and UPA 30 mg (10.8, 9.7, and 11.0 E/100 PY, respectively) versus ADA + MTX (6.4 E/100 PY). Rates of lymphopenia were highest with MTX monotherapy (3.2 E/100 PY). Treatment discontinuations due to laboratory-related events were rare, occurring in 1.1% and 2.2% of patients treated with UPA 15 and 30 mg QD, respectively.
Conclusions
The results of this integrated long-term analysis of laboratory parameters continue to support an acceptable safety profile of UPA 15 mg QD for moderate-to-severe RA.
Similar content being viewed by others


Why carry out this study? |
Upadacitinib is a Janus kinase (JAK) inhibitor that has demonstrated efficacy for the treatment of moderate-to-severe rheumatoid arthritis (RA) across a broad spectrum of patients in the SELECT phase 3 program, with an acceptable safety profile. |
Consistent with the known safety profile of JAK inhibitors, changes in laboratory parameters have been observed in upadacitinib clinical trials, although further long-term data are needed. |
What was learned from the study? |
Long-term upadacitinib treatment was associated with several dose-dependent laboratory abnormalities in patients with RA, with a comparable incidence to those with methotrexate and a generally higher incidence than with adalimumab. |
Results continue to support an acceptable safety profile of upadacitinib 15 mg once daily for the treatment of moderate-to-severe RA. |
Introduction
The goal of therapy for patients with rheumatoid arthritis (RA) is to achieve sustained remission or low disease activity [1, 2]. For patients who fail to achieve these goals with first-line therapy, typically conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs), treatment options include biologic DMARDs (bDMARDs) such as tumor necrosis factor (TNF) inhibitors, or Janus kinase (JAK) inhibitors [2]. Upadacitinib is a JAK inhibitor that has demonstrated efficacy in RA with an acceptable safety profile; after 24/26 weeks of upadacitinib 15-mg treatment, approximately one-quarter of patients with inadequate response to methotrexate (MTX)/csDMARDs achieved clinical remission (Clinical Disease Activity Index [CDAI] ≤ 2.8) [3,4,5,6,7,8,9,10]. While the management of RA should include regular monitoring of disease activity in accordance with treat-to-target principles, consideration of patient safety is equally important, with monitoring of adverse events (AEs) and commonly tested laboratory parameters.
JAK inhibitors are orally administered small molecule drugs that have been shown to be effective for the treatment of moderate-to-severe RA in multiple randomized controlled trials and are recommended treatment options for patients who fail to respond to csDMARDs and/or bDMARDs [2, 11, 12]. JAK inhibitors have been associated with changes in laboratory parameters including decreases in lymphocytes, neutrophils, and hemoglobin, increases in the liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST), increases in serum creatinine and creatine phosphokinase (CPK), and increases in blood cholesterol [12,13,14]. As a result, it is recommended that patients receiving JAK inhibitors undergo routine laboratory monitoring [13, 15,16,17,18].
The JAK inhibitor upadacitinib has been engineered for increased selectivity for JAK1 over JAK2, JAK3, and tyrosine kinase 2 at therapeutic concentrations [18, 19]. Upadacitinib has been evaluated across a broad spectrum of patients in the global phase 3 SELECT program, which included six trials with long-term open-label extensions [18, 20, 21]. The SELECT program included patients naïve to MTX, patients with an inadequate response to MTX/csDMARDs, and patients with an inadequate response to bDMARDs. It assessed upadacitinib both as monotherapy and in combination with MTX [3,4,5,6,7,8].
Consistent with the known safety profile of JAK inhibitors, changes in laboratory parameters have been observed in upadacitinib clinical trials [9, 20]. In this descriptive integrated analysis, we assessed selected long-term laboratory profiles associated with exposure to upadacitinib and active controls (adalimumab and MTX) in patients with RA treated in the SELECT program, with up to 6.5 years of exposure.
Methods
Patients and Studies
Data from six randomized phase 3 trials and their open-label extensions were included in this analysis: SELECT-EARLY (NCT02706873), SELECT-NEXT (NCT02675426), SELECT-MONOTHERAPY (NCT02706951), SELECT-COMPARE (NCT02629159), SELECT-BEYOND (NCT02706847), and SELECT-CHOICE (NCT03086343). Full details of the individual studies have been published previously and are summarized in Supplementary Table S1 [3,4,5,6,7,8].
Eligible patients were ≥ 18 years with active RA, defined as swollen joint count ≥ 6/66, tender joint count ≥ 6/68, and high-sensitivity C-reactive protein ≥ 3 mg/l (≥ 5 mg/l, together with evidence of erosive joint damage and/or autoantibody positivity, for SELECT-COMPARE and -EARLY). Of note, patients were required to have ALT and AST < 2 × upper limit of normal (ULN) at screening.
Patients were MTX-naïve (SELECT-EARLY) or had an inadequate response or intolerance to MTX (SELECT-MONOTHERAPY and -COMPARE), csDMARDs (SELECT-NEXT), or bDMARDs (SELECT-BEYOND and -CHOICE; Supplementary Table S1). All clinical studies were conducted according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines, applicable regulations and guidelines governing clinical study conduct, and the Declaration of Helsinki. As this was an integrated analysis of pooled clinical trial data, IRB approval was not required.
Dosing
Per each study protocol, patients received extended-release upadacitinib (15 mg or 30 mg once daily [QD]), placebo, MTX, subcutaneous adalimumab (40 mg every other week [EOW]) or intravenous abatacept (weight-based dosing), as monotherapy or in combination with background csDMARDs. Patients were not permitted to switch between upadacitinib doses, although following a protocol amendment due to the approval of the upadacitinib 15-mg dose, patients receiving upadacitinib 30 mg were switched to the 15-mg dose (earliest switch at week 108 [SELECT-EARLY]/168 [SELECT-NEXT]/132 [SELECT-MONOTHERAPY]/180 [SELECT-BEYOND]). MTX-naive patients randomized to MTX started oral medication at 10 mg/week (7.5 mg/week in China and Japan) and were titrated to a maximum of 20 mg/week (15 mg/week in Japan) through week 8, according to tolerance (Supplementary Table S1). Of note, patients receiving abatacept in SELECT-CHOICE switched to upadacitinib 15 mg QD at week 24, per the study protocol; thus, long-term data for abatacept are not available for this treatment group and were not included in this analysis.
The proportions of patients experiencing potentially clinically significant laboratory parameters at any time point were summarized for: pooled upadacitinib 15 mg QD, pooled upadacitinib 30 mg QD, adalimumab 40 mg EOW + MTX (SELECT-COMPARE only), and MTX monotherapy (SELECT-EARLY only). Patients who switched from placebo, adalimumab, or MTX to upadacitinib were included in the upadacitinib analysis set from the start of upadacitinib treatment. Those who switched from upadacitinib to adalimumab were included in the adalimumab analysis set from the start of adalimumab and were censored at time of switch. MTX monotherapy was censored at time of rescue to combination therapy (addition of a csDMARD). Serum samples for laboratory testing were collected from fasting patients where possible (minimum 8-h fast); if a patient was unable to fast when necessary, non-fasting status was recorded in the study source documentation.
Safety Analyses and Statistics
AEs and laboratory assessments were measured through August 15, 2022. AEs were defined using standardized Medical Dictionary for Regulatory Activities (MedDRA) query or company MedDRA query search criteria. A treatment-emergent adverse event (TEAE) was defined as any AE with an onset date from the first dose of study drug, up to 30 days after the last dose of either placebo or upadacitinib, or up to 70 days after the last dose of adalimumab (if patients discontinued from the study). Exposure-adjusted event rates (EAERs) were calculated as the total number of events (including multiple events in the same patient) adjusted for total exposure, reported as events per 100 patient-years (PY). Laboratory-related TEAEs of special interest were defined according to investigator-reported AEs (MedDRA query criteria as defined above) as opposed to objective laboratory measures. All events were attributed to treatment at the time of event.
Assessment of laboratory abnormalities included hemoglobin, neutrophils, lymphocytes, ALT, AST, CPK, creatinine, and lipids (low-density lipoprotein [LDL] and high-density lipoprotein [HDL] cholesterol, including the LDL/HDL cholesterol ratio [LDL-C/HDL-C]). Toxicity grading of laboratory-related AEs was based on Outcome Measures in Rheumatology (OMERACT) criteria [22]. For grading of CPK and creatinine abnormalities, National Cancer Institute standard common terminology criteria grading methodology was used [23]. To be included as grade 3 or 4 abnormality, the post-baseline grade must have been higher than baseline. Lipid data are reported as mean change from baseline. The studies were not designed to statistically compare the incidence of laboratory abnormalities and so all analyses are descriptive.
Ethical Approval
All clinical studies were conducted according to the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use guidelines, applicable regulations and guidelines governing clinical study conduct, and the Declaration of Helsinki. As this was an integrated analysis of pooled clinical trial data, IRB approval was not required.
Results
Patient Population
This descriptive analysis included 3209 patients receiving upadacitinib 15 mg QD (10 782.7 PY), 1204 patients receiving upadacitinib 30 mg QD (3162.5 PY), 579 patients receiving adalimumab 40 mg EOW + MTX (1573.2 PY), and 314 patients receiving MTX monotherapy (865.1 PY) across the phase 3 program (Table 1).
Baseline characteristics were generally similar across treatment groups; the majority of patients were female and around half were receiving concomitant glucocorticoids. A lower proportion of patients receiving MTX monotherapy and adalimumab + MTX had a history of bDMARD use, compared with the upadacitinib groups. This was due to the exclusion criteria of SELECT-EARLY and SELECT-COMPARE, ruling out patients with prior exposure to bDMARDs and patients with inadequate response to bDMARDs, respectively. Concomitant use of csDMARDs was more common with upadacitinib 15 mg versus upadacitinib 30 mg (Table 1).
Grade 3 and 4 Changes in Laboratory Parameters
Grade 3 and 4 elevations in ALT and AST were more common with MTX monotherapy and upadacitinib compared with adalimumab + MTX (Table 2). The incidences of grade 3 and 4 decreases in neutrophils were consistent across the treatment groups, whereas lymphocyte and hemoglobin decreases were less common with adalimumab + MTX compared with upadacitinib and MTX monotherapy. CPK elevations were more common with upadacitinib versus the comparators, while creatinine elevations were rare across all treatment groups (Table 2). Grade 3 and 4 changes in hemoglobin and neutrophils were more common with upadacitinib 30 mg versus 15 mg; rates of other events were comparable between the two doses.
Grade 3 and 4 Events by Time Period
Overall, the incidences of grade 3 and 4 laboratory abnormalities were generally low through any 12-month period, with no temporal pattern to their occurrence (Supplementary Fig. S1). In the first 12 weeks of treatment, incidences of grade 3 and 4 abnormalities were generally low and similar between groups, with the exception of grade 3 decreases in hemoglobin and neutrophils, which were numerically higher in patients receiving upadacitinib 30 mg (Supplementary Fig. S2). The incidence of grade 3 decreases in hemoglobin increased between weeks 12 and 24 then decreased through 60 weeks in patients receiving upadacitinib, whilst the incidence of grade 3 decreases in neutrophils reduced over weeks 12–24 then was stable through 60 weeks. Over the first 24-month period, the incidence of grade 3 and 4 hemoglobin decrease stabilized and was lower in the upadacitinib 15 mg QD and adalimumab + MTX groups compared with upadacitinib 30 mg and MTX monotherapy. Of note, the incidence of grade 3 and 4 CPK elevations was slightly higher in both upadacitinib treatment groups compared with adalimumab + MTX and MTX monotherapy in each 12-month period.
Mean Changes Over Time in Laboratory Parameters
Transient decreases in hemoglobin were seen with upadacitinib, which were more pronounced with the 30-mg QD dose than the 15-mg QD dose (Fig. 1a). Decreases in neutrophils were observed in all treatment groups (Fig. 1b), while decreases in lymphocytes were seen with upadacitinib and MTX monotherapy but not adalimumab + MTX (Fig. 1c). Consistent with the frequency of grade 3 and 4 events, greater increases in ALT and AST were seen with upadacitinib and MTX monotherapy compared with adalimumab + MTX (Fig. 1d and e). Increases in CPK were greater with upadacitinib compared with MTX monotherapy and adalimumab + MTX, while creatinine increases were seen in all groups (Fig. 1f and g).


Mean change from baseline in: a hemoglobin, b neutrophils, c lymphocytes, d ALT, e AST, f CPK, and g creatininea. aUPA 30 mg QD, ADA 40 mg EOW, and MTX monotherapy group data are presented per initial randomization and do not include patients who switched treatments. bWeek 24 for UPA 15 mg QD, UPA 30 QD, and MTX monotherapy; week 22 for ADA 40 mg EOW + MTX. cOne data point was omitted due to an erroneous value. ADA adalimumab, ALT alanine aminotransferase, AST aspartate aminotransferase, CPK creatine phosphokinase, EOW every other week, MTX methotrexate, QD once daily, UPA, upadacitinib
Changes in Circulating Lipid Levels
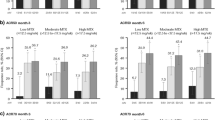
LDL-C levels showed a greater increase from baseline in both the upadacitinib 15 mg and 30 mg groups compared with adalimumab + MTX and MTX monotherapy, while HDL-C levels were generally comparable between treatment groups (Fig. 2a and b). However, few patients remained in the upadacitinib 30 mg treatment group by week 220 which limits comparisons at this time point. The mean LDL-C/HDL-C ratio remained stable for patients receiving upadacitinib 15 mg QD and MTX and increased at later timepoints in patients receiving adalimumab and upadacitinib 30 mg QD (Fig. 2c).

Mean change from baseline in: a LDL-C, b HDL-C, and c LDL/HDL ratioa. aUPA 30 mg QD, ADA 40 mg EOW, and MTX monotherapy group data are presented per initial randomization and do not include patients who switched treatments. bWeek 24 for UPA 15 mg QD, UPA 30 QD, and MTX monotherapy; week 22 for ADA 40 mg EOW + MTX. ADA adalimumab, EOW every other week, HDL-C high-density lipoprotein cholesterol, LDL-C low-density lipoprotein cholesterol, MTX methotrexate, QD once daily, UPA upadacitinib
Laboratory-Related AEs of Special Interest
Anemia occurred at consistent rates between upadacitinib and active comparators (Fig. 3). There were four serious AEs (SAEs) of anemia in the upadacitinib 15 mg group and two in the 30 mg group; two patients from the former group also experienced grade 4 abnormalities in hemoglobin. Neutropenia occurred at consistent rates between the upadacitinib 15-mg dose and active comparators, and lymphopenia rates were highest in the MTX monotherapy group. In the upadacitinib 15 mg group, none of the neutropenia events were serious, and there was no evidence of grade 3 or 4 abnormalities in neutrophils or lymphocytes being associated with serious infection, opportunistic infection, or herpes zoster within 30 days of the abnormality. Hepatic disorders occurred more frequently with upadacitinib and MTX than with adalimumab. No trend was observed regarding time to onset of the peak transaminase elevations in patients treated with either dose of upadacitinib, and most hepatic TEAEs were asymptomatic; no cases of drug-induced liver injury were reported. CPK elevations were more common with upadacitinib than active comparators; these elevations were generally asymptomatic and transient (i.e., lasting no more than two visits before returning to normal or baseline values). For all laboratory-related AEs, rates appeared higher with upadacitinib 30 mg compared with 15 mg (Fig. 3).

Exposure-adjusted event rates for laboratory-related treatment-emergent adverse events of special interest. UPA 15 mg: N = 3209, PY = 10782.7; UPA 30 mg: N = 1204, PY = 3162.5; ADA + MTX: N = 579, PY = 1573.2; MTX monotherapy: N = 314, PY = 865.1. Laboratory-related adverse events of special interest were defined using standardized MedDRA query or company MedDRA query search criteria and reported per investigator’s judgment. ADA adalimumab, CI confidence interval, CPK creatine phosphokinase, E/100 PY event per 100 patient-years, EOW every other week, MedDRA Medical Dictionary for Regulatory Activities, MTX methotrexate, QD once daily, UPA upadacitinib
Two cases of rhabdomyolysis were reported. One case of rhabdomyolysis was reported in a 66-year-old woman with a history of heavy alcohol consumption who was hospitalized on day 1523 of upadacitinib 15-mg treatment following an event of seizure due to alcohol withdrawal. Upon examination, the patient also had elevated troponin, aspiration pneumonia, acute kidney injury, severe malnourishment, and acute hypoxemic respiratory failure. The SAEs of acute kidney injury and rhabdomyolysis resolved within 48 h of treatment with dexamethasone, ampicillin, cefepime, and levetiracetam, and the patient was discharged after 10 days. The additional case of rhabdomyolysis was reported in a 63-year-old man who experienced an SAE of pneumonia and was hospitalized on day 228 of upadacitinib 30 mg treatment. On day 229, the patient also experienced acute renal failure and influenza, with CPK > 691 U/l and creatinine 3.4 mg/dl. Treatment with the study drug was interrupted and the patient was treated with Levaquin, Tamiflu, and vancomycin. The events resolved and treatment with upadacitinib 30 mg was restarted 10 days after initial hospitalization. Both cases of rhabdomyolysis were identified as unrelated to the study drug per the investigator.
Discontinuations of study treatment because of laboratory-related AEs were rare, occurring in ≤ 2.5% of patients across all treatment groups (Table 3). Discontinuations as a result of anemia, lymphopenia, neutropenia, creatinine elevations, and CPK elevations were ≤ 0.3% of patients across all treatment groups. The most frequent laboratory changes resulting in discontinuation were increases in ALT and AST, which were most common in the upadacitinib 30 mg and MTX groups; most of these did not result in study drug discontinuation. Overall, rates of discontinuation due to laboratory-related AEs were 1.1% and 2.2% in the upadacitinib 15 mg and 30 mg groups, respectively, and 0.9% and 2.5% in the adalimumab + MTX and MTX monotherapy groups, respectively (Table 3).
Discussion
This analysis assessed the impact of upadacitinib treatment on laboratory parameters in patients with RA in the context of clinical trials. Consistent with previous safety analyses [24], we observed decreases in hemoglobin, neutrophils, and lymphocytes, and increases in liver enzymes and CPK in a proportion of patients receiving upadacitinib. There was no apparent association of these changes with the rates of serious or opportunistic infection, herpes zoster, or drug-induced liver injury. The incidence of laboratory abnormalities was generally higher with the upadacitinib 30-mg dose compared with upadacitinib 15 mg, supporting the choice of upadacitinib 15 mg as the approved dose for RA given the similar efficacy of the two doses [3–8]. Most patients who experienced abnormalities remained on upadacitinib.
Many treatments used in the management of RA are associated with abnormalities in laboratory parameters and require routine monitoring [25]. MTX and other csDMARDs may result in liver enzyme and creatinine elevations and decreases in neutrophils. Indeed, this analysis reported elevations in liver enzymes in the MTX monotherapy group, although no creatinine elevations and few decreases in neutrophils were observed for this treatment group. The lack of effect on creatinine is consistent with previous results showing that low-dose MTX does not cause kidney-related AEs, in contrast to the high doses of MTX used in oncologic indications [26]. In addition, this analysis reported higher rates of lymphopenia in the MTX-monotherapy group versus the adalimumab + MTX group. While we cannot provide one clear explanation, this could be a result of the smaller sample size (N = 314 versus N = 579, respectively) and PYs reported (865.1 PY versus 1573.2 PY, respectively). Given that AEs were reported at the investigator’s discretion, another explanation could be variability in how lymphopenia events were reported between study investigators. Among bDMARDs, interleukin-6 receptor inhibitors have been associated with neutropenia, thrombocytopenia, elevated liver enzymes, and lipid abnormalities [27, 28]. In contrast, TNF inhibitors appear to have minimal clinically meaningful effects on laboratory parameters and monitoring is not typically recommended, although follow-up laboratory assessments often take place due to the concomitant use of csDMARDs [25]. This is consistent with results from the current analysis, as changes in laboratory parameters observed in the adalimumab + MTX treatment group were generally small.
Across the JAK inhibitor class and within the upadacitinib data in the current analysis, increased serum levels of CPK in patients with inflammatory disorders have been observed. Although typically indicating muscle damage, in vitro data suggest JAK inhibition restores differentiation of myoblasts into mature myocytes and increases CPK expression [29]. While there were two cases of rhabdomyolysis observed in the present study, these were resolved and were identified as not related to the study drug, per investigator. Although high CPK and creatine expression was reported in one of these cases, an alternative etiology to consider is the incidence of influenza and subsequent acute renal failure. Transaminase elevations have also been reported with JAK inhibitors for the treatment of RA, including tofacitinib and baricitinib [15, 16]. Elevations have similarly been reported in patients treated with upadacitinib 15 mg and 30 mg, with most elevations not resulting in treatment discontinuation and being resolved or resolving regardless of upadacitinib discontinuation [24]; results from our analysis are consistent with these findings.
It has been suggested that changes in laboratory parameters may vary according to JAK selectivity. For example, inhibition of JAK2 is thought to be related to hematologic abnormalities due to its involvement in erythropoietin and thrombopoietin signaling, whereas inhibition of JAK3 alters immune cell populations, due to its role in downstream signaling from common gamma chain cytokines [30,31,32,33]. Therefore, the dose-dependent decreases in hemoglobin across upadacitinib treatment groups in the current analysis may be attributed to the inhibitory effect of upadacitinib on JAK2, consistent with results from ex vivo studies using peripheral blood mononuclear cells [34]. Transient decreases in hemoglobin have also been observed in patients receiving baricitinib, which inhibits both JAK1 and JAK2 [35]. However, further research is needed to elucidate the underlying mechanisms of the effect of upadacitinib on hemoglobin.
In addition, changes in circulating lipid levels have been observed with JAK inhibitors including upadacitinib [13, 36]. The LDL-C/HDL-C ratio is considered a reliable predictor of cardiovascular mortality [37] and remained consistent for upadacitinib 15 mg QD in the current analysis. Cardiovascular events are a relevant safety concern with JAK inhibitors, particularly due to the results of the ORAL Surveillance study, which found that tofacitinib was associated with an increased risk of major adverse cardiovascular events (MACE) compared with TNF inhibitors in patients with a history of atherosclerotic cardiovascular disease or other cardiovascular risk factor(s) [38, 39]. In previous analyses of patients receiving upadacitinib, generally increased levels of LDL-C and HDL-C have been observed with no significant impact on cardiovascular disease risk [36], and no apparent association has been found between LDL-C and occurrence of MACE [24]. In this analysis, there was no evidence to suggest that elevated lipids were associated with the occurrence of treatment-emergent MACE. Further research may be needed to explore the association between changes in lipid levels and long-term cardiovascular events.
Overall, changes in laboratory parameters are a known effect of the JAK inhibitor class, and the results reported in this analysis are comparable to those observed in studies of baricitinib, tofacitinib, and filgotinib [40,41,42]. The prescribing information for JAK inhibitors recommends assessing the levels of lymphocytes, neutrophils, hemoglobin, liver enzymes, and lipids at baseline and at regular intervals during treatment [13, 15,16,17,18]. The prescribing information also recommends that treatment should not be initiated, or should be interrupted, in patients with a lymphocyte count of < 500 cells/mm3, a neutrophil count of < 1000 cells/mm3, hemoglobin levels of < 8 g/dl, or if liver injury is suspected, and that elevated lipid levels should be managed according to clinical guidelines for hyperlipidemia [13, 15,16,17,18].
The main limitations of this study are the post hoc nature of the pooled analysis and the lack of a placebo control (although active comparators were included). There were also limited data available for adalimumab and MTX groups compared with upadacitinib. Finally, the studies included in this pooled analysis were not designed for direct or statistical cross-trial comparison between upadacitinib, adalimumab, and MTX, and differences in study design and patient characteristics should be considered and may limit interpretation of the data. The main strength of this study is the large patient population covering six phase 3 trials and > 10 000 PY of exposure to upadacitinib.
Conclusions
In this descriptive, pooled study of the SELECT phase 3 program, the impact of upadacitinib on laboratory parameters was consistent with previous safety analyses. Long-term upadacitinib treatment in patients with RA was associated with several dose-dependent laboratory abnormalities, with a comparable incidence to those with MTX but a generally higher incidence than with adalimumab. Treatment discontinuations related to abnormalities in laboratory parameters were uncommon. The incidence of grade 3 or 4 laboratory abnormalities was generally uncommon over any 12-month period, with no temporal pattern to their occurrence. These results continue to support an acceptable safety profile of upadacitinib 15 mg QD for the treatment of moderate to severely active RA.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan, and execution of a data sharing agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharingwith-qualified-researchers.html.
References
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Smolen JS, Landewé RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2022;82(1):3–18.
Fleischmann R, Pangan AL, Song I-H, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. 2019;71(11):1788–800.
Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139):2513–24.
Rubbert-Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. 2020;383(16):1511–21.
Smolen JS, Pangan AL, Emery P, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393(10188):2303–11.
van Vollenhoven R, Takeuchi T, Pangan AL, et al. Efficacy and safety of upadacitinib monotherapy in methotrexate-naïve patients with moderately-to-severely active rheumatoid arthritis (SELECT-EARLY): a multicenter, multi-country, randomized, double-blind, active comparator-controlled trial. Arthritis Rheumatol. 2020;72(10):1607–20.
Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10139):2503–12.
Burmester GR, Cohen SB, Winthrop KL, et al. Safety profile of upadacitinib over 15000 patient-years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open. 2023;9(1):e002735.
Conaghan PG, Pavelka K, Hsieh SC, et al. Evaluating the efficacy of upadacitinib in patients with moderate rheumatoid arthritis: a post-hoc analysis of the SELECT phase 3 trials. Rheumatol Adv Pract. 2023;7(1):017.
Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatol (Oxford). 2019;58:i17–26.
Angelini J, Talotta R, Roncato R, et al. JAK-inhibitors for the treatment of rheumatoid arthritis: a focus on the present and an outlook on the future. Biomolecules. 2020;10:1002.
Harigai M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatol (Oxford). 2019;58:i34–42.
Charles-Schoeman C, Gonzalez-Gay MA, Kaplan I, et al. Effects of tofacitinib and other DMARDs on lipid profiles in rheumatoid arthritis: implications for the rheumatologist. Semin Arthritis Rheum. 2016;46(1):71–80.
Pfizer. XELJANZ® (tofacitinib) tablets, for oral use. Product information. 2022. https://labeling.pfizer.com/showlabeling.aspx?id=959. Accessed January 18, 2023.
Eli Lilly. OLUMIANT® (upadacitinib) tablets, for oral use. Prescribing information. 2022. https://pi.lilly.com/us/olumiant-uspi.pdf. Accessed January 18, 2023.
Galapagos. JYSELECA (filgotinib). Summary of product information. 2022. https://www.ema.europa.eu/en/documents/product-information/jyseleca-epar-product-information_en.pdf. Accessed January 18, 2023.
AbbVie Inc. RINVOQ® (upadacitinib) extended-release tablets, for oral use. Prescribing information. 2022. https://www.rxabbvie.com/pdf/rinvoq_pi.pdf. Accessed November 21, 2022.
Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2:23.
Tanaka Y. A review of upadacitinib in rheumatoid arthritis. Mod Rheumatol. 2020;30(5):779–87.
Winthrop KL, Nash P, Yamaoka K, et al. Incidence and risk factors for herpes zoster in patients with rheumatoid arthritis receiving upadacitinib: a pooled analysis of six phase III clinical trials. Ann Rheum Dis. 2022;81(2):20613.
Woodworth T, Furst DE, Alten R, et al. Standardizing assessment and reporting of adverse effects in rheumatology clinical trials II: the Rheumatology Common Toxicity Criteria v.2.0. J Rheumatol. 2007;34(6):1401–14.
U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. 2017. Available at: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf. Accessed October 2023.
Cohen SB, van Vollenhoven RF, Winthrop KL, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann Rheum Dis. 2021;80(3):304–11.
Rigby WFC, Lampl K, Low JM, et al. Review of routine laboratory monitoring for patients with rheumatoid arthritis receiving biologic or nonbiologic DMARDs. Int J Rheumatol. 2017;2017:9614241.
Sparks JA, Vanni KMM, Sparks MA, et al. Effect of low-dose methotrexate on eGFR and kidney adverse events: a randomized clinical trial. J Am Soc Nephrol. 2021;32(12):3197–207.
Genentech. Actemra (tocilizumab) injection, for intravenous and subcutaneous use. Prescribing Information 2022. https://www.gene.com/download/pdf/actemra_prescribing.pdf. Accessed February 22, 2023.
Sanofi and Regeneron Pharmaceuticals Inc. Kevzara (sarilumab) injection, for subcutaneous use. Prescribing Information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761037s001lbl.pdf. Accessed February 22, 2023.
Queeney K, Housley W, Sokolov J, et al. FRI0131 elucidating the mechanism underlying creatine phosphokinase upregulation with upadacitinib. Ann Rheum Dis. 2019;78:734–5.
O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii111–5.
O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–28.
Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228(1):273–87.
Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12(1):25–36.
McInnes IB, Byers NL, Higgs RE, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. 2019;21(1):183.
Kay J, Harigai M, Rancourt J, et al. Changes in selected haematological parameters associated with JAK1/JAK2 inhibition observed in patients with rheumatoid arthritis treated with baricitinib. RMD Open. 2020;6(3):e001370.
Makris A, Barkas F, Sfikakis PP, Liberopoulos E, Agouridis AP. The effect of upadacitinib on lipid profile and cardiovascular events: a meta-analysis of randomized controlled trials. J Clin Med. 2022;11(23):6894.
Zhong Z, Hou J, Zhang Q, et al. Assessment of the LDL-C/HDL-C ratio as a predictor of one year clinical outcomes in patients with acute coronary syndromes after percutaneous coronary intervention and drug-eluting stent implantation. Lipids Health Dis. 2019;18(1):40.
Charles-Schoeman C, Buch MH, Dougados M, et al. Risk of major adverse cardiovascular events with tofacitinib versus tumour necrosis factor inhibitors in patients with rheumatoid arthritis with or without a history of atherosclerotic cardiovascular disease: a post hoc analysis from ORAL Surveillance. Ann Rheum Dis. 2023;82(1):119–29.
Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386(4):316–26.
Cohen SB, Tanaka Y, Mariette X, et al. Long-term safety of tofacitinib up to 9.5 years: a comprehensive integrated analysis of the rheumatoid arthritis clinical development programme. RMD Open. 2020;6:001395.
Smolen JS, Genovese MC, Takeuchi T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. 2019;46(1):7–18.
Winthrop KL, Tanaka Y, Takeuchi T, et al. Integrated safety analysis of filgotinib in patients with moderately to severely active rheumatoid arthritis receiving treatment over a median of 1.6 years. Ann Rheum Dis. 2022;81(2):184–92.
Acknowledgements
We thank the participants who took part in the studies we analyzed.
Medical Writing, Editorial and Other Assistance.
Medical writing support was provided by Amy Hall, MSc, of 2 the Nth (Cheshire, UK), and was funded by AbbVie.
Funding
AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, review, and approval of the publication. No honoraria or payments were made for authorship. AbbVie funded the rapid review service and open-access fees.
Author information
Authors and Affiliations
Contributions
All authors had access to the data and participated in the development, review, critique, and approval of the manuscript throughout the editorial process and approved the final manuscript draft submitted for publication. All authors agreed to be accountable for all aspects of the work, ensuring the accuracy and integrity of the publication. All named authors meet the ICMJE criteria for authorship for this article, take responsibility for the integrity of the work, and have given their approval for this version to be published. Research project: (1) study concept and design: all authors; (2) data acquisition: Christina Charles-Schoeman and Gerd R. Burmester; (3) statistical analysis: Nasser Khan and Kassim Rahawi; and (4) data interpretation: Christina Charles-Schoeman, Jon T. Giles, Nancy E. Lane, Ernest Choy, Daniel E. Furst, Jiří Vencovský, Anthony G. Wilson, Gerd R. Burmester, Derek Coombs, Sara K. Penn, Nasser Khan, Jillian B. Yee, Kassim Rahawi, and Iain B. McInnes. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Corresponding author
Ethics declarations
Conflict of Interest
Christina Charles-Schoeman has received research grants from AbbVie, Alexion, Bristol-Myers Squibb, CSL Behring, Pfizer, and Priovant Therapeutics; and consulting fees from AbbVie, Bristol-Myers Squibb, Gilead, Pfizer, Recludix, and Sanofi/Regeneron. Jon T. Giles has received research grants from Pfizer; and consulting fees from AbbVie, Bristol-Myers Squibb, Eli Lilly, Gilead, Pfizer, and UCB. Nancy E. Lane has received consulting fees from Amgen, ANI, GSK, and Mallinckrodt. Ernest Choy has received research grants from Bio-Cancer, Biogen, Novartis, Pfizer, Roche, Sanofi, and UCB; consulting fees from AbbVie, Amgen, Biocon, Biogen, Chugai, Eli Lilly, Fresenius Kabi, Gilead, Janssen, Merck Serono, Novartis, Pfizer, Regeneron, Roche, R-Pharm, and Sanofi; and speaker fees from AbbVie, Amgen, Bristol-Myers Squibb, Chugai, Eli Lilly, Fresenius Kabi, Galapagos, Gilead, Janssen, Novartis, Pfizer, Regeneron, Roche, Sanofi, and UCB. Daniel E. Furst has received research grants from Actelion, Amgen, Bristol-Myers Squibb, Corbus, Galapagos, GSK, NIH, Novartis, Pfizer, Roche/Genentech, and Sanofi; consulting fees from AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, Galapagos, Novartis, and Pfizer; and speaker fees from AbbVie, Continuing Medical Education, and Novartis. Jiří Vencovský has received consulting fees from AbbVie, Boehringer Ingelheim, Eli Lilly, Gilead, and Octapharma; and speaker bureau fees from AbbVie, Biogen, MSD, Pfizer, Roche, Sanofi, and UCB. Anthony G. Wilson has nothing to disclose. Gerd R. Burmester has received consulting fees and speaker bureau fees from AbbVie, Eli Lilly, Gilead, Janssen, MSD, Pfizer, Roche, and UCB. Derek Coombs, Sara K. Penn, Nasser Khan, Jillian B. Yee, and Kassim Rahawi are employees of AbbVie and may own stock or options. Iain B. McInnes has received research grants from Celgene, Eli Lilly, Janssen, Novartis, Pfizer, Roche, and UCB; and consulting fees from AbbVie, Amgen, Bristol-Myers Squibb, Cabaletta, Celgene, Compugen, Eli Lilly, Eyelo, Gilead, Janssen, Novartis, and UCB.
Ethical Approval
All clinical studies were conducted according to the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use guidelines, applicable regulations and guidelines governing clinical study conduct, and the Declaration of Helsinki. As this was an integrated analysis of pooled clinical trial data, IRB approval was not required.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Charles-Schoeman, C., Giles, J.T., Lane, N.E. et al. Impact of Upadacitinib on Laboratory Parameters and Related Adverse Events in Patients with RA: Integrated Data Up to 6.5 Years. Rheumatol Ther 11, 157–175 (2024). https://doi.org/10.1007/s40744-023-00624-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-023-00624-3