
Abstract
Purpose
Mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene cause familial isolated pituitary adenomas (FIPA). AIP mutations have also been found in patients with apparently sporadic pituitary adenomas, particularly in young patients with large adenomas. The aim of this study was to determine the frequency of AIP germline mutations in patients with young-onset sporadic pituitary macroadenomas.
Methods
The AIP gene was sequenced in 218 Portuguese patients with sporadic pituitary macroadenomas diagnosed before the age of 40 years.
Results
Heterozygous rare sequence variants in AIP were identified in 18 (8.3%) patients. However, only four (1.8%) patients had pathogenic or likely pathogenic variants. These consisted of two already known mutations (p.Arg81* and p.Leu115Trpfs*41) and two novel mutations (p.Glu246*, p.Ser53Thrfs*36). All four patients had GH-secreting adenomas diagnosed between the ages of 14 and 25 years. The frequency of AIP pathogenic or likely pathogenic variants in patients under the age of 30 and 18 years was 3.4% and 5.0%, respectively.
Conclusion
The frequency of AIP mutations in this cohort was lower than in other studies. Previous reports may have overestimated the contribution of AIP mutations due to the inclusion of genetic variants of uncertain significance. The identification of novel AIP mutations expands the known spectrum of genetic causes of pituitary adenomas and may help understand the role of AIP mutations in the molecular mechanisms underlying pituitary tumorigenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The vast majority of pituitary adenomas occur in a sporadic context and are likely to occur due to acquired somatic and epigenetic mutations [1, 2]. More rarely, pituitary adenomas occur as part of syndromic diseases (e.g. multiple endocrine neoplasia) or as Familial Isolated Pituitary Adenomas (FIPA), due to inherited germline mutations [3].
Germline mutations in the Aryl Hydrocarbon Receptor-Interacting Protein (AIP) gene have been identified in cases of FIPA [4]. The AIP gene comprises six exons and encodes a 330-amino acid co-chaperone protein, which has a role in the metabolic clearance of dioxin and other toxic carcinogenic agents and in the regulation of the cyclic adenosine monophosphate (cAMP) molecular pathway [5, 6]. The expression of AIP in pituitary cells reduces cell proliferation rate, suggesting that AIP may act as a tumour suppressor gene [7]. Heterozygous inactivating mutations in AIP have been reported along the entire coding region of the gene [8]. Tumours that arise in individuals with AIP mutations usually present at a younger age, have a larger size, show increased invasiveness, and are often resistant to standard treatments [1, 9]. Prospective studies have shown that the identification of patients with AIP mutations may result in improved clinical outcomes [10].
Due to incomplete penetrance, patients with AIP mutations may not have a recognized family history of pituitary adenomas and may be classified as sporadic cases. Studies of patients with apparently sporadic pituitary adenomas have demonstrated that a variable proportion of these are due to germline AIP mutations. The prevalence of AIP mutations has been reported to be around 3.6% in unselected patients with sporadic pituitary adenomas [11]. However, the prevalence of AIP mutations increases to 7.2% in patients diagnosed under the age of 40 years [11], to 11.7% in patients with macroadenomas diagnosed under the age of 30 years [12], and to over 20% in paediatric patients [11, 12]. These findings have led to recommendations for AIP mutation testing in patients with pituitary macroadenomas diagnosed under the age of 30 [12] or 40 years [11].
The aim of this study was to investigate the prevalence of AIP mutations in a large Portuguese cohort of young-onset sporadic pituitary macroadenomas.
Materials and methods
Subjects
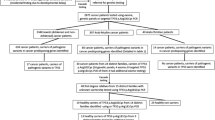
This multicentre study involved 218 Portuguese patients recruited consecutively at the main endocrinology outpatient clinics in Portugal, from 2018 to 2022. Selection criteria were patients with sporadic macroadenomas (tumour greater diameter ≥ 1 cm) diagnosed under the age of 40 years. Patients with a family history of pituitary adenomas (i.e. an affected first or second degree family member) or with evidence of a syndromic form of pituitary adenomas were excluded. Mean age (± standard deviation) at diagnosis was 28.9 ± 7.5 years, 118 patients were under 30 years at diagnosis, and 20 patients were under 18 years at diagnosis. Gender distribution was 113 (52%) females and 105 (48%) males. The type of adenoma was based on histological examination, or in the case of prolactinomas, by clinical, hormonal and radiological examination. Eighty (36.7%) patients had prolactinomas, 61 (28.0%) had somatotropinomas, 34 (15.6%) had non-functioning pituitary adenomas; 14 (6.4%) had corticotropinomas, 15 (6.9%) had mixed-secreting pituitary adenomas; seven (3.2%) had gonadotropinomas, one had a thyrotropinoma, and six (2.8%) had adenomas with undetermined histology. Written informed consent was obtained from all subjects and the study was approved by the Ethics Committee of the Faculty of Health Sciences, University of Beira Interior (Ref. CE-UBI-Pj-2018-027).
Genetic analysis
Genomic deoxyribonucleic acid (DNA) from each patient was extracted from peripheral blood leukocytes using previously described methods [13]. Patients were screened for mutations in AIP by Polymerase Chain Reaction (PCR) amplification of the six coding exons and exon–intron boundaries and through bi-directional sequencing using a semi-automated DNA sequencer (STAB VIDA, Caparica, Portugal; and ABI 3730XL, Applied Biosystems; Thermo Fisher Scientific, Waltham, MA, USA). Primer sequences were previously described by Cazabat et al. [11]. Genomic sequence variants were selected according to the following cumulative criteria: (1) Located in the AIP exons and up to 10 nucleotides from the splice site regions; and (2) Absent or rare (population frequency < 1%) in the Genome Aggregation Database (gnomAD) [14]. Selected variants were then classified by the American College of Medical Genetics and Genomics (ACMG) criteria [15], using the VarSome search engine [16]. Variants classified as pathogenic or likely pathogenic were considered to be causative mutations. The heterozygous frameshift mutations were confirmed by cloning of the PCR product using a CloneJET PCR Cloning Kit (ThermoScientific, Thermo Fisher Scientific, Waltham, MA, USA) followed by DNA sequencing of each allele. Sequence variant nomenclature followed standard guidelines [17] and was based on the cDNA reference sequence for the AIP gene (GenBank accession NM_003977.4). The frequency of AIP mutations was determined in patients diagnosed under the ages of 40, 30 and 18 years, respectively.
Results
Genetic findings
Sequence analysis of the entire coding region of AIP, including the exon–intron boundary regions, revealed that 18 (8.3%) patients had 12 different heterozygous rare sequence variants. However, eight of these variants [c.47G > A (p.Arg16His), c.132C > T (p.Asp44Asp), c.753G > A (p.Leu251Leu), c.891C > A (p.Ala297Ala), c.896C > T (p.Ala299Val), c.*14C > A, c.*60G > C and c.*64G > C] were classified as benign according to ACMG criteria (Table 1). The remaining four variants [c.158_165delGCCGGGCT (p.Ser53Thrfs*36), c.241C > T (p.Arg81*), c.343delC (p.Leu115Trpfs*41), and c.736G > T (p.Glu246*)] were classified as pathogenic or likely pathogenic (Table 1, Fig. 1). Thus, 1.8% (4/218) of the patients with sporadic pituitary macroadenomas were considered to have causative mutations in AIP. All four patients with AIP mutations had been diagnosed before the age of 30 years. In patients diagnosed under the age of 30, the prevalence of AIP mutations in this group was 3.4% (4/118). In the paediatric age group (< 18 years-old), the prevalence of AIP mutations was 5.0% (1/20).

AIP mutations identified in patients. For each mutation (A–D), the DNA sequences of a normal individual (above) and the patient (below) are shown. The positions of the mutations are indicated by asterisks. All mutations were heterozygous. For clarity, the deletion mutations (A, C) are shown only on the cloned sequences of the mutated alleles
Clinical characteristics of patients with AIP mutations
Patient 1
A 20-years-old woman presented with secondary amenorrhea. The amenorrhea had been attributed to anorexia nervosa diagnosed at the age of 15, but did not reverse with the recovery of body weight. She also complained of frontal headaches, excessive sweating and coarse facial features. She denied any visual defects. Her height and weight was 167 cm and 59 kg, respectively. Blood tests revealed basal growth hormone (GH) 14 ng/mL (normal range (NR) < 8), insulin-like growth factor 1 (IGF1) 1100 ng/mL (NR 102–231), follicle-stimulating hormone (FSH) 5.4 mUI/mL (NR 1.2–9), luteinizing hormone (LH) 2.1 mUI/mL (NR 1.1–11.6), estradiol < 5 pg/mL, prolactin 45 ng/mL (NR 4.7–23.0), adrenocorticotropic hormone (ACTH) 30 pg/mL (NR 7.2–63.3), cortisol 15.1 μg/dL (NR 6.2–18.0), thyroid-stimulating hormone (TSH) 1.6 mUI/mL (NR 0.3–4.2) and free thyroxine (FT4) 1.1 ng/dL (NR 0.85–1.7). An oral glucose tolerance test (OGTT) demonstrated failure of GH suppression, with a GH nadir of 1.4 ng/mL. A magnetic resonance imaging (MRI) showed a 20 mm pituitary tumour. A diagnosis of acromegaly was made and the patient underwent transsphenoidal surgery with excision of the pituitary macroadenoma. The histopathology showed a pituitary adenoma with a predominance of acidophilic cells. Post-surgical testing revealed normalization of GH levels, but persistence of the hypogonadotropic hypogonadism. At the age of 29, she underwent fertility treatment and had a twin pregnancy. At the last follow-up, at the age of 37, blood tests revealed basal GH 0.42 ng/mL (NR < 8), IGF1 102 ng/mL (NR 102–231), FSH 1.4 mUI/mL (NR 1.2–9), LH 0.16 mUI/mL (NR 1.1–11.6), estradiol < 5 pg/mL, prolactin 14.8 ng/mL (NR 4.7–23.0), ACTH 22.2 pg/mL (NR 7.2–63.3), cortisol 16.1 μg/dL (NR 6.2–18.0), TSH 0.95 mUI/mL (NR 0.3–4.2) and FT4 1.2 ng/dL (NR 0.85–1.7). There was no known family history of pituitary adenomas. Genetic analysis revealed an AIP heterozygous mutation (c.158_165delGCCGGGCT, p.Ser53Thrfs*36) in the patient. No other family members were available for genetic screening.
Patient 2
A 23-year-old man presented to the emergency department with a history of frequent and severe headaches, without visual defects. An MRI showed a 20 × 18 mm pituitary tumour with suprasellar extension contacting the optic chiasm and evidence of bleeding into the tumour. Acral growth and prognathism were noted. His height and weight were 189 cm and 97 kg, respectively. Serum IGF1 level was 704 ng/mL (NR 115–340), basal GH level was 6.40 ng/mL and prolactin was 17.40 ng/mL (NR 4.04–15.20), whereas FT4, TSH, cortisol and electrolyte levels were within the normal range. An OGTT demonstrated failure of GH suppression with a GH nadir of 5.82 ng/mL. A diagnosis of acromegaly was made and he was proposed for transsphenoidal surgery. In the meantime, he presented to the emergency department with refractory headaches, visual deficit and ptosis. MRI showed a 24 × 22 × 26 mm tumor with suprasellar and cavernous extension and acute bleeding in the tumour. The patient underwent transsphenoidal surgery and histopathology showed a pituitary adenoma with frequent immunoreactive cells for GH and rarer for prolactin and with areas of recent and old intratumoral haemorrhage, suggestive of pituitary apoplexy. Post-surgical testing revealed cortisol and thyroxine deficiencies that were corrected with hydrocortisone 15 mg/day and levothyroxine 50 ug/day. At the last follow-up, 8 months after surgery, prolactin and testosterone levels were normal, IGF1 level was 127 ng/mL (NR 115–340) and the nadir postglucose GH level was 0.1 ng/mL. The patient began reducing the dosages of the replacement therapy. There was no known family history of pituitary adenomas. Genetic analysis revealed an AIP heterozygous mutation (c.241C > T, p.Arg81*) in the patient and in his asymptomatic father, who was referred for clinical, biochemical and MRI assessment.
Patient 3
A 13-year-old boy presented with tall stature. Since the age of 8 years, he demonstrated increased height velocity and started to cross height percentiles and remained above the 97th percentile. The patient also complained of frontal headaches for 2 years. He denied any visual defects. His height and weight was 180.8 cm (+ 3.3 SDS) and 57 kg, respectively. He displayed coarse facial features with frontal bossing and mild prognathism, large hands and feet. His pubertal status was compatible with Tanner stage 5 (testicular volume 20 mL). Past medical history was unremarkable. The mid-parental height was 178.5 cm. Baseline pituitary investigation showed increased levels of IGF1 (587.5 ng/mL, NR 74–450), random GH 11.3 ng/mL (NR 0.12–8.1), hyperprolactinaemia with PRL 35.1 ng/mL (NR 4.4–19), central hypothyroidism with TSH 2.3 µUI/mL (NR 0.51–4.30) and FT4 6.96 pmol/L (NR 12.6–21), morning cortisol of 10.3 µg/dL (NR 6.2–12.5), total testosterone 238 ng/dL (NR 105–545), FSH 3.49 U/L (NR 1.5–8.6), LH 1.64 U/L (NR 1.7–8.6). An OGTT demonstrated failure of GH suppression, with a GH nadir of 8.1 ng/mL. MRI of the sellar region showed a pituitary tumour, measuring 13 × 12 × 14 mm, without signs of cavernous sinus invasion, suprasellar extension or optic chiasm compression. Visual field tests were normal. Bone age was coincident with his chronological age. A diagnosis of pituitary gigantism due to a GH-secreting pituitary macroadenoma and associated central hypothyroidism was made. Treatment with levothyroxine was initiated and the patient underwent transsphenoidal surgery with excision of the pituitary macroadenoma, without complications. Histopathology revealed a mammosomatotroph adenoma with a Ki-67 index of < 1%. Three months after surgery, he presented normalisation of IGF1 levels, GH nadir during OGTT was < 1 ng/mL and no residual tumour was found on the MRI. Recovery of central hypothyroidism was also observed after surgery, and the patient is currently euthyroid without levothyroxine supplementation. He remains in remission 7 years after surgery with serum IGF1 of 308 µg/L (NR 93–449), nadir GH level < 1 ng/mL during OGTT, and a height of 189.5 cm. There was no known family history of pituitary adenomas. Genetic analysis revealed an AIP heterozygous mutation (c.343delC, p.Leu115Trpfs*41) in the patient. No other family members were available for genetic screening.
Patient 4
A 25-year-old woman presented with acromegalic facial features, acral growth and secondary amenorrhea. Her height and weight were 159 cm and 61 kg, respectively. Blood tests revealed basal GH 29.8 ng/mL (NR 0.06–5.0), IGF1 895 ng/mL (NR 117–329), FSH 3.2 mUI/mL (NR 1.2–9), LH 1.69 mUI/mL (NR 1.1–11.6), estradiol 15 pg/mL (NR 15–160), prolactin 42 ng/mL (NR 3.46–19.4), ACTH 30.5 pg/mL (NR 5–46), cortisol 9.8 μg/dL (NR 5–25), TSH 1.95 mUI/mL (NR 0.4–4.0) and FT4 0.8 ng/dL (NR 0.8–1.9). An OGTT failed to suppress the levels of GH. An MRI showed a 28 × 25 × 12 mm pituitary tumour with invasion of the left cavernous sinus, suprasellar extension, and compression of the optic chiasma. She underwent an unsuccessful transsphenoidal surgery followed by a pterional craniotomy, with subtotal excision of the tumour. Histopathology showed a pituitary adenoma with immunoreactivity for GH and Ki-67 < 2%. Two months later, GH levels remained unsuppressed and IGF1 levels were 547 ng/mL (NR 117–329). Testing for other pituitary hormones revealed panhypopituitarism and hormone replacement therapy was initiated. An octreotide test trial resulted in a 63% decrease in GH levels. The patient began treatment with lanreotide 120 mg every 4 weeks and cabergoline 2 mg per week, which resulted in a partial response. Six months later, an MRI showed persistence of a large tumour residue. Eighteen months later, the patient underwent stereotactic radiosurgery and maintained treatment with lanreotide and cabergoline. Two years after radiosurgery, the patient showed clinical, biochemical and imaging improvement, and pharmacological treatment was gradually reduced to lanreotide 120 mg every 9 weeks and cabergoline 0.5 mg per week. At the last follow-up, 9 years after surgery, the patient stopped the treatment with lanreotide and maintained cabergoline 0.25 mg per week and remaining pituitary hormone replacement therapy. Subsequent testing showed a nadir postglucose GH level of 2.87 ng/mL, but with normal IGF1 levels. A head MRI showed residual tumour near the left cavernous sinus. There was no known family history of pituitary adenomas. Genetic analysis revealed an AIP heterozygous mutation (c.736G > T, p.Glu246*) in the patient and in her asymptomatic mother, who was referred for clinical, biochemical and MRI assessment.
Discussion
The prevalence of pathogenic and likely pathogenic AIP variants in our cohort of patients with sporadic pituitary macroadenomas diagnosed under the age of 40 years was 1.8% (4/218). This represents a low prevalence when compared to that of other studies carried out in France (16/222, 7.2%) [11], Brazil (11/132, 8.3%) [18] and Australia (6/34, 17.6%) [19]. Other studies have used lower age cut-offs to analyse the prevalence of AIP mutations. A pan-European collaboration [12] analysed a cohort of 163 patients diagnosed with sporadic pituitary macroadenomas before the age of 30 years and reported an 11.7% (19/163) prevalence of AIP mutations. Likewise, other studies carried out in Turkey [20], Mexico [21], and Spain [22], in patients under 30 years, reported prevalences of AIP mutations of 9% (1/11), 7% (5/55), and 6% (9/148), respectively. In contrast, our study showed that only 3.4% (4/118) of Portuguese patients diagnosed under the age of 30 years had AIP mutations. Furthermore, only 5.0% (1/20) of our paediatric patients presented AIP mutations, which is also a prevalence lower than in other studies [11, 12, 18, 21, 22]. Interestingly, the reported prevalences in the UK (2/100, 2%, under 40 years) [23] and in Germany (2/82, 2.4%, under 30 years) [24] were closer to those observed in our study. More recently, a study in Poland comprising 131 patients with macroadenomas of all ages failed to identify any clearly pathogenic AIP mutations [25].
There may be several explanations for the observed differences across studied populations. First, there may be geographical and ethnic differences in the genetic makeup of the populations and founder effects that increase the frequency of certain mutations. Second, the overall clinical characteristics of the patients may differ between cohorts. For example, as AIP mutations occur more commonly in GH-secreting tumours [26], cohorts that are enriched for patients with acromegaly may present a higher prevalence of these mutations. Lastly, the criteria for classifying genetic variants as pathogenic often vary between studies. Several genetic variants reported as pathogenic or variants of uncertain significance (VUS) in earlier studies are now considered to be benign variants. For example, the benign missense (p.Arg16His and p.Ala299Val) and non-coding (c.*14C > A and c.*64G > A) variants, which we excluded from our study, were included in the prevalence data of other studies [11, 18]. The inclusion of VUS in prevalence studies is particularly problematic because their interpretation is often difficult and, over time, many are reclassified as benign variants [27]. Thus, it is possible that earlier studies have overestimated the prevalence of true pathogenic variants in young-onset pituitary macroadenomas.
We identified four AIP mutations, including two novel [c.158_165delGCCGGGCT (p.Ser53Thrfs*36) and c.736G > T (p.Glu246*)] and two that have been reported before in patients from other countries [c.241C > T (p.Arg81*) and c.343delC (p.Leu115Trpfs*41)] [7, 28]. All of these are frameshift or nonsense mutations that are expected to lead to a premature stop codon and consequently to the formation of a shorter protein or to nonsense-mediated decay [29]. Therefore, these are loss-of-function mutations that abolish protein domains that are important for the binding of AIP to its interaction partners [30].
The mutations identified in this study were all found in patients with GH-secreting adenomas. This is not unexpected, as AIP mutations have been found to be more prevalent in this tumour type [26]. Apart from the young age of the patients, the clinical course of the disease was not particularly unusual and the pituitary surgery was curative in all but one patient. Thus, in this limited group of patients, we were unable to confirm previous reports that suggested a more unfavourable outcome in patients with AIP mutations [1, 9].
None of the patients with AIP mutations reported a family history of pituitary adenomas. However, in two cases, the mutation was also identified in an asymptomatic parent who will now undergo clinical, biochemical and MRI assessment. It remains to be determined if these mutation carriers have clinically silent adenomas or incomplete penetrance of the AIP mutations. It has been estimated that AIP mutations are associated with a disease penetrance of only 20–25% [31] and this explains why family members who share the same mutation often do not express the disease. Nevertheless, our identification of patients with AIP mutations will allow for cascade genetic screening of family members, the identification of additional mutation carriers, the clinical screening of these mutation carriers, and an earlier diagnosis, treatment and, possibly, better long-term outcome of any existing pituitary tumours [10].
Our study has some limitations. First, we did not look for AIP gross deletions that have been reported before [24, 32, 33]. However, studies using Multiplex Ligation-dependent Probe Amplification (MLPA) in large cohorts of patients have not identified any gross gene deletions, indicating that these are unlikely to be frequent [11, 12]. Second, we did not look for mutations in other genes that may be associated with syndromic forms of pituitary adenomas, such as the multiple endocrine neoplasia type 1 (MEN1) gene [34]. Although we excluded patients with evidence of other coexisting endocrine tumours or other features suggestive of syndromic forms of pituitary adenomas, we cannot rule out the possibility of a pituitary adenoma being the first manifestation of an undiagnosed syndrome.
In conclusion, the prevalence of AIP mutations in our cohort is lower than that in previous reports, possibly because we used more stringent criteria to classify variants as pathogenic. No AIP mutations were found in patients diagnosed after the age of 30 years and this suggests that this age cutoff may be the most appropriate for AIP genetic testing in patients with apparently sporadic large pituitary adenomas. Finally, our identification of novel AIP mutations expands the known spectrum of AIP mutations and may contribute to the understanding of the pathogenesis of pituitary adenomas.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Spada A, Mantovani G, Lania AG, Treppiedi D, Mangili F, Catalano R, Carosi G, Sala E, Peverelli E (2022) Pituitary tumors: genetic and molecular factors underlying pathogenesis and clinical behavior. Neuroendocrinology 112(1):15–33. https://doi.org/10.1159/000514862
Chang M, Yang C, Bao X, Wang R (2020) Genetic and epigenetic causes of pituitary adenomas. Front Endocrinol 11:596554. https://doi.org/10.3389/fendo.2020.596554
Coopmans EC, Korbonits M (2022) Molecular genetic testing in the management of pituitary disease. Clin Endocrinol 97(4):424–435. https://doi.org/10.1111/cen.14706
Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, Gundogdu S, De Menis E, Makinen MJ, Launonen V, Karhu A, Aaltonen LA (2006) Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science (New York) 312(5777):1228–1230. https://doi.org/10.1126/science.1126100
Ozfirat Z, Korbonits M (2010) AIP gene and familial isolated pituitary adenomas. Mol Cell Endocrinol 326(1–2):71–79. https://doi.org/10.1016/j.mce.2010.05.001
Hernandez-Ramirez LC, Trivellin G, Stratakis CA (2017) Role of phosphodiesterases on the function of aryl hydrocarbon receptor-interacting protein (AIP) in the pituitary gland and on the evaluation of AIP gene variants. Hormone Metab Res Hormon- und Stoffwechselforschung = Hormones et metabolisme 49(4):286–295. https://doi.org/10.1055/s-0043-104700
Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, Chahal HS, Igreja SC, Jordan S, Rowe J, Stolbrink M, Christian HC, Wray J, Bishop-Bailey D, Berney DM, Wass JA, Popovic V, Ribeiro-Oliveira A Jr, Gadelha MR, Monson JP, Akker SA, Davis JR, Clayton RN, Yoshimoto K, Iwata T, Matsuno A, Eguchi K, Musat M, Flanagan D, Peters G, Bolger GB, Chapple JP, Frohman LA, Grossman AB, Korbonits M (2008) The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab 93(6):2390–2401. https://doi.org/10.1210/jc.2007-2611
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN (2017) The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 136(6):665–677. https://doi.org/10.1007/s00439-017-1779-6
Denes J, Korbonits M (2021) The clinical aspects of pituitary tumour genetics. Endocrine 71(3):663–674. https://doi.org/10.1007/s12020-021-02633-0
Marques P, Caimari F, Hernandez-Ramirez LC, Collier D, Iacovazzo D, Ronaldson A, Magid K, Lim CT, Stals K, Ellard S, Grossman AB, Korbonits M, Consortium F (2020) Significant benefits of AIP testing and clinical screening in familial isolated and young-onset pituitary tumors. J Clin Endocrinol Metab 105(6):e2247–2260. https://doi.org/10.1210/clinem/dgaa040
Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, Young J, Guiochon-Mantel A, Chanson P (2012) Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab 97(4):E663-670. https://doi.org/10.1210/jc.2011-2291
Tichomirowa MA, Barlier A, Daly AF, Jaffrain-Rea ML, Ronchi C, Yaneva M, Urban JD, Petrossians P, Elenkova A, Tabarin A, Desailloud R, Maiter D, Schurmeyer T, Cozzi R, Theodoropoulou M, Sievers C, Bernabeu I, Naves LA, Chabre O, Montanana CF, Hana V, Halaby G, Delemer B, Aizpun JI, Sonnet E, Longas AF, Hagelstein MT, Caron P, Stalla GK, Bours V, Zacharieva S, Spada A, Brue T, Beckers A (2011) High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. Eur J Endocrinol 165(4):509–515. https://doi.org/10.1530/EJE-11-0304
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 16(3):1215. https://doi.org/10.1093/nar/16.3.1215
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database C, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443. https://doi.org/10.1038/s41586-020-2308-7
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A (2019) VarSome: the human genomic variant search engine. Bioinformatics (Oxford) 35(11):1978–1980. https://doi.org/10.1093/bioinformatics/bty897
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE (2016) HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37(6):564–569. https://doi.org/10.1002/humu.22981
Araujo PB, Kasuki L, de Azeredo Lima CH, Ogino L, Camacho AHS, Chimelli L, Korbonits M, Gadelha MR (2017) AIP mutations in Brazilian patients with sporadic pituitary adenomas: a single-center evaluation. Endocr Connect 6(8):914–925. https://doi.org/10.1530/EC-17-0237
De Sousa SMC, McCabe MJ, Wu K, Roscioli T, Gayevskiy V, Brook K, Rawlings L, Scott HS, Thompson TJ, Earls P, Gill AJ, Cowley MJ, Dinger ME, McCormack AI (2017) Germline variants in familial pituitary tumour syndrome genes are common in young patients and families with additional endocrine tumours. Eur J Endocrinol 176(5):635–644. https://doi.org/10.1530/EJE-16-0944
Karaca Z, Taheri S, Tanriverdi F, Unluhizarci K, Kelestimur F (2015) Prevalence of AIP mutations in a series of Turkish acromegalic patients: are synonymous AIP mutations relevant? Pituitary 18(6):831–837. https://doi.org/10.1007/s11102-015-0659-0
Ramirez-Renteria C, Hernandez-Ramirez LC, Portocarrero-Ortiz L, Vargas G, Melgar V, Espinosa E, Espinosa-de-Los-Monteros AL, Sosa E, Gonzalez B, Zuniga S, Unterlander M, Burger J, Stals K, Bussell AM, Ellard S, Dang M, Iacovazzo D, Kapur S, Gabrovska P, Radian S, Roncaroli F, Korbonits M, Mercado M (2016) AIP mutations in young patients with acromegaly and the Tampico Giant: the Mexican experience. Endocrine 53(2):402–411. https://doi.org/10.1007/s12020-016-0930-9
Martinez de LaPiscina I, Portillo Najera N, Rica I, Gaztambide S, Webb SM, Santos A, Moure MD, Paja Fano M, Hernandez MI, Chueca-Guindelain MJ, Hernandez-Ramirez LC, Soto A, Valdes N, Castano L (2021) Clinical and genetic characteristics in patients under 30 years with sporadic pituitary adenomas. Eur J Endocrinol 185(4):485–496. https://doi.org/10.1530/EJE-21-0075
Preda V, Korbonits M, Cudlip S, Karavitaki N, Grossman AB (2014) Low rate of germline AIP mutations in patients with apparently sporadic pituitary adenomas before the age of 40: a single-centre adult cohort. Eur J Endocrinol 171(5):659–666. https://doi.org/10.1530/EJE-14-0426
Schofl C, Honegger J, Droste M, Grussendorf M, Finke R, Plockinger U, Berg C, Willenberg HS, Lammert A, Klingmuller D, Jaursch-Hancke C, Tonjes A, Schneidewind S, Flitsch J, Bullmann C, Dimopoulou C, Stalla G, Mayr B, Hoeppner W, Schopohl J (2014) Frequency of AIP gene mutations in young patients with acromegaly: a registry-based study. J Clin Endocrinol Metab 99(12):E2789-2793. https://doi.org/10.1210/jc.2014-2094
Trofimiuk-Muldner M, Domagala B, Sokolowski G, Skalniak A, Hubalewska-Dydejczyk A (2023) AIP gene germline variants in adult Polish patients with apparently sporadic pituitary macroadenomas. Front Endocrinol 14:1098367. https://doi.org/10.3389/fendo.2023.1098367
Caimari F, Hernandez-Ramirez LC, Dang MN, Gabrovska P, Iacovazzo D, Stals K, Ellard S, Korbonits M, International FC (2018) Risk category system to identify pituitary adenoma patients with AIP mutations. J Med Genet 55(4):254–260. https://doi.org/10.1136/jmedgenet-2017-104957
Burke W, Parens E, Chung WK, Berger SM, Appelbaum PS (2022) The challenge of genetic variants of uncertain clinical significance: a narrative review. Ann Intern Med 175(7):994–1000. https://doi.org/10.7326/M21-4109
Daly AF, Rostomyan L, Betea D, Bonneville JF, Villa C, Pellegata NS, Waser B, Reubi JC, Waeber Stephan C, Christ E, Beckers A (2019) AIP-mutated acromegaly resistant to first-generation somatostatin analogs: long-term control with pasireotide LAR in two patients. Endocr Connect 8(4):367–377. https://doi.org/10.1530/EC-19-0004
Karousis ED, Muhlemann O (2022) The broader sense of nonsense. Trends Biochem Sci 47(11):921–935. https://doi.org/10.1016/j.tibs.2022.06.003
Trivellin G, Korbonits M (2011) AIP and its interacting partners. J Endocrinol 210(2):137–155. https://doi.org/10.1530/JOE-11-0054
Hernandez-Ramirez LC, Gabrovska P, Denes J, Stals K, Trivellin G, Tilley D, Ferrau F, Evanson J, Ellard S, Grossman AB, Roncaroli F, Gadelha MR, Korbonits M, International FC (2015) Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J Clin Endocrinol Metab 100(9):E1242–E1254. https://doi.org/10.1210/jc.2015-1869
Georgitsi M, Heliovaara E, Paschke R, Kumar AV, Tischkowitz M, Vierimaa O, Salmela P, Sane T, De Menis E, Cannavo S, Gundogdu S, Lucassen A, Izatt L, Aylwin S, Bano G, Hodgson S, Koch CA, Karhu A, Aaltonen LA (2008) Large genomic deletions in AIP in pituitary adenoma predisposition. J Clin Endocrinol Metab 93(10):4146–4151. https://doi.org/10.1210/jc.2008-1003
Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, Chapple JP, Trivellin G, Gueorguiev M, Guegan K, Stals K, Khoo B, Kumar AV, Ellard S, Grossman AB, Korbonits M, International FC (2010) Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum Mutat 31(8):950–960. https://doi.org/10.1002/humu.21292
Cuny T, Pertuit M, Sahnoun-Fathallah M, Daly A, Occhi G, Odou MF, Tabarin A, Nunes ML, Delemer B, Rohmer V, Desailloud R, Kerlan V, Chabre O, Sadoul JL, Cogne M, Caron P, Cortet-Rudelli C, Lienhardt A, Raingeard I, Guedj AM, Brue T, Beckers A, Weryha G, Enjalbert A, Barlier A (2013) Genetic analysis in young patients with sporadic pituitary macroadenomas: besides AIP don’t forget MEN1 genetic analysis. Eur J Endocrinol 168(4):533–541. https://doi.org/10.1530/EJE-12-0763
Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, Murat A, Emy P, Gimenez-Roqueplo AP, Tamburrano G, Raverot G, Barlier A, De Herder W, Penfornis A, Ciccarelli E, Estour B, Lecomte P, Gatta B, Chabre O, Sabate MI, Bertagna X, Garcia Basavilbaso N, Stalldecker G, Colao A, Ferolla P, Wemeau JL, Caron P, Sadoul JL, Oneto A, Archambeaud F, Calender A, Sinilnikova O, Montanana CF, Cavagnini F, Hana V, Solano A, Delettieres D, Luccio-Camelo DC, Basso A, Rohmer V, Brue T, Bours V, Teh BT, Beckers A (2007) Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab 92(5):1891–1896. https://doi.org/10.1210/jc.2006-2513
Barlier A, Vanbellinghen JF, Daly AF, Silvy M, Jaffrain-Rea ML, Trouillas J, Tamagno G, Cazabat L, Bours V, Brue T, Enjalbert A, Beckers A (2007) Mutations in the aryl hydrocarbon receptor interacting protein gene are not highly prevalent among subjects with sporadic pituitary adenomas. J Clin Endocrinol Metab 92(5):1952–1955. https://doi.org/10.1210/jc.2006-2702
Lecoq AL, Bouligand J, Hage M, Cazabat L, Salenave S, Linglart A, Young J, Guiochon-Mantel A, Chanson P, Kamenicky P (2016) Very low frequency of germline GPR101 genetic variation and no biallelic defects with AIP in a large cohort of patients with sporadic pituitary adenomas. Eur J Endocrinol 174(4):523–530. https://doi.org/10.1530/EJE-15-1044
Cazabat L, Libe R, Perlemoine K, Rene-Corail F, Burnichon N, Gimenez-Roqueplo AP, Dupasquier-Fediaevsky L, Bertagna X, Clauser E, Chanson P, Bertherat J, Raffin-Sanson ML (2007) Germline inactivating mutations of the aryl hydrocarbon receptor-interacting protein gene in a large cohort of sporadic acromegaly: mutations are found in a subset of young patients with macroadenomas. Eur J Endocrinol 157(1):1–8. https://doi.org/10.1530/EJE-07-0181
Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Makinen MJ, Vierimaa O, Paschke R, Saeger W, van der Luijt RB, Sane T, Robledo M, De Menis E, Weil RJ, Wasik A, Zielinski G, Lucewicz O, Lubinski J, Launonen V, Vahteristo P, Aaltonen LA (2007) Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci USA 104(10):4101–4105. https://doi.org/10.1073/pnas.0700004104
Cai F, Zhang YD, Zhao X, Yang YK, Ma SH, Dai CX, Liu XH, Yao Y, Feng M, Wei JJ, Xing B, Jiao YH, Wei ZQ, Yin ZM, Zhang B, Gu F, Wang RZ (2013) Screening for AIP gene mutations in a Han Chinese pituitary adenoma cohort followed by LOH analysis. Eur J Endocrinol 169(6):867–884. https://doi.org/10.1530/EJE-13-0442
Acknowledgements
We are grateful to the following clinicians for collecting patient samples and clinical data: Ana Agapito (Lisboa), Ana Monteiro (Braga), Ana Palha (Lisboa), Bernardo Pereira (Ponta Delgada), Daniela Cavaco (Lisboa), Davide Carvalho (Porto), Fernando Fonseca (Lisboa), Hélder Simões (Lisboa), Inês Barros (Braga), Inês Damásio (Lisboa), Isabel Inácio (Porto), Joana Pereira (Lisboa), João Duarte (Lisboa), Maria Bugalho (Lisboa), Maria Manuel Silva (Porto), Maria Salomé (Lisboa), Mariana Barbosa (Braga), Olinda Marques (Braga), Sara Donato (Lisboa), Sara Pinheiro (Lisboa), Sílvia Paredes (Braga), Teresa Martins (Coimbra), Teresa Rego (Lisboa), Tiago Silva (Lisboa), Valeriano Leite (Lisboa).
Funding
Open access funding provided by FCT|FCCN (b-on). This research was funded by the Portuguese Foundation for Science and Technology (FCT, project Grants PTDC/MEC-MET/29489/2017 and UIDB/00709/2020). L.M.G. was the recipient of a PhD fellowship from FCT (SFRH/BD/147160/2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. The study was approved by the Ethics Committee of the Faculty of Health Sciences, University of Beira Interior (Ref. CE-UBI-Pj-2018-027).
Informed consent
Written informed consent was obtained from all subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gaspar, L.M., Gonçalves, C.I., Saraiva, C. et al. Low frequency of AIP mutations in patients with young-onset sporadic pituitary macroadenomas. J Endocrinol Invest 46, 2299–2307 (2023). https://doi.org/10.1007/s40618-023-02083-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-023-02083-7