
Abstract
Anal squamous cell carcinoma (ASCC) is a rare gastrointestinal malignancy associated with high-risk human papillomavirus (HPV) and is currently one of the fastest-growing causes of cancer incidence and mortality in developed countries. Although next-generation sequencing technologies (NGS) have revolutionized cancer and immuno-genomic research in various tumor types, a limited amount of clinical research has been developed to investigate the expression and the functional characterization of genomic data in ASCC. Herein, we comprehensively assess recent advancements in “omics” research, including a systematic analysis of genome-based studies, aiming to identify the most relevant ASCC cancer driver gene expressions and their associated signaling pathways. We also highlight the most significant biomarkers associated with anal cancer progression, gene expression of potential diagnostic biomarkers, expression of therapeutic drug targets, and emerging treatment opportunities. This review stresses the urgent need for developing target-specific therapies in ASCC. By illuminating the molecular characteristics and drug-target expression in ASCC, this study aims to provide insights for the development of precision medicine in anal cancer.
Similar content being viewed by others


Access to the latest precision medicine approaches has been limited in anal cancer, where the treatment has remained the same over the last decades with regards to the scope of targeted therapy in clinical practice. |
This article summarizes the current genomic advances and emerging biomarkers for anal squamous cell carcinoma that delineates exciting enormous challenges to move toward to a personalized approach for ASCC |
1 Introduction
Anal squamous cell carcinoma (ASCC) is a rare cancer, accounting for less than 3% of all gastrointestinal neoplasms and less than 1% of all worldwide cancers in both men and women [1]. Worldwide, ASCC is one of the fastest accelerating causes of cancer incidence and mortality in developed countries, particularly in North America and Western Europe [2, 3]. In the USA, 8590 new cases in women and 3350 new cases in men were estimated in 2021. According to the European Cancer Information System (ECIS), there were an estimated 16,600 new cases and 7300 deaths in 2020 [4]. Usually, the average age at diagnosis is around 62 years. However, there has also been an increase in the incidence of anal cancer among younger adults. The age-standardized incidence rate of anal cancer has increased from 0.7 to 1.2 cases per 100,000 populations in 2018, particularly among those aged 20–49 years [5]. The reason for the increase in anal cancer incidence is correlated to an increase in the burden numbers of human papillomavirus (HPV)-related cancers. Other well-known risk factors associated with ASCC are HIV infection, tobacco smoking, immunosuppression following transplantation, and autoimmune diseases such as Crohn’s disease [5, 6].
There are rising levels of awareness over the last few years regarding the prevention of HPV related cancer via vaccination programs and the importance of screening based on an annual systematic Pap test or HPV screening detection, which has been recommended for high-risk groups [7, 8]. Unfortunately, treatment options for non-metastatic ASCC (NM-ASCC) have not evolved significantly over the last 2 decades; concurrent chemoradiotherapy remains the standard of care strategy for non-metastatic diseases. A combination of chemoradiotherapy has yielded a high rate of complete local regression and stands as an efficient strategy allowing anal-surgery preservation and sparing anal functions [8]. Nevertheless, 5-year disease-free survival (DFS) rates in NM-ASCC remain very heterogeneous, ranging from 85% in early tumors to 35% ASCC with lymph node invasion [6, 9, 10]. In clinical practice, prognostic factors of survival in ASCC are the T and N stage, sex, differentiation status, tumor location, HIV-HPV coinfection, and occurrence of a complete response after CRT [11]. Regrettably, these clinical parameters correlating with survival cannot be used to tailor therapy and predict treatment response in individual patients.
In patients with either metastases at diagnosis or who develop metastatic recurrences following chemoradiation therapy, the 5-year survival rate is less than 20% [12]. Thus far, platinum, fluorouracil, and taxanes are the most frequently used anticancer drugs for palliative chemotherapy. Clinical trials using PD1-1/PDL1 either alone or in combination with chemotherapy are ongoing. None of the targeted drugs used in other tumor types have been developed in advanced or metastatic ASCC.
A better understanding of the biology of ASCC may help in developing drug targeted programs. Recently significant progress in genomics research shedding light on the biological mechanisms underlying ASCC has been reported [13,14,15,16,17]. Although, to date, owing to the low incidence of ASCC, there is still a great deal of genomic information to explore, large data sets investigating DNA molecular pathways, epigenetics, tumor microenvironment, and ct-DNA are available and could be used to guide molecular research in ASCC. This review aims to present a comprehensive overview of the main advances achieved in the molecular biology of ASCC to encourage continuous international interest in anal cancer.
2 Genomic Changes in ASCC Development and Progression
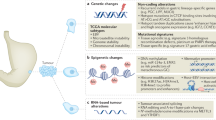
The rare incidence of ASCC has limited the molecular characterization of its mutational landscape compared with other cancers. Analyzing methodically mutational profiles from nine genome-based studies enabled us to identify the primary ASCC cancer driver mutations (Fig. 1). As previously reported, with PIK3CA being the most frequently mutated gene (30–40%), particularly in HPV-positive cases in line with other HPV malignancies [17,18,19]. The E545K variant [c.1633G>A (p.Glu545Lys)] in PIK3CA’s helical domain is prevalent, leading to constitutive Akt signaling activation [20]. Recent studies have also suggested that the APOBEC family of cytidine deaminases, which play a key role in the innate immune response to viral infection, may contribute to generating PIK3CA mutations in HPV-positive ASCC [21]. APOBEC enzymes can deaminate cytidine residues in single-stranded DNA, leading to the accumulation of mutations. HPV infection can induce the expression of APOBEC enzymes, which may contribute to the high mutation burden observed in HPV-positive ASCC. In contrast, tumors exhibiting low APOBEC activity demonstrate an equal likelihood of mutations in the kinase domain hot spot and the helical domain of PIK3CA. These mutations may arise from alternative mutational processes, which contribute to the activation of these mutations and potentially facilitate carcinogenesis [19,20,21,22,23]. The prognostic value of PIK3CA mutations remains controversial in ASCC, as some studies identified PIK3CA mutations as poor prognostic factors, but this was not confirmed in further investigations [22, 24]. On the basis of the high frequency of PIK3CA mutation, targeted agents, such as alpelisib, a PI3K inhibitor, are being studied in other SCC cancers, either alone or in combination [25]. Furthermore, data have suggested that mTOR inhibitors, such as everolimus, could be effective for cases with PIK3CA mutation [25,26,27]. These mutations have also presented a potential resistance to anti-EGFR-based therapies [28].

Heatmap of the most relevant cancer driver mutation identified across nine ASCC cohorts using genomics-based sequencing platforms. Briefly, mutational profiles and their reported frequencies were obtained from nine ASCC studies. The visualization of commonly mutated genes reported in at least two datasets and their associated signaling pathways was performed with the Multi-Experiment Viewer software. ASCC anal squamous cell carcinoma
Other cancer drivers appear to be mutated in a relatively low frequency, such as FBXW7 and KMT2D, ranging between 10% and 20% in most cohorts, respectively [15]. The KMT2D gene encodes a protein known as lysine (K)-specific methyltransferase 2D, also known as MLL2 or MLL4. In addition, KMT2C (also known as MLL3) was found to be mutated in cases of anal cancer [21, 29]. This protein is a histone methyltransferase, which adds a methyl group to histone proteins, leading to changes in chromatin structure and gene expression. KMT2D is also a tumor suppressor gene that is the target of frequent inactivating mutations in several tumor types, including colorectal, pancreatic, and gastric cancer, but their role in anal cancer is not yet fully understood. Also, a new report shows that KMT2D/C loss of function mutations could be associated with tumor-infiltrating lymphocytes and response to immune checkpoint inhibitors in solid tumors [30].
HPV-negative ASCCs are known to display a genomic profile that differs from HPV-positive cases. These tumors have a profile marked mainly by associations with TP53 and CDKN2A variants, which explains their resistance to standard chemotherapy [31]. Owing to their extremely low frequency, a deeper understanding is needed to allow for the use of precision targeted therapies to patients. In theory, we may anticipate similar profiles in copy number aberrations (CNA), such as esophageal squamous cancer cells [32].
Mutations in the RAS pathway have been widely recognized as significant prognostic and predictive biomarkers in colorectal cancer [29]. The aforementioned molecular profiling studies in ASCC have shown that KRAS, BRAF, and NRAS mutations are infrequent, providing a rational for the clinical investigation of anti-EGFR therapies. The randomized phase II CARACAS trial explored dual PD-1 and EGFR blockade in previously treated advanced SCCA patients. Translational analyses in this trial [33] showed that TMB-high and PDL-1 expression were associated with survival benefits in patients treated with anti-PDL1 either with or without cetuximab.
Of note, TMB is being suggested as an agnostic response biomarker in solid tumors for patients treated with pembrolizumab [34]. Although TMB-high proved to be a predictive indicator for immune checkpoint inhibitors in several solid tumors, their use in HPV-positive malignancies is unclear. In head and neck squamous cell carcinoma, HPV infection was associated with increased benefit from PD1/PD-L1 blockade, regardless of TMB. HPV-positive tumors also displayed significantly increased T-cell infiltration and T-cell-inflamed gene expression profiles [35]. These discoveries also suggest that TMB-high is not a promoter of immunogenicity and immune infiltration, but these features are consequences of HPV infection and they are not an effect of any DNA repair mechanism inactivation. In this trial, 12% of patients were found to be TMB-high (10 > mutations/MB), in agreement with a recent report in the largest comprehensive molecular cohort known to date in ASCC that reported 13% (88 with high TMB/from 668 ASCC patients) [35, 36].
The MMR/MSI status, another agnostic somatic signature in solid tumors, was reported in 2% of patients who expressed hypermutant profiles in ASCC [33, 34]. Similar results were observed in the KEYNOTE-158 trial [37], where only one patient with MSI-high and SCCA was included. Despite the weirdness of MSI-H ASCCCA, the agnostic function of this biomarker suggests it is capable of indicating a response to immunotherapies in several tumor types.
3 Epigenomic Changes in ASCC Development and Progression
The progression of HPV-induced precancerous lesions to invasive carcinomas is driven by the accumulation of genomic and epigenomic modifications affecting host cell genes [38]. Epigenetic changes are heritable alterations in gene expression that do not involve changes to the underlying DNA sequences. These changes are an important component of cancer development and progression, and involve alterations in DNA methylation, histone modifications, chromatin remodeling, and the expression of non-coding genes.
DNA methylation is a type of epigenetic modification in which a methyl group is added to a cytosine nucleotide located 5′ of a guanine (CpG) in gene promoter regions and other genomic sites. When the promoter regions of tumor suppressor genes are hypermethylated, this can lead to their inactivation, which can contribute to the development of cancer. Therefore, DNA methylation patterns in specific genes could potentially serve as valuable biomarkers for the detection of anal (pre-)cancer, as well as for monitoring disease progression and treatment response.
Although likely important, the role of DNA methylation in the development of anal cancer remains poorly characterized [39]. Zhang et al. provided the first evidence of aberrant methylation in anal cancer among 11 candidate genes compared with normal tissue using a methylation-specific qPCR-based method. This study suggested that DNA methylation was more common in ASCC and high grade squamous intraepithelial lesions (HGSILs) than in low grade squamous intraepithelial lesions (LGSILs) and normal mucosa. While methylation of IGSF4 and DAPK1 was more prevalent in ASCC and HGSIL, it was absent in LGSIL and normal biopsy samples [39, 40]. Subsequently, an array-based assay analyzing > 1500 CpG sites representing 807 genes reported differences in DNA methylation patterns in 20 genes in the progression from normal anal mucosa to invasive anal carcinoma in a small set of cases (24 patients). Interestingly, 5 out of 20 genes showed no methylation in non-invasive tissues with a significant increase in invasive SCC (CD9, DAPK1, FLT1, HOXA5, and PADI4) [41]. In addition, Siegel et al. reported a total of seven hypermethylated genes (ADAT3, GSG1L, LOC728392, PARD3, SALL3, SFRP2, and SCAMP4) in high- versus low-risk anal cancer cases from a cohort of 121 patients with locally advanced anal cancer using the Illumina HumanMethylation450 array [41]. No biomarker is available for routine clinical practice to determine progression risk of high-grade squamous intraepithelial lesion (HSIL) to ASCC. van der Zee et al. reported a host DNA methylation marker panel (ASCL1, SST, and ZNF582) for the detection of anal pre-cancer lesions (high grade AIN) with a higher risk of progression to ASCC in HIV+ men [42].
In a more recent study, Siegel et al., conducted a genome-wide methylation study of 143 FFPE anal tissues including normal, high-grade pre-neoplastic lesions and anal cancer using the Illumina HumanMethylation 450 array [43]. The authors identified an 84-gene signature differentially methylated between normal anal mucosa and anal cancer. Moreover, this signature segregated anal intraepithelial neoplasias into normal-like or cancer-like groups. Interestingly, our functional enrichment analysis of the Siegel et al. 84-gene methylated signature data showed the involvement of bioprocess related with digestive tract development, transcriptional regulation, cAMP signaling pathway, and neuroactive ligand-receptor interaction, among others (Fig. 2) [43]. Interestingly, neuroactive ligand–receptor interactions have been recently shown to be associated with the development and progression of colorectal and other gastrointestinal cancers [44,45,46]. Whether neuroactive ligand-receptors can directly modulate or affect tumor progression in anal cancer is worthy of further exploration. The development of appropriate regulatory drugs targeting these relevant pathways may contribute to improve the treatment of ASCC.

Gene and pathways network identified among 84 methylated genes in ASCC by Siegel et al. [43]. Differentially methylated genes are colored in green, and their related bioprocesses are colored according to the derived database (KEGG in red, Gene Ontology Biological Process in blue, and MGI Mammalian Phenotype in yellow). ASCC anal squamous cell carcinoma, KEGG pathway Kyoto Encyclopedia of Genes and Genome pathway
Overall, these studies found that significant epigenetic alterations occur in the progression from early to later stage locally advanced ASCC, and genes harboring differentially methylated CpG sites included known tumor suppressor genes and novel targets not previously described in other tumor sites [47]. Previously, studies have described differential methylation patterns across anal squamous neoplastic progression, including normal tissue, precancerous lesions, and anal carcinoma [48]. In addition, HPV may influence the host transcriptome through several epigenetic mechanisms, including HPV E7 oncoprotein-mediated alterations in DNA methyltransferases [49]. Growing evidence suggests that HPV-associated oncogenesis in different organ sites may be associated with common non-random genome-wide methylation events [50]. The differences in methylation may lend clues to understanding the molecular alterations that occur with the malignant progression of anal cancer. Effective methylation-related biomarkers may ultimately guide treatment modification for high-risk patients, including radiation dose intensification, closer monitoring of dose completions and/or gaps in treatment, and developing novel targeted radio-sensitizing agents (Fig. 3).

Diagram of the immune escape pathways modulated by HPV that could contribute to ASCC progression
4 Transcriptomic and Proteomic Changes in ASCC Development and Progression
The development of effective tools, such as DNA microarrays and NGS-based methods, for monitoring global gene expression on a large scale has resulted in the discovery of regulatory pathways in almost all tumor-type processes. In this regard, transcriptomic analysis of cancers has undergone extensive profiling over the last 2 decades, enabling the identification of intrinsic molecular subtypes along with prognostic and predictive gene expression signatures. Not surprisingly, very recent studies evaluating transcriptomic changes that occur during ASCC development and treatment response outcomes are starting to emerge. In this sense, Ye et al. performed the first RNA-seq characterization of differentially expressed transcripts among 12 formalin-fixed paraffin-embedded (FFPE) tumors derived from successful CRT versus recurrent ASCC patients living with HIV. The authors identified 449 differentially expressed coding and non-coding genes among the groups, with a core of immune-related up-regulated genes in the non-recurrent ASCC cases, which suggests a CD4+ T cell-driven immune response. Upregulated in genes in the recurrent cases were related to epidermis development, such as cytokeratin and the hedgehog signaling pathway [51]. Overall, this study suggests that a complex immune-regulatory network may be acting within initial non-recurrent anal cancer isolates which is disrupted upon recurrence.
The extensive molecular profiling of this rare cancer has been hampered by the challenge of acquiring fresh tumor tissue necessary for RNA and protein expression analysis. Recently, Hernandez et al. employed a digital spatial profiling technology on pretreatment anal cancer FFPE specimens to identify biomarkers associated with recurrence after chemoradiation. The authors report that recurrent tumors had higher protein expression of FoxP3, MAPK-activation markers (BRAF, p38-MAPK), and PI3K/Akt activation (phospho-Akt) within the tumor margins. In addition, the tumor microenvironment was characterized by the higher protein expression of immune checkpoint biomarkers, such as PD-1, OX40L, and LAG3. However, no statistically significant differences were identified among the cases compared for RNA expression profile analysis of immune-related gene targets measured using this approach [52].
In addition, a functional proteomics analysis of ASCCs performed by Trilla-Fuertes et al. proposed a molecular classification in two distinctive groups of patients, one group with increased expression of proteins related to cell adhesion, T lymphocytes, and glycolysis; and the other group with increased expression of proteins related to translation and ribosomes bioprocess. However, non-clinicopathological, treatment responses or outcomes were associated with these proteomics-based groups [53]. Further studies on the transcriptomic and proteomic profiling in large ASCC cohorts are needed to extend and corroborate the aforementioned observations.
5 Role of Liquid Biopsy in ASCC Management: HPV-DNA
Circulating tumor DNA (ctDNA) is a fraction of cell-free circulating DNA originating from tumor cells. Serving as a sensitive, real-time biomarker, dynamic ctDNA levels predict treatment response and outcomes across various tumor types [54,55,56,57]. In cancer patients, cell-free DNA (cfcDNA) is mostly derived from apoptotic or necrotic hematopoietic cells, with a portion originating from tumor cells [58]. Distinguishing cfcDNA from ctDNA requires identifying a specific tumor DNA alteration through genome sequencing or PCR-based methods. In contrast, when hrHPV infects a host, its DNA is integrated into the host genome or resides in episomal form, expressing the oncogenes, E6 and E7 to trigger carcinogenesis [59]. The presence of one or more copies of the HPV genome in cfDNA from HPV-related malignancies suggests tumor cell origin [60]. Searching for HPV-DNA in liquid biopsies offers a technological advantage over detecting tumor mutations owing to the size, composition, and numerous copies of each viral DNA in the HPV genome [61, 62]. Compared with tumor DNA with point mutations, HPV DNA released from tumor cells is simpler to detect, potentially eliminating the need to identify tumor DNA alterations [63]. Individuals with HPV-related malignancies exhibit the HPV genome, particularly E6 and E7 DNA, while HPV ctDNA is not detected in healthy controls and HPV16-associated intraepithelial neoplasia [64]. Consequently, HPV-induced tumors serve as an ideal model for monitoring ctDNA. In anal squamous cell carcinoma (ASCC), the necessity for noninvasive markers arises owing to primary tumor chemoradiotherapy, delayed response in some patients achieving complete response after months, and occasional inadequacy of biopsy material [65].
The analytical sensitivity of HPV-DNA tests has significantly improved with advanced technologies such as digital droplet PCR (ddPCR) and NGS. ddPCR exhibits high specificity (97–100%) and sensitivity (89–98.4%) in detecting HPV-DNA in blood samples [61, 66, 67], requiring fewer resources and offering quicker turnaround times than NGS. While some studies focused exclusively on HPV16, the primary cause of HPV-related SCCA, others adopted a comprehensive approach, including other hrHPV genotypes (18, 31, 33, 35, 45, 51, 52, 58, and 73) in proof of concepts [68,69,70]. A multiplex ddPCR method testing five HPV subtypes in a single assay was developed, saving both sample input and detection time [70, 71]. However, ddPCR has a drawback as HPV-DNA analysis cannot be conducted directly in a single step; prior knowledge of viral sequences is necessary to detect them previously in the patient’s cancer tissue. This limitation poses a challenge for ASCC HPV-negative patients [72].
The first NGS-based test for circulating HPV16 DNA demonstrated 100% sensitivity, surpassing ddPCR, even in early tumor stages [73]. The CaptHPV method, utilizing viral genome hybrid-capture-based cHPV-DNA sequencing, provides a comprehensive overview of HPV status, including genotype, quantity, entire sequence, fragment length, and insertion pattern, from a single blood sample [74]. The NGS-based CaptHPV approach captures whole genome sequences of over 200 HPV genotypes, allowing a detailed molecular analysis with high sensitivity and specificity. An ultrasensitive viral capture-based cHPV-DNA assay (HPV-seq) is more sensitive than ddPCR, detecting 0.6 copies of ctDNA, and providing information on ctDNA fragment length and viral integration sites [75]. HPV-seq, with its high sensitivity, is suitable for early detection and minimal residual disease surveillance. For long-term analysis during cancer treatment monitoring, cost-effective ddPCR is a viable option (Table 1). Droplet digital PCR for HPV ctDNA detection appears to be a rapid, noninvasive, and affordable prognostic marker for patients with ASCC [72, 76].
We summarize the main results of this approach in ASCC in Table 2. At first, we outlined that there was a strong correlation observed between higher levels of HPV ctDNA pretreatment and disease burden according to clinical staging. However, the majority of series failed to establish a correlation between pretreatment ctDNA levels and oncological outcomes. Therefore, HPV DNA copies may not serve as a prognostic marker before treatment. This discrepancy is probably attributed to variations in HPV copies in the cancer cells among patients and the limited number of cases in the studies [61, 68, 69, 77,78,79,80]. All series represented in Table 2 have shown good sensitivity and feasibility. The most remarkable finding in all series was that, in the different treatment settings, such as after chemoradiotherapy in NM-ASCC or after chemotherapy in the metastatic approach, the remaining or residual high levels of ctDNA were associated with a worse PFS compared with patients with undetectable levels. The conversion rate after treatment has an impact on the prognosis of ASCC. The prognostic impact of HPV ctDNA appears to be independent of the stage. Thus, these results show a potential role for an early dynamic marker of treatment efficacy and risk of relapse.
Given these findings, HPV ctDNA could serve as a biomarker for rapid responders to CRT, predict sustained response to chemotherapy, and aid in early detection of disease progression during follow-up. It may guide personalized post-CRT therapies, identify those at high risk of relapse, and potentially serve as an efficacy marker to measure the efficacy of immune checkpoint inhibitors in different treatment settings [69, 72, 75, 81,82,83].
Despite the potential of liquid biopsies, challenges include heterogeneous study designs, varying cHPV-DNA assays, and small sample sizes, limiting generalizability [72]. Standardization is crucial for blood collection, cfDNA isolation, sample storage, HPV probe design, and threshold determination. Ongoing studies such as Circa HPV (NCT03739775), INTERACT-ION neoadjuvant (NCT02897427), and PLATO ASCC platform are exploring the role of HPV ctDNA in cancer screening, treatment modulation, and post-therapy surveillance [84,85,86,87,88]. Further research will confirm the clinical utility of HPV ctDNA for optimizing SCCA therapy management.
6 Role of the Tumor Immune Microenvironment in ASCC
Genetic and epidemiological studies have underlined the immunosuppressive potential of HPV-driven oncogenesis [89]. Persistent infection with high-risk HPV subtypes causes malignant transformation due to the activation of HPV E6 and E7 oncogenes, which block the p53 and Rb tumor suppressors, respectively. These oncoproteins primarily enhance angiogenesis, genomic instability, telomere shortening inhibition, apoptosis inhibition, and facilitate the invasion and metastasis process. Further, HPV modifies the tumor microenvironment to produce immune-suppressive and immune evasion conditions that are pro-tumorigenic [89,90,91]. In this sense, HPV malignancies are associated with a genetic profile of germline and somatic mutations that affect critical immune-related pathways, such as antigen presentation and immune checkpoints, associated with the HPV immune evasion and resistance phenotype [92,93,94].
6.1 HPV-Mediated Modulation of the Adaptive and Innate Antitumor Immunity
The activation of CD4+ and CD8+ T cells and T cell-mediated tumor cytotoxicity are dependent on intact antigen presentation by antigen-presenting cells and tumor cells. HPV tumors frequently harbor mutations in genes affecting the antigen presentation pathway of both major histocompatibility complex (MHC) class I and II molecules. Mutations in HLA-A and HLA-B genes that affect antigen presentation, and gain-of-function mutation in CD274 (gene that encodes the immune inhibitory receptor PD-L1) have been recently reported in HPV-positive tumors. Furthermore, oncogenic HPV gene expression is associated with the downregulation of immune-related pathways that affect multiple cellular targets including antiviral genes (IFIT1 and MX1), genes involved in IFN signaling (STAT1), proapoptotic genes (TRAIL and XAF1), and pathogen recognition receptors (TLR3, RIG-I, and MDA5) [43, 95, 96].
HPV modulates innate immune defense mechanisms via pleiotropic effects on immune cell recognition, activation, and migration. Viral E5, E6, and E7 expression can block several pathways that affect antigen processing and HLA presentation of viral peptides at the surface of infected cells, thereby facilitating immune escape [89, 91, 97]. HPV reduces the susceptibility of the infected cells to interferon IFN-β and IFN-λs that impact immune recognition and activation processes. The HPV tumor microenvironment is associated with a reduced number of APCs (Langerhans cells) and low levels of inflammatory chemokines. CCL20, which attracts epidermal APCs that express the CCR6 receptor, is critical for innate immune activation in the skin or mucosa. HPV E6/E7 has a repressive effect on CCL20 transcription, leading to important changes in the migration capacity of Langerhans cells and effector lymphocytes to the anal lesions [98,99,100].
The HPV E5 protein causes alkalization of late endosomes, preventing peptide-bound MHC class I and II molecules from reaching the cell surface cells, and preventing the presentation of viral tumor-associated antigens on MHC molecules and the activation of anti-tumor CD4+ and CD8+ T cells. Overexpression of E5 is a potential mechanism of resistance to immune checkpoint blockade, likely through the acquired loss of antigen presentation [101, 102]. HPV also dysregulates specific HLA molecules and renders natural killer (NK) cells, which normally recognize cells without surface MHC expression, incapable of clearing virally infected cells [103]. A high level of CD8+ TIL infiltration is often associated with a better prognosis, including lower local recurrence rates, better disease-free survival (DFS), and better overall survival (OS). Similar results exist in anal cancer, and it appears that the presence of TILs can effectively estimate the risk of recurrence. A subset of CD4+ T cells, FOXP3+ regulatory T cells (Tregs), can suppress anti-tumor immunity by downregulating induction and proliferation of effector T cells. Tregs play an important role in dampening the host immune response in autoimmune diseases and viral infections. The frequency of Tregs correlated with disease severity, suggesting that Tregs may be associated with interference of HPV immunity. High and interrelated rates of PD-L1+CD14+ antigen-presenting cells and regulatory T cells mark the microenvironment of metastatic lymph nodes from patients with cervical cancer [104].
6.2 Immune Checkpoints and Tumor-Infiltrating Lymphocytes as Biomarkers in ASCC
The PD-1/PD-L1 immune checkpoint is a conserved inhibitory mechanism regulating the immune system that prevents autoimmunity [104, 105]. Multiple cancers, including HPV-related tumors, upregulate this checkpoint to facilitate immune tolerance. The incidence of PDL1-positive tumors is variable, ranging from 56 to 68.8%, and its role as a prognostic factor is still subject to debate. HPV-positive tumors were more likely to have a higher intensity of TIL infiltrate and TAMs in HPV-positive tumors than HPV-negative tumors [90]. In a HPV+ cohort, CD8+ TILs with high expression of PD-L1 were associated with better overall survival than CD8+ TILs with low expression of PD-L1. When PD-1 expression on TILs was analyzed by compartment, an association with survival was found for PD-1 expressing TILs in the tumor, and at the edge of the tumor but not for stromal TILs expressing PD-1 [14]. As discussed above, TILs are more abundant in HPV-positive tumors when compared with HPV-negative tumors. Since PD-L1 is a dynamic biomarker that gets upregulated in response to IFNγ secretion by TILs, the increase in PD-L1 expression in HPV-positive tumors suggests a more inflammatory tumor microenvironment with the recruitment of TILs [35, 106]. The study revealed that patients with tumor PD-L1 ≥ 5% had significantly longer survival than those with PD-L1 less than 5%, with a 10-year survival rate difference of 84% and 49%, respectively. Previous studies have reported better survival in PDL1+ patients. Others have shown an association with worse survival. Discrepancies are partially due to the absence of a standardized methodology for reporting PD-L1 expression (absence of standardized thresholds for positivity, immune cell assessment, and differences in staining between antibodies) and may also be related to the correlation between PD-L1 expression and both a favorable CD8+ TILs microenvironment and the activation of the unfavorable PD-1/PD-L1 immune escape pathway [107].
The presence of tumor-infiltrating lymphocytes (TILs) is more important in ASCC HPV-positive tumors compared with HPV-negative tumors. As PDL1+ is a dynamic biomarker whose expression is stimulated in response to IFN gamma by TILs, it can be hypothesized that PDL1+ expression simply reflects a more inflammatory tumor microenvironment. To date, no further insights have been gained from clinical trials [107, 108]. For instance, in the Keynote 028 phase 1B multi-cohort study, which involved 24 PDL1+ patients, an ORR of 17% was observed [109]. In the Keynote 158 a phase 2 study, which included 112 ASCC patients, the ORR was 11.6%, with 14.7% in patients with CPS > 1 and 6.7% in patients with CPS < 1, suggesting that CPS may have predictive value [110]. A phase II study with retifanlimab showed an ORR of 13.8%, PFS of 2.3 months, and median OS of 10.1 months, with no difference between PDL1 positive or negative [111]. It is challenging to compare these studies since different measurement systems were used, such as CPS or TPS, with varying cutoffs of 1% or 5% for PD-L1 expression levels. Therefore, standardization of the PD-L1 evaluation is required to integrate data from various studies and use this biomarker in the clinical setting. Interestingly, tumors caused by the human papillomavirus (HPV) have a better prognosis. This may be due to several factors. Firstly, the presence of viral antigens can stimulate the immune system and result in infiltration of immune cells, such as CD8+ and PD1+ TILs, into the tumor. Another possible explanation is that HPV-infected cells are frequently p53 wild type, which can activate the external apoptosis program, in contrast to patients with HPV-negative ASCC [112].
ASCC often have mutations in TP53 and CDKN2A, which are associated with poor prognosis [31, 113]. However, there are conflicting data on this topic. HIV infection creates a microenvironment that facilitates the persistence and progression of precancerous lesions The status of PDL1 expression does not appear to be influenced by HIV infection [114]. Also, no association was found between peripheral CD4 count and the expression of checkpoint inhibitors, such as PD-1, PDL1, and LAG-3. Further studies are needed to better understand the relationship between CD4+ cells and PD-L1 expression in HIV-positive patients, as well as the effect of antiretroviral therapies and immunotherapy.
7 Conclusions
Recent advancements in high-throughput sequencing and computational analysis offer unprecedented opportunities to comprehensively explore the molecular and immune landscapes of tumors across diverse cancer types. These approaches have identified genomic alterations and immune-related signatures crucial in driving oncogenesis and immune evasion in ASCC. Early-stage genomic and epigenomic modifications play a pivotal role in progressing HPV-induced precancerous lesions to invasive carcinomas. Mutational biomarkers such as PI3KCA show prognostic and predictive value, guiding targeted therapy use. Agnostic markers, such as TMB and MMR/MSI status, have potential roles, especially in immune checkpoint blockade. DNA methylation patterns in specific genes may serve as valuable biomarkers for detecting anal precancer and monitoring disease progression. Transcriptomic studies reveal immune modulation in nonrecurrent and recurrent ASCC cases, with specific events favoring a pro-tumorigenic immune microenvironment in HPV-associated cancers. HPV-driven malignancies exhibit immunosuppressive conditions, impacting antitumor immunity. Regulatory T cells and PD-L1 further hinder the immune response in the tumor microenvironment. Targeting these immune dysregulations holds promise for effective immunotherapeutic strategies in ASCC. In the metastatic setting, immune checkpoint inhibitors are rapidly evolving, being investigated in second-line, in combination with front-line chemotherapy, and even in the neo-adjuvant setting in ongoing trials. In light of these discoveries, HPV ctDNA has the potential to function as a biomarker for promptly identifying responders to CRT, predicting sustained responses to chemo/chemoradiotherapy, and facilitating the early detection of disease progression during post-treatment monitoring. It could play a crucial role in tailoring post-CRT therapies, identifying individuals with an elevated risk of relapse and a tool to guide decisions on deescalating or escalating. Collaborative research networks focused on this rare pathology, an enhanced understanding of tumor biology and its microenvironment, coupled with the advent of immunotherapies, are essential for a highly promising future in this neglected disease.
References
Islami F, Ferlay J, Lortet-Tieulent J, Bray F, Jemal A. International trends in anal cancer incidence rates. Int J Epidemiol. 2017;46:924–38. https://doi.org/10.1093/ije/dyw276.
Nelson VM, Benson AB 3rd. Epidemiology of anal canal cancer. Surg Oncol Clin N Am. 2017;26:9–15. https://doi.org/10.1016/j.soc.2016.07.001.
Kang Y-J, Smith M, Canfell K. Correction: anal cancer in high-income countries: increasing burden of disease. PLoS One. 2019;14: e0216884. https://doi.org/10.1371/journal.pone.0216884.
Dyba T, Randi G, Bray F, Martos C, Giusti F, Nicholson N, et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur J Cancer. 2021;157:308–47. https://doi.org/10.1016/j.ejca.2021.07.039.
Clifford GM, Georges D, Shiels MS, Engels EA, Albuquerque A, Poynten IM, et al. A meta-analysis of anal cancer incidence by risk group: toward a unified anal cancer risk scale. Int J Cancer. 2021;148:38–47. https://doi.org/10.1002/ijc.33185.
Eng C, Messick C, Glynne-Jones R. The management and prevention of anal squamous cell carcinoma. Am Soc Clin Oncol Educ Book. 2019;39:216–25. https://doi.org/10.1200/EDBK_237433.
Thanasa E, Thanasa A, Kamaretsos E, Paraoulakis I, Balafa K, Gerokostas E-E, et al. Awareness regarding human papilloma virus among health professionals and will to accept vaccination: a systematic review. Cureus. 2022;14: e30855. https://doi.org/10.7759/cureus.30855.
Rao S, Guren MG, Khan K, Brown G, Renehan AG, Steigen SE, et al. Anal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up☆. Ann Oncol. 2021;32:1087–100. https://doi.org/10.1016/j.annonc.2021.06.015.
Ajani JA, Winter KA, Gunderson LL, Pedersen J, Benson AB 3rd, Thomas CR Jr, et al. Prognostic factors derived from a prospective database dictate clinical biology of anal cancer: the intergroup trial (RTOG 98–11). Cancer. 2010;116:4007–13. https://doi.org/10.1002/cncr.25188.
Gunderson LL, Winter KA, Ajani JA, Pedersen JE, Moughan J, Benson AB 3rd, et al. Long-term update of US GI intergroup RTOG 98-11 phase III trial for anal carcinoma: survival, relapse, and colostomy failure with concurrent chemoradiation involving fluorouracil/mitomycin versus fluorouracil/cisplatin. J Clin Oncol. 2012;30:4344–51. https://doi.org/10.1200/JCO.2012.43.8085.
Iseas S, Prost D, Bouchereau S, Golubicki M, Robbio J, Oviedo A, et al. Prognostic factors of long-term outcomes after primary chemo-radiotherapy in non-metastatic anal squamous cell carcinoma: an international bicentric cohort. Biomedicines. 2023. https://doi.org/10.3390/biomedicines11030791.
Nilsson MP, Nilsson ED, Johnsson A, Leon O, Gunnlaugsson A, Scherman J. Patterns of recurrence in anal cancer: a detailed analysis. Radiat Oncol. 2020;15:125. https://doi.org/10.1186/s13014-020-01567-7.
Iseas S, Golubicki M, Robbio J, Ruiz G, Guerra F, Mariani J, et al. A clinical and molecular portrait of non-metastatic anal squamous cell carcinoma. Transl Oncol. 2021;14: 101084. https://doi.org/10.1016/j.tranon.2021.101084.
Guerra GR, Kong JC, Millen RM, Read M, Liu DS, Roth S, et al. Molecular and genomic characterisation of a panel of human anal cancer cell lines. Cell Death Dis. 2021;12:959. https://doi.org/10.1038/s41419-021-04141-5.
Ito T, Takayanagi D, Sekine S, Hashimoto T, Shimada Y, Matsuda M, et al. Comparison of clinicopathological and genomic profiles in anal squamous cell carcinoma between Japanese and Caucasian cohorts. Sci Rep. 2023;13:3587. https://doi.org/10.1038/s41598-023-30624-w.
Mouw KW, Cleary JM, Reardon B, Pike J, Braunstein LZ, Kim J, et al. genomic evolution after chemoradiotherapy in anal squamous cell carcinoma. Clin Cancer Res. 2017;23:3214–22. https://doi.org/10.1158/1078-0432.CCR-16-2017.
Morris V, Rao X, Pickering C, Foo WC, Rashid A, Eterovic K, et al. Comprehensive genomic profiling of metastatic squamous cell carcinoma of the anal canal. Mol Cancer Res. 2017;15:1542–50. https://doi.org/10.1158/1541-7786.MCR-17-0060.
Smaglo BG, Tesfaye A, Halfdanarson TR, Meyer JE, Wang J, Gatalica Z, et al. Comprehensive multiplatform biomarker analysis of 199 anal squamous cell carcinomas. Oncotarget. 2015;6:43594–604. https://doi.org/10.18632/oncotarget.6202.
Chung JH, Sanford E, Johnson A, Klempner SJ, Schrock AB, Palma NA, et al. Comprehensive genomic profiling of anal squamous cell carcinoma reveals distinct genomically defined classes. Ann Oncol. 2016;27:1336–41. https://doi.org/10.1093/annonc/mdw152.
Shin M-K, Payne S, Bilger A, Matkowskyj KA, Carchman E, Meyer DS, et al. Activating mutations in Pik3ca contribute to anal carcinogenesis in the presence or absence of HPV-16 oncogenes. Clin Cancer Res. 2019;25:1889–900. https://doi.org/10.1158/1078-0432.CCR-18-2843.
Cacheux W, Rouleau E, Briaux A, Tsantoulis P, Mariani P, Richard-Molard M, et al. Mutational analysis of anal cancers demonstrates frequent PIK3CA mutations associated with poor outcome after salvage abdominoperineal resection. Br J Cancer. 2016;114:1387–94. https://doi.org/10.1038/bjc.2016.144.
Cacheux W, Dangles-Marie V, Rouleau E, Lazartigues J, Girard E, Briaux A, et al. Exome sequencing reveals aberrant signalling pathways as hallmark of treatment-naive anal squamous cell carcinoma. Oncotarget. 2018;9:464–76. https://doi.org/10.18632/oncotarget.23066.
Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014;7:1833–41. https://doi.org/10.1016/j.celrep.2014.05.012.
Mondaca S, Chatila WK, Bates D, Hechtman JF, Cercek A, Segal NH, et al. FOLFCIS treatment and genomic correlates of response in advanced anal squamous cell cancer. Clin Colorectal Cancer. 2019;18:e39–52. https://doi.org/10.1016/j.clcc.2018.09.005.
Dunn LA, Riaz N, Fury MG, McBride SM, Michel L, Lee NY, et al. A phase 1b study of cetuximab and BYL719 (Alpelisib) concurrent with intensity modulated radiation therapy in stage III–IVB head and neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2020;106:564–70. https://doi.org/10.1016/j.ijrobp.2019.09.050.
Alqahtani A, Ayesh HSK, Halawani H. PIK3CA gene mutations in solid malignancies: association with clinicopathological parameters and prognosis. Cancers. 2019. https://doi.org/10.3390/cancers12010093.
Jung K, Kang H, Mehra R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers Head Neck. 2018;3:3. https://doi.org/10.1186/s41199-018-0030-z.
Parseghian CM, Napolitano S, Loree JM, Kopetz S. Mechanisms of innate and acquired resistance to anti-EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin Cancer Res. 2019;25:6899–908. https://doi.org/10.1158/1078-0432.CCR-19-0823.
Armstrong SA, Malley R, Wang H, Lenz H-J, Arguello D, El-Deiry WS, et al. Molecular characterization of squamous cell carcinoma of the anal canal. J Gastrointest Oncol. 2021;12:2423–37. https://doi.org/10.21037/jgo-20-610.
Liu R, Niu Y, Liu C, Zhang X, Zhang J, Shi M, et al. Association of KMT2C/D loss-of-function variants with response to immune checkpoint blockades in colorectal cancer. Cancer Sci. 2023;114:1229–39. https://doi.org/10.1111/cas.15716.
Meulendijks D, Tomasoa NB, Dewit L, Smits PHM, Bakker R, van Velthuysen M-LF, et al. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br J Cancer. 2015;112:1358–66. https://doi.org/10.1038/bjc.2015.20.
Hirata H, Niida A, Kakiuchi N, Uchi R, Sugimachi K, Masuda T, et al. The evolving genomic landscape of esophageal squamous cell carcinoma under chemoradiotherapy. Cancer Res. 2021;81:4926–38. https://doi.org/10.1158/0008-5472.CAN-21-0653.
Prete AA, Manca P, Messina M, Formica V, Frassineti GL, Zampino MG, et al. Extensive molecular profiling of squamous cell anal carcinoma in a phase 2 trial population: translational analyses of the “CARACAS” study. Eur J Cancer. 2023;182:87–97. https://doi.org/10.1016/j.ejca.2022.12.025.
Marcus L, Fashoyin-Aje LA, Donoghue M, Yuan M, Rodriguez L, Gallagher PS, et al. FDA approval summary: pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin Cancer Res. 2021;27:4685–9. https://doi.org/10.1158/1078-0432.CCR-21-0327.
Wang J, Sun H, Zeng Q, Guo X-J, Wang H, Liu H-H, et al. HPV-positive status associated with inflamed immune microenvironment and improved response to anti-PD-1 therapy in head and neck squamous cell carcinoma. Sci Rep. 2019;9:13404. https://doi.org/10.1038/s41598-019-49771-0.
Wang J, Xiu J, Farrell A, Baca Y, Arai H, Battaglin F, et al. Mutational analysis of microsatellite-stable gastrointestinal cancer with high tumour mutational burden: a retrospective cohort study. Lancet Oncol. 2023;24:151–61. https://doi.org/10.1016/S1470-2045(22)00783-5.
Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord J-P, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38:1–10. https://doi.org/10.1200/JCO.19.02105.
Soto D, Song C, McLaughlin-Drubin ME. Epigenetic alterations in human papillomavirus-associated cancers. Viruses. 2017. https://doi.org/10.3390/v9090248.
Zhang J, Martins CR, Fansler ZB, Roemer KL, Kincaid EA, Gustafson KS, et al. DNA methylation in anal intraepithelial lesions and anal squamous cell carcinoma. Clin Cancer Res. 2005;11:6544–9. https://doi.org/10.1158/1078-0432.CCR-05-0374.
Hernandez JM, Siegel EM, Riggs B, Eschrich S, Elahi A, Qu X, et al. DNA methylation profiling across the spectrum of HPV-associated anal squamous neoplasia. PLoS One. 2012;7: e50533. https://doi.org/10.1371/journal.pone.0050533.
Siegel EM, Eschrich S, Winter K, Riggs B, Berglund A, Ajidahun A, et al. Epigenomic characterization of locally advanced anal cancer: a radiation therapy oncology group 98–11 specimen study. Dis Colon Rectum. 2014;57:941–57. https://doi.org/10.1097/DCR.0000000000000160.
van der Zee RP, Richel O, van Noesel CJM, Ciocănea-Teodorescu I, van Splunter AP, ter Braak TJ, et al. Cancer Risk stratification of anal intraepithelial neoplasia in human immunodeficiency virus-positive men by validated methylation markers associated with progression to cancer. Clin Infect Dis. 2020;72:2154–63. https://doi.org/10.1093/cid/ciaa397.
Siegel EM, Ajidahun A, Berglund A, Guerrero W, Eschrich S, Putney RM, et al. Genome-wide host methylation profiling of anal and cervical carcinoma. PLoS One. 2021;16: e0260857. https://doi.org/10.1371/journal.pone.0260857.
Yao H, Li C, Tan X. An age stratified analysis of the biomarkers in patients with colorectal cancer. Sci Rep. 2021;11:22464. https://doi.org/10.1038/s41598-021-01850-x.
Yu J, Zhang Q, Wang M, Liang S, Huang H, Xie L, et al. Comprehensive analysis of tumor mutation burden and immune microenvironment in gastric cancer. 2021. Biosci Rep. https://doi.org/10.1042/BSR20203336.
Shi L, Zhang Y, Wang H. Prognostic prediction based on histopathologic features of tumor microenvironment in colorectal cancer. Front Med. 2023;10:1154077. https://doi.org/10.3389/fmed.2023.1154077.
Lorincz AT, Nathan M, Reuter C, Warman R, Thaha MA, Sheaff M, et al. Methylation of HPV and a tumor suppressor gene reveals anal cancer and precursor lesions. Oncotarget. 2017;8:50510–20. https://doi.org/10.18632/oncotarget.17984.
van der Zee RP, Richel O, van Noesel CJM, Novianti PW, Ciocanea-Teodorescu I, van Splunter AP, et al. Host Cell deoxyribonucleic acid methylation markers for the detection of high-grade anal intraepithelial neoplasia and anal cancer. Clin Infect Dis. 2019;68:1110–7. https://doi.org/10.1093/cid/ciy601.
Chaiwongkot A, Phanuphak N, Pankam T, Bhattarakosol P. Human papillomavirus 16 L1 gene methylation as a potential biomarker for predicting anal intraepithelial neoplasia in men who have sex with men (MSM). PLoS One. 2021;16: e0256852. https://doi.org/10.1371/journal.pone.0256852.
Phillips S, Cassells K, Garland SM, Machalek DA, Roberts JM, Templeton DJ, et al. Gene methylation of CADM1 and MAL identified as a biomarker of high grade anal intraepithelial neoplasia. Sci Rep. 2022;12:3565. https://doi.org/10.1038/s41598-022-07258-5.
Ye Y, Maroney KJ, Wiener HW, Mamaeva OA, Junkins AD, Burkholder GA, et al. RNA-seq analysis identifies transcriptomic profiles associated with anal cancer recurrence among people living with HIV. Ann Med. 2023;55:2199366. https://doi.org/10.1080/07853890.2023.2199366.
Hernandez S, Das P, Holliday EB, Shen L, Lu W, Johnson B, et al. Differential spatial gene and protein expression associated with recurrence following chemoradiation for localized anal squamous cell cancer. Cancers. 2023. https://doi.org/10.3390/cancers15061701.
Trilla-Fuertes L, Ghanem I, Gámez-Pozo A, Maurel J, Pastrián LG, Mendiola M, et al. Genetic profile and functional proteomics of anal squamous cell carcinoma: proposal for a molecular classification. Mol Cell Proteom. 2020;19:690–700. https://doi.org/10.1074/mcp.RA120.001954.
Arisi MF, Dotan E, Fernandez SV. Circulating tumor DNA in precision oncology and its applications in colorectal cancer. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23084441.
Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med. 2018;379:1754–65. https://doi.org/10.1056/NEJMra1706174.
Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat Rev Clin Oncol. 2021;18:297–312. https://doi.org/10.1038/s41571-020-00457-x.
Schrock AB, Pavlick D, Klempner SJ, Chung JH, Forcier B, Welsh A, et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with advanced cancers of the gastrointestinal tract or anus. Clin Cancer Res. 2018;24:1881–90. https://doi.org/10.1158/1078-0432.CCR-17-3103.
Stejskal P, Goodarzi H, Srovnal J, Hajdúch M, van ‘t Veer LJ, Magbanua MJM. Circulating tumor nucleic acids: biology, release mechanisms, and clinical relevance. Mol Cancer. 2023;22:15. https://doi.org/10.1186/s12943-022-01710-w.
Valmary-Degano S, Jacquin E, Prétet J-L, Monnien F, Girardo B, Arbez-Gindre F, et al. Signature patterns of human papillomavirus type 16 in invasive anal carcinoma. Hum Pathol. 2013;44:992–1002. https://doi.org/10.1016/j.humpath.2012.08.019.
Krasniqi E, Barba M, Venuti A, Pizzuti L, Cappuzzo F, Landi L, et al. Circulating HPV DNA in the management of oropharyngeal and cervical cancers: current knowledge and future perspectives. J Clin Med Res. 2021. https://doi.org/10.3390/jcm10071525.
Damerla RR, Lee NY, You D, Soni R, Shah R, Reyngold M, et al. Detection of early human papillomavirus-associated cancers by liquid biopsy. JCO Precis Oncol. 2019. https://doi.org/10.1200/PO.18.00276.
Hilke FJ, Muyas F, Admard J, Kootz B, Nann D, Welz S, et al. Dynamics of cell-free tumor DNA correlate with treatment response of head and neck cancer patients receiving radiochemotherapy. Radiother Oncol. 2020;151:182–9. https://doi.org/10.1016/j.radonc.2020.07.027.
Wang Y, Li Y, Liang X, Xin S, Yang L, Cao P, et al. The implications of cell-free DNAs derived from tumor viruses as biomarkers of associated cancers. J Med Virol. 2022;94:4677–88. https://doi.org/10.1002/jmv.27903.
Rettig EM, Faden DL, Sandhu S, Wong K, Faquin WC, Warinner C, et al. Detection of circulating tumor human papillomavirus DNA before diagnosis of HPV-positive head and neck cancer. Int J Cancer. 2022;151:1081–5. https://doi.org/10.1002/ijc.33996.
Jones CM, Goh V, Sebag-Montefiore D, Gilbert DC. Biomarkers in anal cancer: from biological understanding to stratified treatment. Br J Cancer. 2017;116:156–62. https://doi.org/10.1038/bjc.2016.398.
Siravegna G, O’Boyle CJ, Varmeh S, Queenan N, Michel A, Stein J, et al. Cell-free HPV DNA provides an accurate and rapid diagnosis of HPV-associated head and neck cancer. Clin Cancer Res. 2022;28:719–27. https://doi.org/10.1158/1078-0432.CCR-21-3151.
Chera BS, Kumar S, Beaty BT, Marron D, Jefferys S, Green R, et al. Rapid Clearance profile of plasma circulating tumor HPV type 16 DNA during chemoradiotherapy correlates with disease control in HPV-associated oropharyngeal cancer. Clin Cancer Res. 2019;25:4682–90. https://doi.org/10.1158/1078-0432.CCR-19-0211.
Lefèvre AC, Pallisgaard N, Kronborg C, Wind KL, Krag SRP, Spindler K-LG. The clinical value of measuring circulating HPV DNA during chemo-radiotherapy in squamous cell carcinoma of the anus. Cancers. 2021. https://doi.org/10.3390/cancers13102451.
Bernard-Tessier A, Jeannot E, Guenat D, Debernardi A, Michel M, Proudhon C, et al. Clinical validity of HPV circulating tumor DNA in advanced anal carcinoma: an ancillary study to the epitopes-HPV02 trial. Clin Cancer Res. 2019;25:2109–15. https://doi.org/10.1158/1078-0432.CCR-18-2984.
Cabel L, Bonneau C, Bernard-Tessier A, Héquet D, Tran-Perennou C, Bataillon G, et al. HPV ctDNA detection of high-risk HPV types during chemoradiotherapy for locally advanced cervical cancer. ESMO Open. 2021;6: 100154. https://doi.org/10.1016/j.esmoop.2021.100154.
Hanna GJ, Supplee JG, Kuang Y, Mahmood U, Lau CJ, Haddad RI, et al. Plasma HPV cell-free DNA monitoring in advanced HPV-associated oropharyngeal cancer. Ann Oncol. 2018;29:1980–6. https://doi.org/10.1093/annonc/mdy251.
Lin G, Li J. Circulating HPV DNA in HPV-associated cancers. Clin Chim Acta. 2023;542: 117269. https://doi.org/10.1016/j.cca.2023.117269.
Lee JY, Garcia-Murillas I, Cutts RJ, De Castro DG, Grove L, Hurley T, et al. Predicting response to radical (chemo)radiotherapy with circulating HPV DNA in locally advanced head and neck squamous carcinoma. Br J Cancer. 2017;117:876–83. https://doi.org/10.1038/bjc.2017.258.
Sastre-Garau X, Diop M, Martin F, Dolivet G, Marchal F, Charra-Brunaud C, et al. A NGS-based blood test for the diagnosis of invasive HPV-associated carcinomas with extensive viral genomic characterization. Clin Cancer Res. 2021;27:5307–16. https://doi.org/10.1158/1078-0432.CCR-21-0293.
Leung E, Han K, Zou J, Zhao Z, Zheng Y, Wang TT, et al. HPV sequencing facilitates ultrasensitive detection of HPV circulating tumor DNA. Clin Cancer Res Off J Am Assoc Cancer Res. 2021. https://doi.org/10.1158/1078-0432.CCR-19-2384.
Spehner L, Boustani J, Cabel L, Doyen J, Vienot A, Borg C, et al. Present and future research on anal squamous cell carcinoma. Cancers. 2021. https://doi.org/10.3390/cancers13153895.
Cabel L, Jeannot E, Bieche I, Vacher S, Callens C, Bazire L, et al. Prognostic impact of residual hpv ctdna detection after chemoradiotherapy for anal squamous cell carcinoma. Clin Cancer Res. 2018;24:5767–71. https://doi.org/10.1158/1078-0432.CCR-18-0922.
Lee JY, Cutts RJ, White I, Augustin Y, Garcia-Murillas I, Fenwick K, et al. Next generation sequencing assay for detection of circulating HPV DNA (cHPV-DNA) in patients undergoing radical (chemo)radiotherapy in anal squamous cell carcinoma (ASCC). Front Oncol. 2020;10:505. https://doi.org/10.3389/fonc.2020.00505.
Azzi G, Tavallai M, Aushev VN, Koyen Malashevich A, Botta GP, Tejani MA, et al. Using tumor-informed circulating tumor DNA (ctDNA)-based testing for patients with anal squamous cell carcinoma. Oncologist. 2023;28:220–9. https://doi.org/10.1093/oncolo/oyac249.
Alvarez J, Cercek A, Mohan N, Cuaron JJ, Zinovoy M, Reyngold M, et al. Circulating tumor DNA (ctDNA) for response assessment in patients with anal cancer treated with definitive chemoradiation. J Clin Orthod. 2023;41:1–1. https://doi.org/10.1200/JCO.2023.41.4_suppl.1.
Haring CT, Brummel C, Bhambhani C, Jewell B, Neal MH, Bhangale A, et al. Implementation of human papillomavirus circulating tumor DNA to identify recurrence during treatment de-escalation. Oral Oncol. 2021;121: 105332. https://doi.org/10.1016/j.oraloncology.2021.105332.
Eng C, Ciombor KK, Cho M, Dorth JA, Rajdev LN, Horowitz DP, et al. Anal cancer: emerging standards in a rare disease. J Clin Oncol. 2022;40:2774–88. https://doi.org/10.1200/JCO.21.02566.
Cabel L, Bidard F-C, Servois V, Cacheux W, Mariani P, Romano E, et al. HPV circulating tumor DNA to monitor the efficacy of anti-PD-1 therapy in metastatic squamous cell carcinoma of the anal canal: a case report. Int J Cancer. 2017;141:1667–70. https://doi.org/10.1002/ijc.30863.
Anti-PD-1 and mDCF followed by chemoradiotherapy in patients with stage III squamous cell anal carcinoma. Full Text View—ClinicalTrials.Gov. https://classic.clinicaltrials.gov/ct2/show/NCT04719988. Accessed 13 July 2023.
Shakir R, Adams R, Cooper R, Downing A, Geh I, Gilbert D, et al. Patterns and predictors of relapse following radical chemoradiation therapy delivered using intensity modulated radiation therapy with a simultaneous integrated boost in anal squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2020;106:329–39. https://doi.org/10.1016/j.ijrobp.2019.10.016.
Throat and Other HPV-Related Cancers in Men: Identifying Them Early—Full Text View—ClinicalTrials.Gov. https://classic.clinicaltrials.gov/ct2/show/NCT02897427. Accessed 13 July 2023
Sebag-Montefiore D, Adams R, Bell S, Berkman L, Gilbert DC, Glynne-Jones R, et al. The development of an umbrella trial (PLATO) to address radiation therapy dose questions in the locoregional management of squamous cell carcinoma of the anus. Int J Radiat Oncol Biol Phys. 2016;96:E164–5.
Circulating Tumor DNA for the Early Detection of HPV-positive Pelvic Cancer Relapses—Full Text View—ClinicalTrials.Gov. https://classic.clinicaltrials.gov/ct2/show/NCT03739775. Accessed 13 July 2023.
Roden RBS, Stern PL. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat Rev Cancer. 2018;18:240–54. https://doi.org/10.1038/nrc.2018.13.
Shamseddine AA, Burman B, Lee NY, Zamarin D, Riaz N. Tumor immunity and immunotherapy for HPV-related cancers. Cancer Discov. 2021;11:1896–912. https://doi.org/10.1158/2159-8290.CD-20-1760.
Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–60. https://doi.org/10.1038/nrc2886.
Westrich JA, Warren CJ, Pyeon D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017;231:21–33. https://doi.org/10.1016/j.virusres.2016.11.023.
Zhou C, Tuong ZK, Frazer IH. Papillomavirus immune evasion strategies target the infected cell and the local immune system. Front Oncol. 2019;9:682. https://doi.org/10.3389/fonc.2019.00682.
Elmusrati A, Wang J, Wang C-Y. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma. Int J Oral Sci. 2021;13:24. https://doi.org/10.1038/s41368-021-00131-7.
Minu Chandra Muddabhaktuni B, Koyyala VPB. The Cancer Genome Atlas. Indian J Med Paediatr Oncol. 2021;42:353–5. https://doi.org/10.1055/s-0041-1735440.
Cancer Genome Atlas Research Network, Albert Einstein College of Medicine, Analytical Biological Services, Barretos Cancer Hospital, Baylor College of Medicine, Beckman Research Institute of City of Hope, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543: 378–384. https://doi.org/10.1038/nature21386
Moerman-Herzog A, Nakagawa M. Early defensive mechanisms against human papillomavirus infection. Clin Vaccine Immunol. 2015;22:850–7. https://doi.org/10.1128/CVI.00223-15.
Karim R, Meyers C, Backendorf C, Ludigs K, Offringa R, van Ommen G-JB, et al. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS One. 2011;6:e17848. https://doi.org/10.1371/journal.pone.0017848.
Guess JC, McCance DJ. Decreased migration of Langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3α production. J Virol. 2005;79:14852–62. https://doi.org/10.1128/jvi.79.23.14852-14862.2005.
Sperling T, Ołdak M, Walch-Rückheim B, Wickenhauser C, Doorbar J, Pfister H, et al. Human papillomavirus type 8 interferes with a novel C/EBPβ-mediated mechanism of keratinocyte CCL20 chemokine expression and Langerhans cell migration. PLoS Pathog. 2012;8: e1002833. https://doi.org/10.1371/journal.ppat.1002833.
Miyauchi S, Sanders PD, Guram K, Kim SS, Paolini F, Venuti A, et al. HPV16 E5 mediates resistance to PD-L1 blockade and can be targeted with rimantadine in head and neck cancer. Cancer Res. 2020;80:732–46. https://doi.org/10.1158/0008-5472.CAN-19-1771.
Hemmat N, Baghi HB. Human papillomavirus E5 protein, the undercover culprit of tumorigenesis. Infect Agent Cancer. 2018;13:31. https://doi.org/10.1186/s13027-018-0208-3.
Wagner S, Wittekindt C, Reuschenbach M, Hennig B, Thevarajah M, Würdemann N, et al. CD56-positive lymphocyte infiltration in relation to human papillomavirus association and prognostic significance in oropharyngeal squamous cell carcinoma. Int J Cancer. 2016;138:2263–73. https://doi.org/10.1002/ijc.29962.
Stern PL. Harnessing immunity for therapy in human papillomavirus driven cancers. Tumour Virus Res. 2021;11: 200212. https://doi.org/10.1016/j.tvr.2021.200212.
Dutta S, Ganguly A, Chatterjee K, Spada S, Mukherjee S. Targets of immune escape mechanisms in cancer: basis for development and evolution of cancer immune checkpoint inhibitors. Biology. 2023. https://doi.org/10.3390/biology12020218.
Liu C, Lu J, Tian H, Du W, Zhao L, Feng J, et al. Increased expression of PD-L1 by the human papillomavirus 16 E7 oncoprotein inhibits anticancer immunity. Mol Med Rep. 2017;15:1063–70. https://doi.org/10.3892/mmr.2017.6102.
Chan AM, Roldan Urgoiti G, Jiang W, Lee S, Kornaga E, Mathen P, et al. The prognostic impact of PD-L1 and CD8 expression in anal cancer patients treated with chemoradiotherapy. Front Oncol. 2022;12:1000263. https://doi.org/10.3389/fonc.2022.1000263.
Zhang Y, Wu J, Zhao C, Zhang S, Zhu J. Recent advancement of PD-L1 detection technologies and clinical applications in the era of precision cancer therapy. J Cancer. 2023;14:850–73. https://doi.org/10.7150/jca.81899.
Frenel J-S, Le Tourneau C, O’Neil B, Ott PA, Piha-Paul SA, Gomez-Roca C, et al. Safety and efficacy of pembrolizumab in advanced, programmed death ligand 1-positive cervical cancer: results from the phase Ib KEYNOTE-028 trial. J Clin Oncol. 2017;35:4035–41. https://doi.org/10.1200/JCO.2017.74.5471.
Chung HC, Piha-Paul SA, Lopez-Martin J, Schellens JHM, Kao S, Miller WH Jr, et al. Pembrolizumab after two or more lines of previous therapy in patients with recurrent or metastatic SCLC: results from the KEYNOTE-028 and KEYNOTE-158 studies. J Thorac Oncol. 2020;15:618–27. https://doi.org/10.1016/j.jtho.2019.12.109.
Rao S, Anandappa G, Capdevila J, Dahan L, Evesque L, Kim S, et al. A phase II study of retifanlimab (INCMGA00012) in patients with squamous carcinoma of the anal canal who have progressed following platinum-based chemotherapy (POD1UM-202). ESMO Open. 2022;7: 100529. https://doi.org/10.1016/j.esmoop.2022.100529.
Yuan C-H, Filippova M, Duerksen-Hughes P. Modulation of apoptotic pathways by human papillomaviruses (HPV): mechanisms and implications for therapy. Viruses. 2012;4:3831–50. https://doi.org/10.3390/v4123831.
Zhu X, Jamshed S, Zou J, Azar A, Meng X, Bathini V, et al. Molecular and immunophenotypic characterization of anal squamous cell carcinoma reveals distinct clinicopathologic groups associated with HPV and TP53 mutation status. Mod Pathol. 2021;34:1017–30. https://doi.org/10.1038/s41379-020-00729-y.
Yanik EL, Kaunitz GJ, Cottrell TR, Succaria F, McMiller TL, Ascierto ML, et al. Association of HIV status with local immune response to anal squamous cell carcinoma: implications for immunotherapy. JAMA Oncol. 2017;3:974–8. https://doi.org/10.1001/jamaoncol.2017.0115.
Acknowledgements
This research was funded by Foundation Nelia and Amadeo Barletta (FNAB) and NIH Grant CA221208.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This research was funded by Foundation Nelia and Amadeo Barletta (FNAB) and NIH Grant CA221208.
Availability of data and materials
The data presented in this study are available on request from the corresponding author.
Conflict of interest
The authors S.I., M.G, L.G., V.D.P., J.A., M.C.A., and E.R declare no conflicts of interest.
Ethical approval
Not applicable.
Authors contributions
All the authors have directly participated in the preparation of this manuscript and have read and approved the final version submitted and declare no ethical conflicts of interest. S.I., M.G, L.G., V.D.P., J.A., M.C.A., and E.R.: conceptualization and methodology; S.I., M.G, L.G., V.D.P., J.A., M.C.A., and E.R.: validation, visualization, and formal analysis; S.I., M.G., L.G., V.D.P., J.A., M.C.A., and E.R.: investigation and data curation; S.I., M.G., L.G., V.D.P., J.A., M.C.A., and E.R.: writing—original draft. All authors: S.I., M.G., L.G., V.D.P., J.A., M.C.A., and E.R. All authors have read and agreed to the published version of the manuscript.
Code availability
Not applicable.
Consent
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Iseas, S., Mariano, G., Gros, L. et al. Unraveling Emerging Anal Cancer Clinical Biomarkers from Current Immuno-Oncogenomics Advances. Mol Diagn Ther 28, 201–214 (2024). https://doi.org/10.1007/s40291-023-00692-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00692-9