
Abstract
Spinal muscular atrophy (SMA) is a neurodegenerative disease primarily characterized by a loss of spinal motor neurons, leading to progressive paralysis and premature death in the most severe cases. SMA is caused by homozygous deletion of the survival motor neuron 1 (SMN1) gene, leading to low levels of SMN protein. However, a second SMN gene (SMN2) exists, which can be therapeutically targeted to increase SMN levels. This has recently led to the first disease-modifying therapy for SMA gaining formal approval from the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). Spinraza (nusinersen) is a modified antisense oligonucleotide that targets the splicing of SMN2, leading to increased SMN protein levels, capable of improving clinical phenotypes in many patients. In addition to Spinraza, several other therapeutic approaches are currently in various stages of clinical development. These include SMN-dependent small molecule and gene therapy approaches along with SMN-independent strategies, such as general neuroprotective factors and muscle strength-enhancing compounds. For each therapy, we provide detailed information on clinical trial design and pharmacological/safety data where available. Previous clinical studies are also discussed to provide context on SMA clinical trial development and the insights these provided for the design of current studies.
Similar content being viewed by others
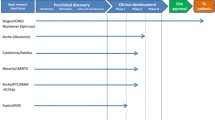
The approval of Spinraza for the treatment of spinal muscular atrophy is a major milestone in motor neuron disease and translational research. |
Numerous additional therapies, both survival motor neuron (SMN)-dependent and SMN-independent, are currently in development and will likely expand therapeutic possibilities in coming years. |
Future research will have a strong focus on identifying combinatorial therapeutic strategies by combining SMN-targeting therapy with other, SMN-independent therapies. |
1 Introduction
Spinal muscular atrophy (SMA) is a devastating neurological disease characterized primarily by the degeneration of lower motor neurons in the spinal cord [1]. SMA is the most common genetic cause of infant mortality and occurs in approximately one in 10,000 live births [2]. The monogenetic cause of SMA has long been known [3], which has allowed basic research on SMA to always have a strong focus on therapy development. These research efforts have led to the development and recent approval of the first disease-modifying therapy for SMA, Spinraza (nusinersen), an important milestone in SMA and neuromuscular disease research [4,5,6]. Initial experiences with Spinraza have already provided important lessons for the development of therapies for related motor neuron diseases and other monogenetic neurological disorders. Several additional therapies for SMA are now in various stages of clinical development, and basic research has identified further promising preclinical leads. In this review, we first provide an overview of the clinical, genetic and pathogenic background of SMA. Next, we detail current clinical studies, including pharmacological and safety data where available. Finally, we discuss future directions and priorities for SMA therapy development.
2 Clinical, Genetic and Pathogenic Background of Spinal Muscular Atrophy (SMA)
2.1 Clinical Background of SMA
Despite its established monogenetic cause, the clinical phenotype of SMA is highly variable [1, 7]. SMA patients with the most severe (type 0, neonatal onset) and common (type I, approximately 50% of all cases) forms are characterized by disease onset before 6 months of age. These patients do not reach any major motor milestones and die before the age of 2 years. SMA type II patients present at a later age (between 6 and 18 months), usually learn to sit unaided, but are never able to stand or walk and have a severely reduced life expectancy. SMA type III is clinically very heterogeneous, ranging from patients with severe neuromuscular disability to patients who are relatively mildly affected. Both age of onset and life expectancy are equally variable in these forms of the disease. Finally, SMA type IV is the mildest form of SMA, with disease onset not normally occurring before the third decade, comparatively minor disability and normal life expectancy. Based on underlying genetics, motor milestones and clinical phenotype, further subdivisions into clinical subtypes (type Ia, Ib and Ic and type IIa, IIb, IIIa and IIIb) can be made [8].
2.2 Genetics of SMA
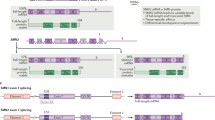
SMA is caused by a homozygous deletion of the survival motor neuron 1 (SMN1) gene in ~ 95% of cases and other deleterious variants in SMN1 in the remaining ~ 5% of cases [3]. These variants unanimously lead to low levels of functional SMN protein [9]. Humans have a second copy of this gene, known as SMN2, which is almost identical to SMN1. However, in SMN2, a synonymous nucleotide substitution at the 5′ end of exon 7 leads to exon skipping and translation into unstable, quickly degraded truncated SMN protein (SMN∆7) for the majority of transcripts [10, 11]. The number of SMN2 copies is variable, and because each copy produces low levels of full-length, functional SMN, the number of SMN2 copies correlates well with a patient’s clinical phenotype [12]. However, both between patients with the same number of SMN2 copies and within families, considerable variation in clinical phenotypes can occur. Indeed, research on discordant families has led to the identification of several genetic modifiers of SMA, including plastin-3 (PLS3) and neurocalcin delta (NCALD) (see also Sect. 4.1) [13,14,15]. It is likely, therefore, that further genetic and other disease-modifying factors are still to be discovered.
2.3 Cellular Mechanisms of SMA
Because of its central role in the pathogenesis of SMA, SMN’s cellular role has been extensively studied. SMN is a ubiquitously expressed, 32-kDa protein. SMN has an important housekeeping role in all cells with regard to regulating the biogenesis of ribonucleoprotein (RNP) complexes, of which its role in the formation of small nuclear RNPs (snRNPs) has been studied in most detail [16,17,18,19,20,21]. By interacting with GEMIN2–8 and unrip, SMN regulates the binding of small nuclear ribonucleic acids (snRNAs) to Sm proteins and subsequently guides these RNA–protein complexes from the cytoplasm into the nucleus. There, the newly formed snRNPs mature in nuclear Cajal bodies, after which they move into nuclear speckles to be recruited into active spliceosomes. Recent research indicates that SMN has in fact a general role in RNP formation, as illustrated by defects in the formation of small nucleolar RNPs (snoRNPs) (required for the post-translational modification of non-coding RNAs), messenger RNPs (mRNPs) (involved in the transport of mRNAs), and signal recognition particles (SRPs) (regulating the transport of newly synthetized proteins) when SMN levels are low [19, 22, 23].
SMN depletion affects a number of other cellular pathways that are of particular interest for the maintenance of neuronal homeostasis. For example, actin dynamics, ubiquitin homeostasis, mitochondrial function and endocytosis are all disrupted in SMA [24]. When SMN expression is reduced to pathological levels, decreased SMN-profilin interaction leads to increased formation of profilin–Rho-associated protein kinase (ROCK) complexes that cause activation of Ras homologue gene family, member A (RhoA), a negative regulator of axon outgrowth [25, 26]. Moreover, increased levels of actin-binding protein PLS3 are protective in both SMA disease models and patients [13, 14]. Furthermore, decreased levels of the E1 ubiquitin-activating enzyme, ubiquitin-like modifier activating enzyme 1 (UBA1), are a central feature of SMA pathogenesis across a range of models, and therapeutic targeting of UBA1 rescues a range of SMA-associated phenotypes [27, 28]. In addition, a number of studies have indicated defects in mitochondrial function and transport in SMA, including decreased expression of specific mitochondrial proteins such as phosphoglycerate kinase 1 (PGK1) [29, 30]. Finally, the role of NCALD as a genetic modifier illustrates how, by interacting with actin and clathrin, endocytotic processes can be affected in SMA and influence disease phenotypes [15]. Whether the above pathways are related or whether they are independently affected in SMA remains to be determined. Interestingly, however, local protein translation (both globally and at the level of specific mRNAs) is defective in several SMA models [31,32,33,34]. Careful spatiotemporal regulation of local protein translation is vital for maintaining neuronal homeostasis and has been linked to many of the above pathways [35, 36], thereby providing a possible cellular pathway that links and unites these molecular mechanisms in the pathogenesis of SMA.
3 Current SMA Therapy Developments
Because severely reduced levels of SMN have long been known to be the cause of SMA, therapy development has so far largely focused on approaches that aim to increase full-length SMN protein levels [37]. These approaches include antisense oligonucleotides (ASOs) and small molecules that target SMN2, and gene therapy to restore SMN1 and cellular SMN expression. Each of these approaches has shown great promise in preclinical studies, often leading to significant amelioration of disease phenotypes in a range of animal models [38,39,40,41,42]. The same preclinical studies, however, have also indicated several limitations and requirements when treating SMA by targeting SMN. First, the timing of SMN-targeted therapies in SMA mouse models has been shown to be essential for the efficient rescue of disease phenotypes; the earlier treatment is started, the more robustly the disease phenotype is rescued [43]. Data emerging from recent clinical trials have suggested that this is also an important aspect of therapy in SMA patients. This finding might limit the efficacy of SMN-targeted therapies, as initial diagnosis of SMA following symptom onset might suggest that the optimal therapeutic window for therapy delivery has already passed. Moreover, although lower motor neurons are the primary pathogenic target in SMA, studies in animal models indicate that other, non-central nervous system (non-CNS) tissues are also affected [44,45,46,47,48,49]. Although not all organs are affected to the same extent in all animal models of SMA, preclinical studies have indicated that restoration of SMN expression is required in all tissues and not just in the CNS to provide robust rescue of the SMA phenotype [50]. Several studies and case reports in SMA patients indicate that, indeed, peripheral tissues can be affected in addition to motor neuron degeneration [45, 51], confirming that preclinical findings are also relevant for patient studies.
3.1 Survival Motor Neuron (SMN)-Targeted Therapies
3.1.1 Spinraza
Spinraza (nusinersen, Biogen/Ionis) is a modified 18-mer ASO that has been approved for treatment of all types of SMA in Europe, Canada, Japan, Australia and the USA (Tables 1 and 2) [5, 52]. It works by binding to the ISS-N1 regulatory motif in the intron downstream of exon 7 on SMN2 pre-mRNA, preventing the binding of factors such as heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1). This promotes the inclusion of exon 7, thereby increasing the amount of full-length SMN2 mRNA and, subsequently, full-length SMN protein [6]. Spinraza is administered by intrathecal injection at an equivalent dose of 12 mg (4–5 mL based on age), with three initial loading doses at intervals of 14 days and a fourth loading dose 30 days later [53]. The loading phase is followed by maintenance doses once every 4 months [5, 53]. Following intrathecal injection, Spinraza is distributed from the cerebrospinal fluid (CSF) throughout the CNS, including into motor neurons, glia and vascular endothelial cells [53]. Spinraza is cleared from the CSF into systemic circulation consistent with normal CSF turnover, with the mean time to maximum plasma concentration ranging from 1.7 to 6 h. Consistent with this, Spinraza was detected in kidney, liver and skeletal muscle along with the CNS at autopsy [53]. Spinraza leads to increased levels of SMN protein, as detected in CSF [54], increased inclusion of SMN exon 7 and, subjectively, increased levels of SMN protein as determined by immunohistochemistry on spinal cord [53]. Spinraza was still detectable in the CSF 15–168 days after dosing, with a mean terminal elimination half-life of 135–177 days in the CSF and 63–87 days in the plasma, indicating prolonged exposure of the CNS and peripheral tissues to Spinraza [53, 55].
A phase III clinical trial of Spinraza for SMA type I (ENDEAR) was performed on 121 patients who presented with SMA before the age of 7 months. ENDEAR was a multinational, randomized, double-blind, sham-controlled clinical trial. Following the final analysis of this study, 51% of Spinraza-treated patients were motor milestone responders [as assessed by Sect. 2 of the Hammersmith Infant Neurological Examination (HINE)] compared to 0% of controls; however, only 8% of Spinraza-treated patients were able to sit independently. Moreover, there was a significant improvement in survival of Spinraza-treated patients compared to controls [56]. Also, motor function was improved in Spinraza-treated infants who required permanent ventilation compared to ventilation-dependent, sham-controlled infants. The most frequent adverse events in this study were respiratory infections and constipation [5, 56]. In a similar phase III clinical trial (CHERISH), Spinraza was studied in type II and III SMA patients who presented with clinical symptoms after 6 months of age. Interim analysis of 126 patients enrolled on the CHERISH study revealed a significant improvement in motor function at 15 months from baseline [assessed by the Hammersmith Functional Motor Scale Expanded (HFMSE)] in Spinraza-treated patients compared to controls [5, 56, 57]. Here, adverse events were largely related to the lumbar puncture procedure (headache, back pain and post-lumbar puncture syndrome) or were similar to those observed in the ENDEAR trial [5, 56]. Following favourable interim analyses of both the ENDEAR and CHERISH studies, the trials were stopped and the patients were transitioned onto SHINE, an open-label extension study in which the long-term efficacy of treatment with Spinraza will be assessed [56,57,58].
Alongside these studies, a phase II clinical trial (NURTURE) aims to address the effect of Spinraza treatment on pre-symptomatic SMA patients. Here, SMN1 deletion carriers (with two or three copies of SMN2, SMA type I/II genotypes) receive Spinraza treatment before 6 weeks of age, in advance of the onset of overt disease symptoms. The NURTURE trial is an open-label, single-arm study comparing pre-symptomatic Spinraza efficacy to a control group of affected siblings and natural history data [59]. Initial results from the second interim analysis indicate that motor milestones (such as sitting and crawling) are generally achieved at age-appropriate time points, indicating a significantly greater effect of a pre-symptomatic treatment compared to treatment starting after symptom onset [5, 59, 60]. Despite this, however, only three of nine patients were standing unaided following 1 year of treatment [60], indicating that even pre-symptomatic treatment with Spinraza does not represent a complete ‘cure’ for SMA.
In summary, these promising initial results have been greeted with enthusiasm by clinicians, research scientists and the patient community alike. It will be of great interest to follow the results from current follow-up trials as they become available in order to ascertain more about the long-term efficacy and safety of Spinraza for treatment of SMA. Moreover, ongoing preclinical studies have revealed that next-generation oligonucleotides can be developed that have a therapeutic effect comparable to that of Spinraza, but which can be delivered through less invasive (e.g. intravenously) delivery routes and at lower doses [61].
3.1.2 Gene Therapy (AVXS-101)
A more direct approach to increase SMN protein levels is restoring SMN expression by gene therapy. In preclinical studies in SMA mouse models, adeno-associated virus (AAV)-mediated SMN1 gene replacement resulted in widespread expression of SMN in the spinal cord and significantly increased survival [39,40,41, 62]. Further studies in primates indicated that systemically delivered AAV9-SMN1 efficiently crossed the blood–brain barrier, resulting in transgene expression in brain, spinal motor neurons, dorsal root ganglia neurons and glial cells throughout the CNS, in addition to widespread expression in the liver, heart and skeletal muscle [63]. Based on these promising preclinical studies, a phase I study was initiated to test the safety and efficacy of SMN1 gene therapy (AVXS-101, AveXis) using a self-complimentary adeno-associated virus serotype 9 (scAAV9) delivered as a single dose intravenously to type I SMA patients (with two copies of SMN2) (Tables 1 and 2). The first cohort of patients (n = 3) received the virus at a dose of 6.7 × 1013 vg/kg, and the second cohort of patients (n = 12) received the proposed therapeutic dose of 2.0 × 1014 vg/kg [64]. Patients 2–15 also received prednisolone at a daily dose of 1 mg/kg for approximately 30 days, starting 1 day before viral delivery, to suppress the initial immune response after delivery of a high viral load [64]. Initial reports of AVXS-101 treatment indicate an improvement in survival along with achievement of motor milestones, such as maintaining head control, sitting unassisted (11 of 12 cohort 2 patients) and oral feeding (11 of 12 cohort 2 patients) [64].
As a follow-up study, an open-label, single-arm, single-dose pivotal trial (STR1VE) has been designed to test the efficacy of AVXS-101 in a minimum of 15 SMA type I patients (with one or two SMN2 copies) who are symptomatic and less than 6 months old at the time of gene therapy, across multiple trial centres. Patients will receive an intravenous infusion of AVXS-101 at a dose of 1.1 × 1014 vg/kg (equivalent to the proposed therapeutic dose received by cohort 2 in the phase I clinical trial), with at least a 4-week dosing interval between the first three participants to review the safety profile of the treatment. Along with treatment safety, the trial will evaluate the efficacy as assessed by the achievement of developmental milestones (sitting for 30 seconds at 18 months) and event-free survival at 14 months [65]. Future developments are eagerly awaited, as AVXS-101 provides an opportunity to substantially improve SMA via one-off virus dosing. However, although some promising advancements have recently been made, viral gene therapy approaches are still not commonplace in the clinic and careful monitoring of gene therapy-treated patients will be required to assess long-term safety and durability of the treatment.
3.1.3 SMN2 Splice-Modifying Small Molecules (RG7916 and LMI070)
In addition to these ASO and gene therapy approaches to increase SMN levels, more traditional pharmacological, small molecule-based approaches to target SMN2 splicing have also been developed. Initial screens in mouse models of SMA indicate that orally available small molecules can efficiently increase the production of full-length SMN2 mRNA and SMN protein. These studies demonstrated an improvement in motor function, protection of the neuromuscular system from degeneration and increased survival in SMA model mice [42]. After these promising preclinical results, two independently developed drugs are being tested in phase I and II clinical trials. The first of these drugs, branaplam (formerly LMI070, Novartis) is based on a weekly drug dose, and initial studies investigated the safety and tolerability over a 13-week trial period, with a subsequent 13-month extension to continue safety monitoring and assess efficacy in SMA type I patients with two copies of SMN2 (Tables 1 and 2). Initial results suggested some improvements in motor function and indicated adverse events that were mostly mild and reversible [66]. However, enrolment onto the trial was temporarily paused because of the occurrence of nerve injury in a parallel chronic preclinical toxicology study. This problem has now been addressed, and enrolment on the trial has been resumed with the implementation of additional nerve tests and the option of the weekly branaplam dose administrated orally rather than by feeding tube only [67, 68].
The second small molecule SMN2-splicing modifier in phase II clinical trials is RG7916 (RO7034067, Roche) (Tables 1 and 2). In a single ascending dose study in healthy volunteers, it was shown that RG7916 was safe, well-tolerated and increased full-length SMN2 mRNA levels. Following this, several phase II clinical trials have been initiated in which RG7916 is administered orally on a daily basis to SMA patients with two or more copies of SMN2. Two of these trials are two-part trials (FIREFISH and SUNFISH), in which the first part of the trial assesses the safety of two dose levels of RG7916 and the second part aims to investigate the efficacy of the most appropriate dose [69]. FIREFISH is an open-label trial in which RG7916 is administered to SMA type I patients aged between 1 and 7 months old, while the SUNFISH trial investigates RG7916 in type II and type III SMA patients aged 2–25 years (ambulatory and non-ambulatory). In contrast to FIREFISH, SUNFISH is a randomized, double-blind, placebo-controlled clinical trial. Initial reports of both FIREFISH and SUNFISH suggest some benefit of the therapy for SMA patients [69]. Following successful selection of the appropriate dose, the SUNFISH trial has now progressed to the pivotal second part of the study on non-ambulatory type II and III SMA patients [70]. The final, phase II clinical trial of RG7916 (JEWELFISH) is an open-label trial investigating safety, tolerability and efficacy in patients previously treated with other SMN2-targeting small molecule therapies [69].
Although the initial results from these trials indicate modest functional improvement, the oral delivery of these drugs makes them more flexible and easier and safer to administer than both CNS-delivered ASOs and viral gene therapy. Therefore, these drugs, if and when formally approved, are likely to play a significant role in the clinic in coming years.
3.1.4 SMN Protein-Enhancing Molecules (Valproic Acid)
Finally, initial studies that preceded the development of ASOs, gene therapy and small molecule splice enhancers, aimed to upregulate SMN2 transcription by inhibiting histone deacetylases (HDACs). Of these HDAC inhibitors, valproic acid (VPA) has been studied in particularly great detail and is one of the only therapeutic strategies for SMA with published results from multiple clinical trials (Tables 1 and 2). Although the consensus now is that VPA does not provide significant benefits for SMA patients, the trials that showed this preceded many current studies and have provided important lessons for current clinical trials.
An initial phase II, open-label trial of VPA in type I, II and III SMA patients aged 2–31 years indicated that VPA was safe and well-tolerated [71]. The patients received VPA in the form of divalproex sodium-coated particles (125 mg per capsule) in divided doses, two to three times daily, to maintain overnight trough levels of 50–100 mg/dL; indeed, at the three treatment assessments (3, 6 and 12 months), mean overnight trough levels were between 50 and 65 mg/dL. This trial identified common adverse events of weight gain and reductions in total or free plasma carnitine, which caused a worsening of gross motor function in two participants [71]. While there was no significant change in full-length SMN transcript levels following treatment with VPA, a significant improvement in motor function, primarily in patients under 5 years of age, was observed. Importantly, the primary conclusions of this study were that subsequent VPA clinical trials should be conducted on more restricted cohorts of patients (e.g. patients with the same type of SMA or similar age of onset) and that carnitine should be given as a supplement to all subjects [71].
Following this initial phase II trial, several subsequent trials on defined cohorts of SMA patients were conducted. The first of these was the two-part CARNI-VAL trial; both cohorts received VPA at the same dose as the previous phase II trial as well as l-carnitine supplement (100 mg/mL liquid) at a dosage of 50 mg/kg/day (maximum of 1000 mg) divided into two daily doses [72, 73]. CARNI-VAL part 1 was a double-blind, placebo-controlled, randomized clinical trial on non-ambulatory SMA patients aged 2–8 years [73], whereas part 2 was an open-label trial in ambulatory SMA patients aged 3–17 years [72]. A similar double-blind, placebo-controlled, cross-over clinical trial of VPA (VALIANT; no carnitine supplement) was performed in ambulatory adults with SMA [74]. In all three trials, VPA did not improve strength or motor function in SMA patients compared to baseline or placebo controls; however, some younger patients on the CARNI-VAL part 1 trial did show improvement in motor function [72,73,74]. These results were confirmed by an independent trial of VPA in type II and III SMA patients in which no benefit was observed in type III SMA patients, but a significant improvement in motor function in type II SMA patients was seen [75]. Finally, in another clinical trial, l-carnitine and VPA were administered to SMA type I patients between the ages of 2 weeks and 12 months in an open-label study. Again, there was no impact on motor function and no improvement in survival in the treated cohort [76].
Interestingly, a cellular mechanism that might be related to these disappointing results has since been identified. In a study investigating the responsiveness to VPA in SMA patients, it was found that only one-third of SMA patients treated with VPA responded to treatment and had increased SMN2 transcript levels [77]. Transcriptome-wide profiling of SMA fibroblasts identified that CD36 RNA expression was fivefold higher in non-responders compared to VPA-responders, and indeed, subsequent analysis revealed that CD36 overexpression prevented VPA-induced SMN expression [77]. This finding makes it potentially interesting to speculate about the usefulness of VPA for SMA patients who are ineligible for SMN-targeted therapies but have low levels of CD36. Despite the lack of positive outcomes in VPA clinical trials, being early SMA clinical trials, they provided valuable insights into how to structure clinical trials in SMA cohorts. One of the key outcomes of the initial phase II VPA trial was that clinical trials should be conducted on restricted cohorts of SMA patients [71], something that has been accepted for more recent SMA clinical trials. Furthermore, VPA trials in restricted cohorts of SMA patients identified a range of feasible and reliable outcome measures for different populations of SMA patients that can be readily used in future clinical trials [72,73,74, 76]. Moreover, these clinical trials also highlighted pitfalls of current clinical trial design. For example, SMN protein and transcript levels were measured in peripheral blood, where no difference was detected [71,72,73]. However, blood SMN expression levels might differ from those measured in the CNS and therefore might not completely reflect expression changes in more disease-affected tissues. Finally, from these analyses, the benefits of VPA were only evident in young SMA type II patients [73, 75], suggesting that early treatment will be necessary to prevent effects of SMN deficiency on motor function and that if treating older or ambulatory SMA patients, a longer course of treatment may be necessary to reverse the disease process.
3.2 SMN-Independent Therapies
3.2.1 Neuroprotection (Olesoxime)
While the first-generation of SMN-targeting therapies progress through the various phases of clinical research, several second-generation, SMN-independent therapies are already under clinical development. These second-generation therapies are initially centred around administering neuroprotective factors or enhancing muscle strength. Olesoxime (Hoffman-La Roche/Trophos SA) is a neuroprotective factor which exerts its neuroprotective effects by binding to components of the mitochondrial permeability pore, thereby preventing excessive permeability under stress conditions [78,79,80,81] (Tables 1 and 2). In animal models of neurodegeneration, olesoxime has shown neuroprotective effects by preventing release of pro-apoptotic factors from mitochondria and maintaining energy production [78, 81]. The efficacy of olesoxime was assessed in a randomized, double-blind, placebo-controlled phase II clinical trial for type II and non-ambulatory type III SMA patients aged 3–25 years [82]. Olesoxime was administered daily at a dose of 10 mg/kg in an oral liquid suspension (100 mg/mL) for a trial duration of 24 months. The average individual plasma trough concentration of olesoxime (Cavg) was 8590 ng/mL, with a range of 4130–16,567 ng/mL. The dose of olesoxime was safe for the duration of the study, with an adverse event profile similar to the placebo; the most common adverse events in the treatment group were vomiting, cough, pyrexia and nasopharyngitis. The outcomes of this clinical trial suggest that olesoxime supported the maintenance of motor function, particularly in a subgroup of patients aged 6–15 years and in patients who had high exposure to olesoxime (defined as Cavg ≥ 7500 ng/mL) [82]. An open-label, single-arm study is currently ongoing to evaluate long-term safety, efficacy and tolerability of olesoxime in patients previously enrolled on the phase II study. As olesoxime is a general neuroprotective agent, the outcomes of these studies are potentially of interest for the treatment of other neurodegenerative diseases.
3.2.2 Muscle-Enhancing Drugs (CK-107 and SRK-015)
Another group of therapies currently in development for the treatment of SMA is aimed at improving neuromuscular function, muscular weakness and muscle fatigue. CK-107 (formerly CK-2127107, Cytokinetics) is a troponin complex activator that slows calcium release from fast skeletal muscle troponin to sensitize the sarcomere to calcium, leading to an increased force output at submaximal frequencies of motor nerve stimulation (Table 1). Indeed, preclinical studies indicated that CK-107 reduces fatigability of rat skeletal muscle in vivo [83]. Following promising preclinical and safety phase I pharmacokinetic and pharmacodynamic profiles in healthy volunteers, CK-107 is currently in phase II clinical trials. In this double-blind, randomized, placebo-controlled clinical trial, type II, III and IV SMA patients aged 12 years and older will receive 150 mg CK-107 (or placebo) twice daily for 8 weeks in cohort 1. Following analysis of safety and pharmacokinetics, a dose of up to 450 mg CK-107 will be administered twice daily to cohort 2 [84]. The outcome of this trial is expected in the near future.
In addition, the myostatin inhibitor SRK-015 (Scholar Rock) has been shown to increase muscle mass and force in healthy mice (Table 1). SRK-015 acts by binding to latent myostatin and hindering protease cleavage which is thought to prevent latent myostatin activation, leading to increased muscle cell growth and differentiation [85]. After preclinical studies showed that SRK-015 improved muscle function in SMA mice, it was announced that SRK-015 is due to enter clinical trials for SMA in 2018 as both a standalone therapy and in combination with SMN-targeted therapies [86]. However, due to dysregulation of components of the myostatin signalling pathway in SMA [87], the therapeutic effect of SRK-015 may be limited. Although the efficacy of neuroprotective and muscle-enhancing drugs remains to be determined, positive results in these trials will provide interesting opportunities to enhance the efficacy of SMN-targeted trials.
4 Future Directions for SMA Therapy Development
4.1 Combination of SMN-Targeted and Neuroprotective Approaches
The approval of Spinraza, alongside other ongoing clinical trials, holds great promise for the treatment of SMA over the coming years. However, when considering results from preclinical studies and initial results from clinical trials, it is likely that SMN-targeted therapy on its own will not be sufficient to halt or restore all disease-associated phenotypes [43, 50, 88]. One major line of research attempts to directly address these issues by exploring the possibility of developing combinatorial therapeutic approaches. These approaches aim to combine SMN-targeted therapies with additional, SMN-independent strategies. Indeed, the recently announced trial for myostatin inhibitor SRK-015 will consist of groups of patients that both have or have not concurrently received Spinraza. Of interest, several recent preclinical studies have used innovative approaches to study the effect of combining SMN-targeted therapies with further preclinical targets. In these studies, mouse models of SMA were treated with suboptimal doses of Spinraza, or comparable ASOs, to generate intermediate models of SMA [13, 89]. In intermediate models, a partial rescue of the disease phenotype by ASO treatment is thought to better represent the clinical improvements being observed in patients enrolled in clinical trials for Spinraza [88]. Targeting of known modifiers of human disease subsequently leads to an improvement in disease progression that is vastly better than when SMN is targeted alone. For example, studies in discordant SMA families have previously identified PLS3 as a potent modifier of SMA pathology: SMN1 deletion carriers were completely protected from developing SMA by high expression levels of PLS3 [14]. When increasing PLS3 levels in intermediate mouse models of SMA, either by gene therapy approaches or genetically, survival and overall phenotype vastly improved [13, 89]. Similarly, depletion of NCALD, another modifier of SMA in discordant families, vastly improved SMA-associated phenotypes in a number of animal models, including an intermediate mouse model [15]. Functionally, these proteins are linked to actin dynamics and endocytosis, suggesting an important role for these pathways in SMA pathogenesis. These findings further illustrate the need to develop therapies that go beyond SMN targeting and build on the basic understanding of disease pathogenesis to optimize therapy development (Fig. 1).

Overview of potential combinatorial approaches for SMA therapy. Most therapies currently under development for the treatment for SMA work by targeting SMN levels. Additional approaches being explored include drugs designed to enhance muscle function. Other therapies, including olesoxime and several alternative promising preclinical leads, support the neuromuscular system (and potentially other non-neuronal cells and tissues) by targeting a number of cellular pathways, such as the mitochondrion (olesoxime), actin dynamics (PLS3, fasudil and Y-27632), endocytosis (NCALD) and/or ubiquitin homeostasis (UBA1, ML372). Combinations of these various approaches will likely be required to provide robust rescue of all SMA-associated pathologies across a patient’s lifespan. NCALD neurocalcin delta, PLS3 plastin-3, SMA spinal muscular atrophy, SMN survival motor neuron, UBA1 ubiquitin-like modifier activating enzyme 1
4.2 Promising Preclinical Leads
For the development of agents that show promise for combinatorial approaches, it is of interest to note that many disease-modifying molecular pathways have been discovered in SMA. Targeting each of these pathways holds potential promise for further therapy development in SMA. For example, gene therapy approaches that restore UBA1 expression levels have been shown to lead to broad improvement of pathology and a modest increase in survival in an SMA mouse model, independent of SMN targeting [27]. Similarly, the compound ML372, which selectively inhibits the ubiquitylation of SMN, has been shown to increase SMN protein levels and improve survival and motor function of SMA mice [90]. Also, treatment with the beta-catenin inhibitor quercetin, a plant extract that is widely available as a food supplement, significantly improves neuromuscular function in animal models of SMA [28]. In addition, other preclinical work has also identified fasudil and Y-27632 as promising therapeutic leads for SMA. In these studies, inhibition of the RhoA/Rho kinase (ROCK) pathway was shown to improve the phenotype in mouse models of SMA by partially restoring actin dynamics that lead to improved function at the neuromuscular junction [91, 92]. Finally, recent work has identified changes in ribosome biology as an important cellular pathway that might be associated with motor neuron-specific degeneration in SMA [31]. Considering the central role of local protein translation in maintaining neuronal homeostasis these findings warrant further investigation of these pathways, including how they can be effectively therapeutically targeted.
Discussing all proteins and pathways that have been shown to modulate SMA phenotypes in detail would go beyond the scope of this review. However, the above examples aim to illustrate that recent basic research into disease mechanisms of SMA has not stopped at the level of understanding SMN function and replacing SMN expression, and is likely to lead to further insights in disease pathogenesis, including the identification of novel therapeutic targets.
4.3 Further Considerations
Despite the positivity and promise that surround the current developments in the SMA field, several factors complicate further research into current and novel therapies for SMA [93]. First, basic research indicates that the timing of SMN-targeted therapies is essential for maximum efficacy, which is confirmed by initial trial results [5, 43, 53]. Delays in diagnosis might not always make quick therapeutic intervention possible, particularly in milder cases of the disease, which calls for the development of further, SMN-independent therapies. Moreover, strategies such as neonatal screening should be discussed to minimize diagnostic delay [94, 95]. Second, despite a shared genetic basis, clinical heterogeneity between patients exists. Indeed, although Spinraza has been approved for the treatment of all types of SMA, data on the efficacy of Spinraza in type III and IV SMA are still relatively sparse. Finding therapies that are efficient across SMA clinical phenotypes will be a complex challenge, and results, particularly for milder forms of the disease, are likely to take a long time to establish. Furthermore, it is to be expected that future trial design will be complex, as the effect of Spinraza on the clinical progress of SMA is still incompletely understood and yet, many patients are likely to begin treatment soon. Comparing the efficacy of Spinraza to other SMN-targeted therapies will be challenging, as delivery routes can be complex and washout periods can be long. Indeed, the design of future studies investigating the efficacy of combinatorial therapies will be even more challenging, as many patients will likely have already received one or more SMN-targeting drug. In addition to the practical issues around study design and patient selection, a number of ethical and financial issues are now becoming increasingly relevant. For example, the pricing of Spinraza might prevent some groups of patients from gaining access to treatment. Moreover, although drugs like Spinraza and AVXS-101 lead to increased survival, little is known as yet about the quality of life as perceived by the treated patients and their families. Each of these issues will require consideration for the successful further development of therapies aiming to provide robust, life-long treatments for SMA.
5 Concluding Remarks
The recent approval of Spinraza is a milestone development for the SMA research and patient communities. The significance of the availability of an approved, disease-modifying therapy for the treatment of a monogenetic motor neuron disease for future research, both basic and clinical, cannot be understated. Developments in the SMA field will no doubt be very carefully monitored by scientists and clinicians studying related disorders and are likely to provide important lessons for the study of other monogenetic, neurological diseases and motor neuron diseases. Increasing our understanding of the long-term efficacy of Spinraza, including that in type III and IV SMA patients, and that of other drugs that are currently in development for SMA will be of great interest in the years to come and provide further hope to SMA patients and their families for efficient treatment of this devastating disorder.
References
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–33.
Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy—a literature review. Orphanet J Rare Dis. 2017;12(1):124.
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65.
Aartsma-Rus A. FDA approval of nusinersen for spinal muscular atrophy makes 2016 the year of splice modulating oligonucleotides. Nucleic Acid Ther. 2017;27(2):67–9.
Hoy SM. Nusinersen: first global approval. Drugs. 2017;77(4):473–9.
Singh NN, Howell MD, Androphy EJ, Singh RN. How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017;24(9):520–6.
Wadman RI, Stam M, Gijzen M, Lemmink HH, Snoeck IN, Wijngaarde CA, et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0-4. J Neurol Neurosurg Psychiatry. 2017;88(4):365–7.
Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 2012;11(5):443–52.
Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–9.
Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–11.
Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–83.
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–68.
Hosseinibarkooie S, Peters M, Torres-Benito L, Rastetter RH, Hupperich K, Hoffmann A, et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet. 2016;99(3):647–65.
Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–7.
Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Lohr H, Bradler C, et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet. 2017;100(2):297–315.
Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 2015;35(23):8691–700.
Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90(6):1023–9.
Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90(6):1013–21.
Pellizzoni L, Baccon J, Charroux B, Dreyfuss G. The survival of motor neurons (SMN) protein interacts with the snoRNP proteins fibrillarin and GAR1. Curr Biol. 2001;11(14):1079–88.
Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133(4):585–600.
Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298(5599):1775–9.
Donlin-Asp PG, Fallini C, Campos J, Chou CC, Merritt ME, Phan HC, et al. The survival of motor neuron protein acts as a molecular chaperone for mRNP assembly. Cell Rep. 2017;18(7):1660–73.
Piazzon N, Schlotter F, Lefebvre S, Dodre M, Mereau A, Soret J, et al. Implication of the SMN complex in the biogenesis and steady state level of the signal recognition particle. Nucleic Acids Res. 2013;41(2):1255–72.
Hosseinibarkooie S, Schneider S, Wirth B. Advances in understanding the role of disease-associated proteins in spinal muscular atrophy. Expert Rev Proteom. 2017;14(7):581–92.
Bowerman M, Shafey D, Kothary R. Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J Mol Neurosci. 2007;32(2):120–31.
Nolle A, Zeug A, van Bergeijk J, Tonges L, Gerhard R, Brinkmann H, et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum Mol Genet. 2011;20(24):4865–78.
Powis RA, Karyka E, Boyd P, Come J, Jones RA, Zheng Y, et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight. 2016;1(11):e87908.
Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, et al. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J Clin Invest. 2014;124(4):1821–34.
Boyd PJ, Tu WY, Shorrock HK, Groen EJN, Carter RN, Powis RA, et al. Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genet. 2017;13(4):e1006744.
Miller N, Shi H, Zelikovich AS, Ma YC. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum Mol Genet. 2016;25(16):3395–406.
Bernabo P, Tebaldi T, Groen EJ, Lane FM, Perenthaler E, Mattedi F, et al. In vivo translatome profiling in spinal muscular atrophy reveals a role for SMN protein in ribosome biology. Cell Rep. 2017;21(4):953–65.
Fallini C, Donlin-Asp PG, Rouanet JP, Bassell GJ, Rossoll W. Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J Neurosci. 2016;36(13):3811–20.
Sanchez G, Bondy-Chorney E, Laframboise J, Paris G, Didillon A, Jasmin BJ, et al. A novel role for CARM1 in promoting nonsense-mediated mRNA decay: potential implications for spinal muscular atrophy. Nucleic Acids Res. 2016;44(6):2661–76.
Sanchez G, Dury AY, Murray LM, Biondi O, Tadesse H, El Fatimy R, et al. A novel function for the survival motoneuron protein as a translational regulator. Hum Mol Genet. 2013;22(4):668–84.
Holt CE, Schuman EM. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron. 2013;80(3):648–57.
Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, et al. Dynamic axonal translation in developing and mature visual circuits. Cell. 2016;166(1):181–92.
Lorson MA, Lorson CL. SMN-inducing compounds for the treatment of spinal muscular atrophy. Future Med Chem. 2012;4(16):2067–84.
Zhou H, Janghra N, Mitrpant C, Dickinson RL, Anthony K, Price L, et al. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum Gene Ther. 2013;24(3):331–42.
Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20(4):681–93.
Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28(3):271–4.
Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2(35):35ra42.
Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345(6197):688–93.
Kariya S, Obis T, Garone C, Akay T, Sera F, Iwata S, et al. Requirement of enhanced Survival motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest. 2014;124(2):785–800.
Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med. 2013;19(1):40–50.
Somers E, Lees RD, Hoban K, Sleigh JN, Zhou H, Muntoni F, et al. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann Neurol. 2016;79(2):217–30.
Szunyogova E, Zhou H, Maxwell GK, Powis RA, Francesco M, Gillingwater TH, et al. Survival motor neuron (SMN) protein is required for normal mouse liver development. Sci Rep. 2016;04(6):34635.
Thomson AK, Somers E, Powis RA, Shorrock HK, Murphy K, Swoboda KJ, et al. Survival of motor neurone protein is required for normal postnatal development of the spleen. J Anat. 2017;230(2):337–46.
Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19(20):3895–905.
Deguise MO, De Repentigny Y, McFall E, Auclair N, Sad S, Kothary R. Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum Mol Genet. 2017;26(4):801–19.
Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478(7367):123–6.
Wijngaarde CA, Blank AC, Stam M, Wadman RI, van den Berg LH, van der Pol WL. Cardiac pathology in spinal muscular atrophy: a systematic review. Orphanet J Rare Dis. 2017;12(1):67.
Gillingwater TH. Dawn of a new therapeutic era for spinal muscular atrophy. Lancet. 2016;388(10063):2964–5.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–26.
Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86(10):890–7.
Luu KT, Norris DA, Gunawan R, Henry S, Geary R, Wang Y. Population pharmacokinetics of nusinersen in the cerebral spinal fluid and plasma of pediatric patients with spinal muscular atrophy following intrathecal administrations. J Clin Pharmacol. 2017;57(8):1031–41.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–32.
Mercuri E, Finkel R, Kirschner J, Chiriboga C, Kuntz N, Sun P, Gheuens S, Bennett C, Schneider E, Farwell W, CHERISH Study Group. Efficacy and safety of nusinersen in children with later-onset spinal muscular atrophy (SMA): end of study results from the phase 3 CHERISH study. Neuromuscul Disord. 2017;27(Supplement 2):S210. https://doi.org/10.1016/j.nmd.2017.06.418.
Scoto M, Finkel RS, Mercuri E, Muntoni F. Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther. 2017;24(9):514–9.
Crawford TO. Efficacy and safety of nusinersen in genetically diagnosed infants with presymptomatic spinal muscular atrophy (SMA): results from the second interim analysis of the ongoing, phase 2 NURTURE study. In: 21st annual spinal muscular atrophy researcher meeting 2017, Orlando, Florida; 2017.
Hwu W, De D, Bertini E, Foster R, Gheuens S, Farwell W, Reyna S. Outcomes after 1-year in presymptomatic infants with genetically diagnosed spinal muscular atrophy (SMA) treated with nusinersen: interim results from the NURTURE study. Neuromuscul Disord. 2017;27(Supplement 2):S212. https://doi.org/10.1016/j.nmd.2017.06.424.
Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci USA. 2016;113(39):10962–7.
Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, O’Riordan CR, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120(4):1253–64.
Bevan AK, Duque S, Foust KD, Morales PR, Braun L, Schmelzer L, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19(11):1971–80.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–22.
AveXis. AveXis announces plan to initiate pivotal trial of AVXS-101 in SMA type 1 using new product from New GMP Commercial process. FDA notified AveXis it may initiate pivotal trial based on a review of data provided by the company following CMC Type B meeting. 2017. http://investors.avexis.com/phoenix.zhtml?c=254285&p=irol-newsArticle&ID=2303674. Accessed 30 Sept 2017.
Charnas L. Safety and efficacy findings in the first-in-human trial of the oral splice modulator branaplam in type 1 spinal muscular atrophy (SMA): interim results. In: 21st Annual Spinal Muscular Atrophy Researcher Meeting 2017; 2017; Orlando, Florida; 2017.
Charnas L. Safety and efficacy findings in the first-in-human trial (FIH) of the oral splice modulator branaplam in type 1 spinal muscular atrophy (SMA): interim results. In: 22nd International Congress of the World Muscle Society; 2017; Saint Malo, France: Neuromuscular Disorders; 2017. pp S207–S8.
Novartis. Novartis releases update on LMI070 (branaplam) clinical trial. 2017. http://www.curesma.org/news/novartis-branaplam-update.html. Accessed 30 Sept 2017.
Mercuri E. Clinical studies of RG7916 in patients with spinal muscular atrophy: study update. In: 21st Annual Spinal Muscular Atrophy Researcher Meeting 2017; 2017; Orlando, Florida; 2017.
Jethwa S. Start of Part 2 of the SUNFISH study. 2017. http://www.sma-europe.eu/wp-content/uploads/2017/09/sunfish_part2start_12.09.17.pdf. Accessed 27 Sept 2017.
Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. 2009;4(5):e5268.
Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, et al. SMA CARNIVAL trial part II: a prospective, single-armed trial of l-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One. 2011;6(7):e21296.
Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, et al. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of l-carnitine and valproic acid in spinal muscular atrophy. PLoS One. 2010;5(8):e12140.
Kissel JT, Elsheikh B, King WM, Freimer M, Scott CB, Kolb SJ, et al. SMA valiant trial: a prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle Nerve. 2014;49(2):187–92.
Darbar IA, Plaggert PG, Resende MB, Zanoteli E, Reed UC. Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid. BMC Neurol. 2011;24(11):36.
Krosschell KJ, Kissel JT, Townsend EL, Simeone SD, Zhang RZ, Reyna SP, et al. Clinical trial of l-carnitine and valproic acid in spinal muscular atrophy type I. Muscle Nerve. 2018;57(2):193–9.
Garbes L, Heesen L, Holker I, Bauer T, Schreml J, Zimmermann K, et al. VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum Mol Genet. 2013;22(2):398–407.
Bordet T, Berna P, Abitbol JL, Pruss RM. Olesoxime (TRO19622): a novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals (Basel). 2010;3(2):345–68.
Gouarne C, Giraudon-Paoli M, Seimandi M, Biscarrat C, Tardif G, Pruss RM, et al. Olesoxime protects embryonic cortical neurons from camptothecin intoxication by a mechanism distinct from BDNF. Br J Pharmacol. 2013;168(8):1975–88.
Gouarne C, Tracz J, Paoli MG, Deluca V, Seimandi M, Tardif G, et al. Protective role of olesoxime against wild-type alpha-synuclein-induced toxicity in human neuronally differentiated SHSY-5Y cells. Br J Pharmacol. 2015;172(1):235–45.
Sunyach C, Michaud M, Arnoux T, Bernard-Marissal N, Aebischer J, Latyszenok V, et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology. 2012;62(7):2346–52.
Bertini E, Dessaud E, Mercuri E, Muntoni F, Kirschner J, Reid C, et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16(7):513–22.
Hwee DT, Kennedy AR, Hartman JJ, Ryans J, Durham N, Malik FI, et al. The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J Pharmacol Exp Ther. 2015;353(1):159–68.
Rubnicki S. Clinical trial update of CY 5021: CK-2127107, a selective activator of the fast skeletal muscle troponin complex, for the potential treatment of spinal muscular atrophy. In: 21st Annual Spinal Muscular Atrophy Researcher Meeting 2017; 2017; Orlando, Florida; 2017.
Long K. Inhibition of myostatin activation by SRK-015 promotes muscle strength in a therapeutic mouse model of SMA. In: 21st Annual Spinal Muscular Atrophy Researcher Meeting 2017; Orlando, Florida; 2017.
ScholarRock. SRK-015 for spinal muscular atrophy (SMA). 2017. http://www.scholarrock.com/pipeline/srk-015-for-sma/intro/. Accessed 30 Aug 2017.
Mariot V, Joubert R, Hourde C, Feasson L, Hanna M, Muntoni F, et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun. 2017;8(1):1859.
Zhou H, Meng J, Marrosu E, Janghra N, Morgan J, Muntoni F. Repeated low doses of morpholino antisense oligomer: an intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum Mol Genet. 2015;24(22):6265–77.
Kaifer KA, Villalon E, Osman EY, Glascock JJ, Arnold LL, Cornelison DD, et al. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight. 2017;2(5):e89970.
Abera MB, Xiao J, Nofziger J, Titus S, Southall N, Zheng W, et al. ML372 blocks SMN ubiquitination and improves spinal muscular atrophy pathology in mice. JCI Insight. 2016;1(19):e88427.
Bowerman M, Beauvais A, Anderson CL, Kothary R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet. 2010;19(8):1468–78.
Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 2012;07(10):24.
Farrar MA, Park SB, Vucic S, Carey KA, Turner BJ, Gillingwater TH, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355–68.
Dobrowolski SF, Pham HT, Downes FP, Prior TW, Naylor EW, Swoboda KJ. Newborn screening for spinal muscular atrophy by calibrated short-amplicon melt profiling. Clin Chem. 2012;58(6):1033–9.
Taylor JL, Lee FK, Yazdanpanah GK, Staropoli JF, Liu M, Carulli JP, et al. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin Chem. 2015;61(2):412–9.
Acknowledgements
The authors would like to thank Dr. Kiterie Faller for critical reading of the manuscript. E.J.N.G. is supported by the Wellcome Trust (Grant 106098/Z/14/Z). H.K.S. is supported by a Euan MacDonald Centre for MND Research PhD studentship to T.H.G. Research in the Gillingwater laboratory relevant to this review is funded by the UK SMA Research Consortium (SMA Trust), MND Scotland and SMA Europe.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the publication of this review.
Conflict of interest
T.H.G. is Chair of the Scientific and Clinical Advisory Panel of the SMA Trust and is a panel member for SMA Europe and AFM. H.K.S. and E.J.N.G. have no conflicts of interest to declare.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shorrock, H.K., Gillingwater, T.H. & Groen, E.J.N. Overview of Current Drugs and Molecules in Development for Spinal Muscular Atrophy Therapy. Drugs 78, 293–305 (2018). https://doi.org/10.1007/s40265-018-0868-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0868-8