
Abstract
Introduction and Objective
Serous retinopathy can be associated with MEK inhibitors, including cobimetinib. We present results of an integrated safety analysis to further characterize ocular functional and structural changes due to serous retinopathy.
Methods
Four studies evaluating cobimetinib at the approved dose and schedule in combination with other oncology drugs were included. Study CO39721 incorporated standardized ophthalmologic assessments to fully characterize serous retinopathy events over time and was the primary study for analysis. Supporting information was provided by studies GO28141, WO29479, and GO30182.
Results
In total, 655 patients received one or more doses of cobimetinib and comprised the safety-evaluable population. Overall, 117 patients (17.9%) had one or more serous retinopathy events, 24 (3.7%) had two or more events, and four (0.6%) had three or more events. Grade 3 events occurred in < 2.5% of patients. In CO39721, the median time to onset was 15 days (range 7–111); median time to resolution of first occurrence was 26 days (range 6–591 + days). Twelve of 25 patients (48.0%) recovered without a dose modification and 4/25 (16.0%) were recovered/recovering following a dose modification. The most frequent presentation of serous retinopathy was focal subretinal fluid on optical coherence tomography (62.8% of cases); in some instances (25.7% of cases), subretinal fluid was multifocal. There was no loss of visual function or visual acuity at serous retinopathy onset or resolution.
Conclusions
Results from this integrated safety analysis indicate that cobimetinib-associated serous retinopathy can be managed with or without a dose modification of cobimetinib at the discretion of the treating physician. No visual loss or permanent retinal damage was identified on comprehensive ophthalmologic assessments.
Clinical Trial Registration
ClinicalTrials.gov identifiers: NCT03178851, NCT01689519, NCT02322814, and NCT02788279.
Similar content being viewed by others


Among patients treated with cobimetinib at the approved dose and schedule in combination with other oncology drugs, the incidence of serous retinopathy was 17.9%; most events were asymptomatic or mildly symptomatic and recurrence was infrequent. |
Serous retinopathy typically occurred within the first few cycles after initiation of cobimetinib treatment and resolved within a few weeks after onset with either a dose modification or without any change in dosing. |
Comprehensive ophthalmologic assessments to characterize the visual and anatomical impact of cobimetinib-associated serous retinopathy did not identify any visual function loss or permanent retinal damage in this analysis. |
1 Introduction
The mitogen-activated protein kinase, or RAS–RAF–MEK–ERK, pathway regulates cell proliferation, differentiation, and survival and is aberrantly activated in more than a third of all cancers and approximately 90% of cutaneous melanomas [1]. Several MEK inhibitors have been developed and have been evaluated in a variety of cancers, but focus has increasingly turned to the evaluation of combinatorial strategies because of limited single-agent efficacy [2]. Cobimetinib is a reversible inhibitor of MEK1 and MEK2 that is approved for use in combination with vemurafenib for the treatment of BRAFV600-mutated locally advanced or metastatic melanoma [3, 4].
The mitogen-activated protein kinase pathway plays an important role in the maintenance, protection, and repair of the retina [5], and serous retinopathy (SR) has been identified as a class effect with MEK inhibitors [6, 7]. Symptomatic ocular toxicity has been reported in 0.5–19% of patients with other MEK inhibitors (trametinib, binimetinib, or refametinib) [8,9,10,11,12]. Higher incidences have been reported in studies that incorporated prospective screening for ocular toxicity, which frequently detects asymptomatic events [7, 13]. Although initially described as resembling central SR, clinical and morphologic characteristics of MEK inhibitor-associated SR are distinct from those of classic central serous chorioretinopathy [6, 7]. Although the mechanism of MEK inhibitor-associated SR is not well understood, evidence suggests that mitogen-activated protein kinase signaling can regulate the density of aquaporins between retinal pigment epithelium cells [14]. Inhibition of MEK may alter the permeability of the retinal pigment epithelium, allowing accumulation of subretinal fluid that can lead to retinal pigment epithelial detachment (RPED) [5].
In a previous analysis of the phase III coBRIM study, which incorporated a prospective evaluation of SR signs and symptoms, cobimetinib in combination with vemurafenib was associated with symptomatic or asymptomatic SR in 26% of patients with BRAFV600-mutated melanoma [15]. Previous studies of cobimetinib, including coBRIM, have not incorporated standardized eye examination schedules for patients who developed SR. Consequently, the CO39721 study was designed to include a comprehensive ocular examination schedule with fixed eye exams to fully characterize SR events and their sequelae [16]. We present the results of an integrated safety analysis to further characterize SR/RPED with cobimetinib at the approved dose and schedule in combination with various oncology drugs across different cancer types.
2 Methods
2.1 Included Studies
Four studies were included in the analysis (Table 1). Detailed methods for each of the studies have been previously reported. In brief, CO39721 (NCT03178851) was a phase lb, multicenter, open-label study evaluating cobimetinib in combination with atezolizumab in patients with BRAFV600 wild-type advanced melanoma who had progressed on prior anti-programmed death 1 therapy [16]. GO28141 (coBRIM; NCT01689519) was a phase III, multicenter, randomized, double-blind study evaluating cobimetinib in combination with vemurafenib in patients with previously untreated, BRAFV600 mutation-positive, advanced melanoma [3, 4]. WO29479 (COLET; NCT02322814) was a phase II, multicenter, randomized, open-label study evaluating cobimetinib in combination with paclitaxel, or atezolizumab and paclitaxel or nab-paclitaxel, in patients with previously untreated advanced triple-negative breast cancer [17]. Finally, GO30182 (COTEZO; NCT02788279) was a phase III, multicenter, randomized, open-label study evaluating cobimetinib in combination with atezolizumab in patients with previously untreated, advanced colorectal cancer [18]. In all four studies, cobimetinib was administered at the approved dose and schedule (60 mg once daily for 21 days followed by 7 days off in each 28-day cycle). Dose modifications for the management of SR were mandated by the protocol in each of the included studies (Table 1).
To fulfill a post-marketing requirement to the US FDA, CO39721 was designed to incorporate standardized ophthalmologic assessments to fully characterize the visual and anatomical impact of SR events over time and was considered the primary study for this analysis. Detailed ophthalmologic data were collected using prespecified standardized eye exams for patients who presented with SR, including visual acuity, indirect ophthalmoscopy, spectral-domain optical coherence tomography (OCT), and automated visual field testing (10-2 threshold if SR was detected only within the macula; 24-2 threshold if SR was extramacular or extended beyond the macula) until resolution of the SR event. Supporting information was provided by studies GO28141, WO29479, and GO30182, which incorporated prospective monitoring of SR and/or RPED, but on a less rigorous and well-defined schedule than study CO39721 (Table 1).
Each of the included studies was approved by the institutional review board or ethics committee at each participating institution and were conducted in accordance with the provisions of the Declaration of Helsinki and the International Conference on Harmonisation guidelines for Good Clinical Practice. All patients provided written informed consent.
2.2 Outcomes and Analysis
The analysis population for evaluation of SR and/or RPED comprised all patients in each study who received any amount of cobimetinib. Serous retinopathy and/or RPED reported as adverse events in any of the included studies was identified using a standardized list of Medical Dictionary for Regulatory Activities preferred terms for SR and/or RPED (Table S1 of the Electronic Supplementary Material [ESM]). Only SR and/or RPED events that occurred during or after the first dose of study treatment were included in the analysis.
Verbatim descriptions of adverse events were mapped to Medical Dictionary for Regulatory Activities thesaurus terms and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Grade 1 events were defined as mild (asymptomatic or mild symptoms; clinical or diagnostic observations only; intervention not indicated). Grade 2 events were defined as moderate (minimal, local, or noninvasive intervention indicated; limiting age-appropriate instrumental Activities of Daily Living). Grade 3 events were defined as severe (severe or medically significant but not life-threatening; hospitalization or prolongation of hospitalization indicated; disabling; limiting self-care Activities of Daily Living). Grade 4 events were defined as life threatening (urgent intervention indicated), and grade 5 events were defined as events leading to death. Multiple occurrences of the same event were counted only once using the highest grade.
Time to onset of SR and/or RPED was defined as the time from the first dose of the study drug to the start date of SR and/or RPED. Duration of SR and/or RPED was defined as the time from the event start date to the resolution date. Time to first occurrence and duration of the first occurrence of SR and/or RPED were analyzed separately from subsequent occurrences. Median time to resolution was estimated by Kaplan–Meier methodology with censoring of patients whose events did not resolve. Serous retinopathy and/or RPED events leading to a dose modification or a discontinuation of cobimetinib were summarized descriptively.
Potential risk factors for SR and/or RPED were assessed in the total pooled safety population and were summarized using descriptive statistics. Baseline ophthalmology confounders were identified through a review of ocular medical and surgical history, and baseline medical conditions were reviewed for risk factors identified as associated with retinopathy in a literature review (Table S2 of the ESM).
3 Results
Across the four included studies, 655 patients received at least one dose of cobimetinib and comprised the safety-evaluable population for analysis.
3.1 Incidence and Severity
A total of 117 patients (17.9%) had at least one SR and/or RPED event, with a total of 164 events reported (Table 2). Second and third occurrences were infrequent, with 24 patients (3.7%) having two or more events, and four patients (0.6%) having three or more events. The most frequently reported events by Medical Dictionary for Regulatory Activities preferred terms were chorioretinopathy (n = 42; 6.4%), MEK inhibitor-associated SR (n = 17; 2.6%), retinal detachment (n = 15; 2.3%), retinopathy (n = 13; 2.0%), and detachment of retinal pigment epithelium (n = 15; 2.3%).
The majority of SR/RPED events were grade 1 or 2. Grade 3 events were uncommon, occurring in ten patients (1.5%), and included chorioretinopathy (n = 4), retinal detachment (n = 2), retinopathy (n = 2), macular detachment (n = 1), and detachment of the retinal pigment epithelium (n = 1). There were no grade 4 or 5 events. Serious events occurred in four patients (0.6%): grade 1 retinal detachment (n = 1), grade 2 chorioretinopathy (n = 2), and grade 3 chorioretinopathy (n = 1).
3.2 Time to Onset of First SR/RPED Event
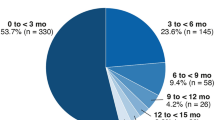
First occurrence of SR/RPED typically occurred within the first two cycles of treatment (Table 3). Median time to onset was 15 days (range 7–111) in the CO39721 study and 25–80 days (range 2–988) across the three supporting studies. In the CO39721 study, median time to onset for the second occurrence was 18.5 days (range 15–126) and median time to third occurrence was 20.5 days (range 14–27).
3.3 Management and Resolution
Most SR/RPED events resolved without treatment or with a modification of dosing (Table 4). Of 117 patients with one or more occurrences of SR/RPED, 40 patients (34.1%) had a dose modification (dose reduction or interruption) of cobimetinib and nine patients (7.7%) discontinued cobimetinib. At the time of the clinical data cut-off, SR/RPED events were resolved in 67/117 patients (54.7%). More than a third of patients (45/117; 38.5%) recovered or were recovering following no change in dosing. In study CO39721, 12/25 patients (48%) recovered with no dose modification, and 4/25 patients (16%) were either recovered or recovering following dose modification.
Among patients with resolution of SR/RPED, the median duration of first occurrence was 14 days (range 6–42) in the CO39721 study. Median time to resolution of the first occurrence was 26 days (range 6–591 +) in the CO39721 study and 34–97 days (range 3–2234 +) across the three supporting studies.
3.4 Risk Factors
In a univariate analysis, hypertension, autoimmune disease, pre-existing ocular confounders, male sex and age ≥50 years were identified as significant risk factors for the development of SR/RPED (Table 5).
3.5 Ophthalmologic Assessments in Patients with SR in Study CO39721
In study CO39721, a total of 31 SR events were reported in 25 patients, with four patients having a second occurrence and two patients having a third occurrence. Serous retinopathy was bilateral in 15 patients and unilateral in five patients, for a total of 35 eyes affected.
Development of SR had no impact on best-corrected mean visual acuity. Mean visual acuity was similar at baseline (79.1 letters; n = 35 eyes), the time of first SR onset (79.1 letters; n = 35 eyes), and at the last result regardless of SR resolution (80.7 letters; n = 33 eyes).
At baseline, 23/35 eyes (65.7%) had normal indirect ophthalmoscopy results and 12/35 (34.3%) had baseline abnormalities unrelated to SR. In the 23 eyes with normal readings at baseline, indirect ophthalmoscopy results were normal in ten eyes and abnormal in 13 eyes at SR onset. Serous retinopathy resolved in 7/13 eyes; at the time of the last SR resolution, indirect ophthalmoscopy findings returned to normal in two eyes and remained abnormal in five eyes. At the last available indirect ophthalmoscopy, findings were normal in 12 eyes and abnormal in 11 eyes.
Optical coherence tomography findings of subretinal fluid were similar to those previously illustrated [15]. Thirty-four of 35 eyes (97.1%) had OCT findings related to SR, mostly as focal subretinal fluid in the fovea (22/35 eyes, 62.8%; Table S3 of the ESM). Subretinal fluid was multifocal at SR onset in 9/35 eyes (25.7%). Subretinal fluid was still present at the last available OCT in 4/35 eyes (11.4%).
At screening, 7/35 eyes (20.0%) had visual field defects (data missing for five eyes). No new visual field defects were reported during the study.
4 Discussion
Previously, an incidence of SR of 26% was reported with cobimetinib in combination with vemurafenib [15] but the longitudinal impact on visual function and retinal anatomy from SR was not comprehensively studied. The current report expands on those findings in a larger group of patients including cobimetinib in combination with other oncology drugs. Based on the evaluation of 655 patients who were treated with cobimetinib in combination with other agents across four studies, the frequency of SR and/or RPED ranged from 6.4 to 26.6%. Reported incidences of MEK inhibitor-associated SR with other MEK inhibitors (alone or in combination with other oncology drugs) vary widely, with a range from 1 to 2% with trametinib, ~ 2% with refametinib, 10–40% with selumetinib, and 9–62% with binimetinib [8,9,10,11,12,13, 19]. However, a comparison of the incidence of SR across MEK inhibitors is complicated by the lack of prospective screening for retinopathy in the majority of studies, as well as a lack of uniformity in the description, diagnosis, and reporting of ocular toxicity across studies. Given the lack of randomized studies that include a MEK inhibitor monotherapy arm, it remains unclear whether the incidence of MEK inhibitor-associated SR is altered by combinations with other oncology drugs. However, SR is infrequently reported with immune checkpoint inhibitor or BRAF inhibitor monotherapy [20], and evidence to date suggests that SR observed with combined BRAF plus MEK inhibition is largely caused by the MEK inhibitor [6, 13, 15, 21] .
In the CO39721 study, which incorporated standardized ocular examinations (visual acuity, indirect ophthalmoscopy, OCT, and automated visual field testing) to fully characterize the visual and anatomical impact of SR events over time, the most frequent presentation of SR during cobimetinib treatment was focal subretinal fluid on OCT (62.8% of cases), mostly in the fovea, which resolved in a large majority of cases. Multifocal spots of subretinal fluid were less common, present in 25.7% of cases. There was no impact on visual function by the accumulation of subretinal fluid, either at the occurrence of the event or at the time of resolution; more specifically, there was no visual loss on visual acuity testing and no development of scotomas on automated visual field testing. Most first events of SR/RPED occurred within the first few cycles of treatment and resolved within a few weeks after onset with either a dose modification or without any change in the dosing regimen. The majority of patients did not require discontinuation or a dose modification of cobimetinib.
Risk factors identified as associated with the development of SR were male sex, hypertension, autoimmune disease, age ≥ 50 years, and pre-existing ocular confounders. Male sex, hypertension, and autoimmune disease are known risk factors for the development of central SR in the general population, which is the most similar ocular pathology to cobimetinib-induced SR [22,23,24,25,26,27]. In addition, having pre-existing ocular confounding factors such as retinal pathologies associated with the development of subretinal fluid increased the risk of developing SR.
Although this analysis comprises a large cohort of patients treated with cobimetinib, the analysis is limited by the less rigorous assessment schedule in the three supporting studies relative to the CO39721 study. The more rigorous assessment schedule for CO39721 may have resulted in the identification of the first events of SR/RPED ~ 2 weeks earlier than in the supporting studies. Nevertheless, results of ocular assessments and clinical outcomes were consistent across the four studies regardless of the monitoring schedule used and the overall management of patients was not impacted by the frequency of assessments.
5 Conclusions
In this integrated safety analysis, no visual function loss or permanent retinal damage was detected in patients with cobimetinib-associated SR who underwent comprehensive ophthalmologic assessments. Some patients may have a higher risk of developing SR, especially men and those with systemic hypertension, an autoimmune disease, age ≥ 50 years, and pre-existing ocular confounders that could lead to subretinal fluid accumulation. It is advised that patients undergo an ophthalmologic examination before receiving cobimetinib as a baseline assessment. After initiation of cobimetinib treatment, patients who experience new or worsening visual disturbances should be referred to an ophthalmologist to check for the development of SR using conventional functional and structural tests and should be followed longitudinally until resolution of the event. These events can often be managed without any change to dosing or with a dose modification of cobimetinib at the discretion of the treating physician.
References
Barbosa R, Acevedo LA, Marmorstein R. The MEK/ERK network as a therapeutic target in human cancer. Mol Cancer Res. 2021;19(3):361–74. https://doi.org/10.1158/1541-7786.MCR-20-0687.
Tatli O, Dinler DG. Recent developments in targeting RAS downstream effectors for RAS-driven cancer therapy. Molecules. 2021;26(24):7561. https://doi.org/10.3390/molecules26247561.
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76. https://doi.org/10.1056/NEJMoa1408868.
Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17(9):1248–60. https://doi.org/10.1016/S1470-2045(16)30122-X.
van der Noll R, Leijen S, Neuteboom GH, Beijnen JH, Schellens JH. Effect of inhibition of the FGFR-MAPK signaling pathway on the development of ocular toxicities. Cancer Treat Rev. 2013;39(6):664–72. https://doi.org/10.1016/j.ctrv.2013.01.003.
Francis JH, Habib LA, Abramson DH, Yannuzzi LA, Heinemann M, Gounder MM, et al. Clinical and morphologic characteristics of MEK inhibitor-associated retinopathy: differences from central serous chorioretinopathy. Ophthalmology. 2017;124(12):1788–98. https://doi.org/10.1016/j.ophtha.2017.05.038.
Urner-Bloch U, Urner M, Jaberg-Bentele N, Frauchiger AL, Dummer R, Goldinger SM. MEK inhibitor-associated retinopathy (MEKAR) in metastatic melanoma: long-term ophthalmic effects. Eur J Cancer. 2016;65:130–8. https://doi.org/10.1016/j.ejca.2016.06.018.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14. https://doi.org/10.1056/NEJMoa1203421.
Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773–81. https://doi.org/10.1016/S1470-2045(12)70270-X.
Bendell JC, Javle M, Bekaii-Saab TS, Finn RS, Wainberg ZA, Laheru DA, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer. 2017;116(5):575–83. https://doi.org/10.1038/bjc.2017.10.
Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–56. https://doi.org/10.1016/S1470-2045(13)70024-X.
Weekes CD, Von Hoff DD, Adjei AA, Leffingwell DP, Eckhardt SG, Gore L, et al. Multicenter phase I trial of the mitogen-activated protein kinase 1/2 inhibitor BAY 86–9766 in patients with advanced cancer. Clin Cancer Res. 2013;19(5):1232–43. https://doi.org/10.1158/1078-0432.CCR-12-3529.
Urner-Bloch U, Urner M, Stieger P, Galliker N, Winterton N, Zubel A, et al. Transient MEK inhibitor-associated retinopathy in metastatic melanoma. Ann Oncol. 2014;25(7):1437–41. https://doi.org/10.1093/annonc/mdu169.
Jiang Q, Cao C, Lu S, Kivlin R, Wallin B, Chu W, et al. MEK/ERK pathway mediates UVB-induced AQP1 downregulation and water permeability impairment in human retinal pigment epithelial cells. Int J Mol Med. 2009;23(6):771–7. https://doi.org/10.3892/ijmm_00000191.
de la Cruz-Merino L, Di Guardo L, Grob JJ, Venosa A, Larkin J, McArthur GA, et al. Clinical features of serous retinopathy observed with cobimetinib in patients with BRAF-mutated melanoma treated in the randomized coBRIM study. J Transl Med. 2017;15(1):146. https://doi.org/10.1186/s12967-017-1246-0.
Sandhu SK, Atkinson VG, Cao MG, Medina T, Rivas AS, Caro I, et al. 1358P—interim analysis of a phase Ib study of cobimetinib plus atezolizumab in patients with advanced BRAFV600 wild type melanoma progressing on prior anti-PD-L1 therapy. Ann Oncol. 2019;30(Suppl._5):554. https://doi.org/10.1093/annonc/mdz255.046.
Brufsky A, Kim SB, Zvirbule Ž, Eniu A, Mebis J, Sohn JH, et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): primary analysis. Ann Oncol. 2021;32(5):652–60. https://doi.org/10.1016/j.annonc.2021.01.065.
Eng C, Kim TW, Bendell J, Argiles G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20(6):849–61. https://doi.org/10.1016/S1470-2045(19)30027-0.
Méndez-Martínez S, Calvo P, Ruiz-Moreno O, Pardiñas Barón N, Leciñena Bueno J, Gil Ruiz MDR, et al. Ocular adverse events associated with MEK inhibitors. Retina. 2019;39(8):1435–50. https://doi.org/10.1097/IAE.0000000000002451.
Fortes BH, Tailor PD, Dalvin LA. Ocular toxicity of targeted anticancer agents. Drugs. 2021;81(7):771–823. https://doi.org/10.1007/s40265-021-01507-z.
Gogas HJ, Flaherty KT, Dummer R, Ascierto PA, Arance A, Mandala M, et al. Adverse events associated with encorafenib plus binimetinib in the COLUMBUS study: incidence, course and management. Eur J Cancer. 2019;119:97–106. https://doi.org/10.1016/j.ejca.2019.07.016.
Liu B, Deng T, Zhang J. Risk factors for central serous chorioretinopathy: a systematic review and meta-analysis. Retina. 2016;36(1):9–19. https://doi.org/10.1097/IAE.0000000000000837.
Ojaimi E, Nguyen TT, Klein R, Islam FM, Cotch MF, Klein BE, et al. Retinopathy signs in people without diabetes: the multi-ethnic study of atherosclerosis. Ophthalmology. 2011;118(4):656–62. https://doi.org/10.1016/j.ophtha.2010.08.007.
Chao JR, Lai MY, Azen SP, Klein R, Varma R. Retinopathy in persons without diabetes: the Los Angeles Latino Eye Study. Investig Ophthalmol Vis Sci. 2007;48(9):4019–25. https://doi.org/10.1167/iovs.07-0206.
Jeganathan VS, Cheung N, Tay WT, Wang JJ, Mitchell P, Wong TY. Prevalence and risk factors of retinopathy in an Asian population without diabetes: the Singapore Malay Eye Study. Arch Ophthalmol. 2010;128(1):40–5. https://doi.org/10.1001/archophthalmol.2009.330.
van Leiden HA, Dekker JM, Moll AC, Nijpels G, Heine RJ, Bouter LM, et al. Risk factors for incident retinopathy in a diabetic and nondiabetic population: the Hoorn study. Arch Ophthalmol. 2003;121(2):245–51. https://doi.org/10.1001/archopht.121.2.245.
Gunnlaugsdottir E, Halldorsdottir S, Klein R, Eiriksdottir G, Klein BE, Benediktsson R, et al. Retinopathy in old persons with and without diabetes mellitus: the Age, Gene/Environment Susceptibility-Reykjavik Study (AGES-R). Diabetologia. 2012;55(3):671–80. https://doi.org/10.1007/s00125-011-2395-y.
Acknowledgements
The authors thank the participating investigators, patients, and current and past members of the Roche and Genentech cobimetinib and vemurafenib teams. Third-party medical writing assistance was provided by Melanie Sweetlove, MSc (ApotheCom, San Francisco, CA, USA) and was funded by F. Hoffmann-La Roche Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
F. Hoffmann-La Roche Ltd sponsored each of the included studies.
Conflicts of interest/competing interests
All authors are current or former employees of Roche/Genentech.
Ethics approval
Each of the included studies was approved by the institutional review board or ethics committee at each participating institution and was conducted in accordance with the provisions of the Declaration of Helsinki and the International Conference on Harmonisation guidelines for Good Clinical Practice.
Consent to participate
All patients provided written informed consent before participation in each of the included studies.
Consent for Publication
Not applicable.
Availability of data and material
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Code availability
Not applicable.
Authors’ contributions
GB and SM conceived the analysis and wrote the manuscript. GB, GRG, YP, IC, CX, and SM analyzed the data. All authors critically reviewed and approved the final manuscript.
Additional information
Yogesh Patel: At the time of the analysis; no current affiliation.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Barteselli, G., Goodman, G.R., Patel, Y. et al. Characterization of Serous Retinopathy Associated with Cobimetinib: Integrated Safety Analysis of Four Studies. Drug Saf 45, 1491–1499 (2022). https://doi.org/10.1007/s40264-022-01248-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-022-01248-2