
Abstract
Introduction
Radiopharmaceuticals may cause adverse events. Knowledge about adverse events from a patient’s perspective could help healthcare professionals to detect, understand, and manage adverse events more efficiently when using radiopharmaceuticals. Researchers need a validated questionnaire that can be used in patients to assess adverse events with radiopharmaceuticals.
Objective
The aim of this study was to develop, validate the content of, and perform initial testing of a questionnaire assessing patient-reported adverse events of radiopharmaceuticals.
Methods
Based on existing literature, six professionals drafted and evaluated a first version of the questionnaire. Further content validation was performed using cognitive interviews with six patients undergoing a nuclear medicine examination. After adaptations, the questionnaire was developed into a web-based questionnaire. One hundred patients undergoing nuclear examination tested this version, and the results were used to assess its acceptability and evaluate reported adverse events.
Results
Questions and answer options were revised in the initial questionnaire to improve clarity. In addition, some questions were removed. The final version consisted of 18 questions. In the test phase, the acceptability of the questionnaire was demonstrated (e.g. 79% of the patients who received the questionnaire completed it, and the median time to complete the questionnaire was 12 min for patients who reported an adverse event). Of the 100 patients (53% men, median age 64 years), 12 reported a total of 22 adverse events. One of these adverse events had a high causal association.
Conclusion
After validation and testing, the developed questionnaire to study patient-reported adverse events of radiopharmaceuticals is a suitable and valid instrument which can be used in future research.
Similar content being viewed by others


There are unique aspects inherent in the use of radiopharmaceuticals in nuclear medicine departments that need to be included in a questionnaire assessing patient-reported adverse events of these products. |
A questionnaire was developed that can be used for research purposes to assess patient-reported adverse events of radiopharmaceuticals. |
1 Introduction
Radiopharmaceuticals are used in nuclear medicine for diagnosis and therapy [1, 2]. While it is known that radiopharmaceuticals can cause adverse events, it is assumed that the frequency of adverse events in diagnostic radiopharmaceuticals is relatively low compared to events caused by other types of drugs [3,4,5,6,7,8]. This can be explained by the low dose of the tracer with a subsequent absence of pharmacological effect, and the limited use of the tracer in an individual patient—usually only once [3, 4]. However, underreporting of adverse events—also described for other types of drugs—is likely to play a role in this low frequency [9,10,11]. In addition, adverse events of radiopharmaceuticals may be left undetected, as follow-up contact seldom occurs between the patient and the nuclear medicine department after the examination is completed.
In order to investigate the frequency of adverse events and partially overcome the issues of underreporting, information provided by the patients themselves can be useful. Such information may shed a different light on the frequency of the adverse events, and more detailed information aids in assessing the causal relationship between the radiopharmaceutical and the reported event. Furthermore, information that patients provide may differ from information that healthcare professionals provide. Physicians generally focus more on serious, often rare adverse events, while patients report milder but more frequent adverse events such as tiredness [12]. Patients can also provide information about the impact of adverse events on their quality of life [13, 14]. More knowledge about the frequency of adverse events and the perspective of patients could help healthcare professionals to inform patients and to better prepare them in managing any adverse event that may arise.
Previous research has shown that questionnaires can be useful instruments in obtaining information from patients about adverse events and about, for instance, the time course, severity, and outcome of the adverse events [15, 16]. Even though researchers have developed several questionnaires in the past [15,16,17], they were developed for other types of drugs and are not suitable for radiopharmaceuticals because of aspects that are unique to the use of these products in nuclear medicine departments. Examples are the specific preparation of the patient before the nuclear medicine examination or additional steps during the procedure such as physical exercise or the concomitant administration of interventional agents, like agents that induce stress in the case of the assessment of myocardial perfusion. Information about these aspects will be essential when assessing reported adverse events. Furthermore, the use of radiological contrast agents in the case of combined techniques, such as positron emission tomography/computed tomography (PET/CT), and the fact that the nuclear medicine department usually has no follow-up contact with the patient after the examination is completed requires questions to be asked about the experience of patients at several specific moments. To our knowledge, only one study about radiopharmaceuticals assessed adverse events from the perspective of patients. In this study, researchers developed and validated a questionnaire, which 55 patients using Tc-99m medronic acid completed. However, this study involved one specific radiopharmaceutical and the researchers did not specify detailed information about the method of validation [18].
Therefore, we aimed to develop and validate a questionnaire dedicated to assessing adverse events with radiopharmaceuticals from the patient’s perspective, which can be used in future research. This paper describes the development, content validation, and initial testing of the questionnaire.
2 Methods
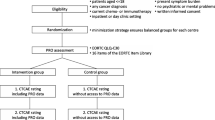
This study consisted of three phases: (1) the development of a questionnaire in the Dutch language, assessing adverse events from the perspective of patients undergoing an examination using radiopharmaceuticals, (2) the validation of its content, and (3) initial testing of the questionnaire (Fig. 1). We obtained ethical exemption in writing from the Medical Ethics Committee of the Isala Hospital, in Zwolle in The Netherlands (reference number 16.08138), as this study did not require formal approval, according to Dutch law.

Adapted with permission from de Vries et al. (2013) [15]
Process of development and validation of the questionnaire.
2.1 Phase 1: Questionnaire Development
Based on existing literature [3,4,5,6,7, 19, 20], two researchers (Q.d.H., N.S.) drafted a first version of the questionnaire containing the main questions in the following sections: (1) patient characteristics, (2) health status, (3) past nuclear medicine examinations and occurrence of adverse events, (4) preparation by the patient before a nuclear medicine examination, (5) administration of the radiopharmaceutical and occurrence of adverse events, (6) the period after the nuclear medicine examination and occurrence of adverse events, and (7) any further comments by the patient.
In section one, we obtained demographic data using closed-ended questions (i.e. assessing gender, education—grouped according to International Standard Classification of Education (ISCED) 2011 [21]—and use of over-the-counter medicines) and open-ended questions (i.e. assessing age, weight, and height). Subsequently, in section two, we measured the patient’s health status with the EuroQol-5-dimensions-3 levels (EQ-5D-3L) questionnaire [22]. The EQ-5D-3L is a qualified instrument to measure quality of life, including a descriptive system and a visual analogue score (EQ-VAS) [23]. Permission for its use was obtained. Sections three, five, and six contained open-ended and closed-ended questions about adverse events experienced during past nuclear medicine examinations, those associated with the administration of the radiopharmaceutical in the current nuclear medicine examination, and those experienced in the period after the examination, respectively. Section four contained closed-ended (both single-answer and multiple-answer) questions about the preparation by the patient before the nuclear medicine examination. In section seven, the patient could provide additional remarks about both the questionnaire and the nuclear medicine examination.
When patients indicated in the questionnaire that they experienced one or more adverse events, we asked additional closed-ended and open-ended questions, including aspects such as experienced symptoms, status of recovery, whether patients contacted a healthcare professional, and the type of professional (i.e. general practitioner, hospital staff, nuclear physician, nurse, pharmacist, and referring physician hospital). The additional questions also contained items to perform a causality assessment. This concerned the time of onset of the adverse event, previous experiences with nuclear medicine examinations, administration of interventional agents, or, in the case of combined techniques such as PET/CT, the use of contrast agents and other possible causes of the adverse event. We based these questions on the Naranjo algorithm [24], which is commonly used for causality assessment in pharmacovigilance, and the Silberstein algorithm [7], which specifically focuses on causality assessment of adverse events with radiopharmaceuticals.
A separate section in the questionnaire was meant only for the researchers to provide additional information obtained from medical records, such as the name of the radiopharmaceutical, its dose (in megabecquerel), type of examination, renal function, co-medication, and indications for use.
2.2 Phase 2: Content Validation
Previously, it has been determined that five experts are the minimum requirement for content validation. Moreover, is has been suggested that these experts are from all relevant disciplines to cover the content domain being assessed [25]. We selected six experts with expertise in the field of questionnaire development (n = 1), pharmacovigilance (n = 2), and nuclear medicine (n = 3) to form the expert panel in this study. Members of the expert panel independently reviewed the first paper-based version of the questionnaire. Two researchers (Q.d.H., N.S.) identified issues, which were used to draft a second version.
We subjected the second version of the questionnaire to cognitive interviewing in order to get insight into the way patients understand the questions and how they interpret the answer options, highlighting any ambiguities [26]. Six patients undergoing a nuclear medicine examination at the Isala Hospital in Zwolle participated in this part of the study. We selected consecutive patients willing to participate on the day of their nuclear medicine examination until we reached six participants. They were 18 years old or older and were able to read and speak the Dutch language. One researcher (Q.d.H.) conducted the interviews and audio-recorded them, with approval of the interviewees. In the interview, the researcher used a set of proactive and reactive so-called ‘probes’, while the patients were completing the questionnaire. Probes are questions specifically designed to obtain detailed information that the interviewee may not otherwise share [27]. Examples include ‘How did you come to an answer?’ ‘Can you repeat the question in your own words?’ and ‘How sure are you of the answer given?’ We transcribed the interviews using transcription software (Atlas.ti v7.5.12) and analysed the transcripts (Q.d.H.) to identify issues where the interviewee had difficulties answering the question. The identified issues were coded according to a dedicated system containing the following five categories: comprehension/communication, recall-based, bias/sensitivity, response categories, and logical/structural problems [28]. Two researchers (Q.d.H., NS) discussed the identified issues, which led to a third and enhanced version of the questionnaire.
Because the questionnaire is to be sent out to patients after a certain time interval after patients have left the nuclear medicine department and is to be used in a larger group of patients, we converted the paper-based questionnaire into a web-based questionnaire. Furthermore, this enabled the automation of sending the questionnaire after a time interval of 7 days after the nuclear medicine examination and allowed digital and faster processing of the data. Additionally, web-based questionnaires have shown some advantages in the past, such as a lower number of unanswered questions and more detailed answers to open questions [29]. The web-based version of the questionnaire was created using an online data manager (De Researchmanager®) [30]. The expert panel tested the web-based questionnaire on user friendliness and comprehensiveness. This led to revisions of the web-based version.
2.3 Phase 3: Testing of the Questionnaire
We then tested the revised web-based version of the questionnaire in a larger population of 100 patients to evaluate its acceptability and record adverse events. This is the number of patients recommended in literature [31]. Patients undergoing nuclear examination at Isala hospital were invited to participate in this test phase until we reached 100 participants who completed the questionnaire. Prior to the nuclear examination they received an invitation letter with a participation form. Patients were excluded when data were missing on the participation form that were required to initiate the web-based questionnaire, such as e-mail address or signature. We obtained written permission of patients willing to participate. Those patients received a link to the web-based questionnaire 7 days after their nuclear medicine examination. A reminder was sent after another 7 days when patients had not completed the questionnaire, but access to the questionnaire was limited to 21 days after the nuclear medicine examination. These time spans were chosen for two reasons. First, we would expect possible adverse events to occur within a few days after the nuclear medicine examination [8]. Second, longer recall periods may introduce bias due to patients forgetting information or patients bringing up information from other sequential doctor visits or examinations [32,33,34].
To assess acceptability in patients, we evaluated three characteristics: the percentage of patients completing the questionnaire, the time in which they completed the questionnaire, and their ability to answer all questions. We recorded the number of patients completing the questionnaire and the time in which they completed it. To assess the ability to answer all questions, we added—only during the test phase—at the end of each section a question asking whether there were any issues with answering the questions or with the wording, and, if so, what those issues were.
We evaluated reported adverse events and the time course. Reported adverse events were coded according to a Preferred Term of the Medical Dictionary for Regulatory Activities (MedDRA®) terminology. MedDRA is the international medical terminology developed under the auspices of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) [35]. Furthermore, we performed a causality assessment on all reported cases using the Naranjo [24] and Silberstein [7] algorithms. For the causality assessment, we used data obtained with the questionnaire on the time of onset of the adverse event, adverse events during previous nuclear medicine examinations, the recovery status of the patient, other possible causes of the adverse event such as administration of interventional agents, or other patient-reported possible causes. To determine if there were previous conclusive reports on the reaction or if it was a known response pattern, we used data from the literature [8] and the summary of product characteristics of the products. Two researchers (Q.d.H., N.S.) independently conducted the coding and assessed the causality. When results syntheses were not in agreement, the results were discussed with a third researcher (E.v.P.) to resolve discrepancies. After inclusion of the targeted 100 patients, we analysed the results descriptively using Excel version 1808 (Microsoft) and discussed the analysis to come to a final questionnaire (Q.d.H., N.S.).
3 Results
3.1 Phase 1: Questionnaire Development
The first draft of the questionnaire contained 30 main questions distributed over seven sections and additional questions about adverse events, which were posed when patients indicated they experienced an adverse event.
3.2 Phase 2: Content Validation
The expert panel provided feedback on the content of the first version of the questionnaire. Most of the comments related to clarity and wording. Of the 30 questions in the first version, we removed six questions, added one question, changed four questions, and left 20 questions unchanged. Of the six removed questions, five were related to the waiting period for the patient before and after the examination, which we deemed irrelevant. The sixth question that was removed was about the changes the patients experienced during the examination and was found to be repetitive. This resulted in a revised, second version of the questionnaire (Fig. 1) with 25 main questions.
Thereafter, six patients participated in the cognitive interviews. The patients were between 51 years old and 76 years old and had varying levels of education. One patient had trouble reading the questions due to partial visual impairment. As the interviewer read the questions out loud for this patient, the patient was still able to participate. Patients mentioned a total of 67 issues, mostly related to the categories comprehension/communication (52%) and response (22%) (Table 1). Of the second version, with 25 main questions, we left eleven unchanged, removed eight questions (because they were difficult to interpret by patients and on closer inspection were not considered necessary), revised five questions or answer options to improve clarity, combined one question with another, and added two new questions. The results of the cognitive interviews resulted in a third version of the questionnaire (Fig. 1) with a total of 18 questions. We converted this third version of the questionnaire to a web-based questionnaire.
Next, five of the six members of the expert panel evaluated the web-based version of the questionnaire and made 66 comments. The most mentioned comments were related to spelling, layout, or accompanying texts. One term was simplified (i.e. ‘medical professional’ to ‘caregiver’), and we changed the wording of two questions. The final questionnaire (Fig. 1) contained 18 main questions in seven sections and 12 additional questions (see the Electronic Supplementary Material [ESM], Supplementary Material 1), and the expert panel considered it suitable for further testing in patients. An English translation of the questions in this questionnaire is presented in Table 2. All questions removed during the content validation as well as an English translation can be found in Supplementary Material 2 in the ESM.
3.3 Phase 3: Testing of the Questionnaire
Over the course of 2 months, 650 patients received an invitation letter. Of those, 127 patients provided valid written permission to participate in the test phase of the web-based questionnaire (Fig. 2). Of these 127 patients receiving the questionnaire, 100 completed it (79%). This test population consisted of 53% men and 46% women. One patient (1%) indicated a different gender or did not want to specify gender. The median (interquartile range [IQR]) age was 64 (56–71) years old (Table 3). Radiopharmaceuticals most used were Tc-99m oxidronic acid, Tc-99m tetrofosmin, and F-18 fludeoxyglucose. Of the test population, 88% of the patients reported no adverse events, and 12% reported one or more adverse events. Patients reporting no adverse event completed the questionnaire in a median time of 8 min (IQR 6–12), and patients who reported an adverse event needed 12 min (IQR 9–16). With respect to the ability to answer the questions, two patients made two comments about the web-based questionnaire. One patient commented that she did not know her exact weight and made an estimation. Another patient indicated that she was not sure if a radiopharmaceutical was administered. All other patients stated they had no problem in answering the questions and understood all words.

Inclusion process of patients in the test phase
In total, 12 patients reported 22 adverse events. Adverse events reported were fatigue (n = 7), nausea (n = 3), abdominal discomfort (n = 2), chest discomfort (n = 2), feeling hot (n = 2), back pain (n = 1), dyspnoea (n = 1), limb discomfort (n = 1), pain in extremity (n = 1), paraesthesia (n = 1), and sense of oppression (n = 1). Patients reported 15 adverse events to have occurred within 1 h after administration of the radiopharmaceutical and reported the other seven adverse events to have occurred within 7 days after leaving the nuclear department. Patients reported eight adverse events with Tc-99m tetrofosmin, two with F-18 fludeoxyglucose, and other adverse events with Ra-223 dichloride, Tc-99m exametazime–labelled cells, and Tc-99m oxidronic acid. We found one adverse event—back pain with Ra-223 chloride after 7 days—to be probably (Naranjo) and possibly (Silberstein) causally related. The other 21 adverse events were possibly (Naranjo) or unlikely (Silberstein) to be causally related. More detail on the adverse events of the radiopharmaceutical, and the causality assessment of the adverse events can be found in Supplementary Material 3.
4 Discussion
In this study, a questionnaire to assess adverse events of radiopharmaceuticals from the perspective of the patient was developed, its content was validated, and initial testing was conducted. During the test phase, the questionnaire had good acceptability in patients. We found that the majority of the patients completed the questionnaire, that respondents completed the questionnaire in a reasonable time of 8 min for those not reporting an adverse event and 12 min for those reporting an adverse event, and that a vast majority of the patients indicated they had no problem in answering the questions and understood all words.
In the content validation part of our study, the expert panel and the cognitive interviews with patients identified several issues. This shows the importance of involving patients in the development of a questionnaire, as has been noted previously [36]. During the content validation, we found that the difference between the radiopharmaceutical, the interventional drug, and contrast media is not always clear to patients. After improving the questionnaire by clarifying the questions and putting the questions in a clear order, this issue did not occur again. However, this aspect might not be completely elucidated, and further research could reveal more detail about the reasons why patients do not always know the difference between the different pharmaceuticals and whether specific information provided to patients could improve this understanding.
In the test phase of our study, 12% of the patients reported an adverse event of radiopharmaceuticals—with only one adverse event assessed to have a higher causal relationship. This frequency seems to be higher than the number observed in a previous study about radiopharmaceuticals in which one out of 55 patients (1.8%) reported three adverse reactions [18]. However, that study involved only one specific radiopharmaceutical, which was not used in our study. Further research in a larger group of patients is needed to establish more insight into the frequency and types of adverse effects in nuclear medicine.
We performed a causality assessment using two algorithms and found that the categories of the two algorithms differ. For example, we found one adverse event to be probably (Naranjo) and possibly (Silberstein) causally related. This difference is inherent to the setup of each of the algorithms. Naranjo’s algorithm uses ten questions with a scoring system assigning causality on the basis of a total score in categories ‘definite’, ‘probable’, ‘possible’, or ‘doubtful’. Whereas Silberstein’s algorithm uses categories ‘not related’, ‘unlikely’, ‘possible’, or ‘probable’, with specific conditions to be met for each category. Also, Naranjo’s algorithm includes questions on aspects such as re-challenge, reappearance of the reaction with placebo, drug detection in toxic concentrations, and response after dose adjustment which are less relevant for radiopharmaceuticals, and which are not included in Silberstein’s algorithm. It might be worthwhile to compare both algorithms and examine the agreement and correlation of both methods in future research. In general, it should be noted that establishing a causal relationship between suspected drug and adverse event is difficult and that despite the fact that algorithms are often used in pharmacovigilance this cannot replace a thorough medical examination for an individual case.
Our questionnaire is a useful addition to the already existing questionnaires assessing adverse events for other types of drugs [15,16,17] since it focuses on adverse events of radiopharmaceuticals and includes aspects that are unique to the nuclear medicine examination. The questionnaire not only asks the patient about adverse events shortly after the examination but also has the possibility to ask about adverse events that occur later. The questionnaire includes relevant questions to support causality assessment. Although the design and validation of this questionnaire was done with a Dutch population with its specific cultural characteristics and in the Dutch language, we expect that the questionnaire is also useful in other populations or languages. However, validation of a translated version is required. Also, validation of the Dutch version has not ended, as it is a continuous process with, for instance, the possibility of changes in the interpretation of questions over time [37].
The strength of our study is the systematic development and validation of the questionnaire and the testing in a large number of patients. However, we must acknowledge several limitations of our study. One limitation is that we interviewed a limited number of six patients during the content validation. Although research indicates that a small group will reveal most critical problems [38], we cannot dismiss the possibility that a larger number of patients might have revealed additional issues. Another limitation is that data obtained in the test phase might be prone to bias [39]. Of the patients initially invited, 15% completed the questionnaire, and we did not ask patients the reasons for not participating. Furthermore, the education of the population in the test phase seems somewhat higher (37% having a bachelor’s degree, master’s degree, or equivalent level) in comparison with the general population (30% having a bachelor’s degree, master’s degree, or equivalent level [40]). Because of the choice to use a web-based questionnaire, some patients may not have been able to participate. On the other hand, internet access is rapidly increasing, which will enable more and more patients to participate in web-based questionnaires [41].
5 Conclusion
We developed a questionnaire to be used for studies to assess adverse events of radiopharmaceuticals from the perspective of the patient. After extensive validation and testing by experts and patients, this questionnaire proved to be suitable and valid. Researchers could use the questionnaire in further studies to learn more about adverse events of radiopharmaceuticals in a larger population, and this could be helpful for identifying potential adverse events of new radiopharmaceuticals.
References
Maltby P, Theobald T. Survey of current diagnostic radiopharmaceuticals. In: Theobald T, editor. Sampson’s textbook of radiopharmacy. 4th ed. London: Pharmaceutical Press; 2011. p. 277–306.
Chan P, Croasdale J. Survey of current therapeutic radiopharmaceuticals. In: Theobald T, editor. Sampson’s textbook of radiopharmacy. 4th ed. London: Pharmaceutical Press; 2011. p. 303–23.
Hesslewood SR, Keeling DH. Frequency of adverse reactions to radiopharmaceuticals in Europe. Eur J Nucl Med. 1997;24:1179–82.
Silberstein EB. Prevalence of adverse events to radiopharmaceuticals from 2007 to 2011. J Nucl Med. 2014;55:1308–10.
Bagheri H, Galian ME, Bastie D, et al. Enquête prospective sur les effets indésirables des médicaments radiopharmaceutiques. Thérapie. 1996;51:550–3.
Silberstein EB. Prevalence of adverse reactions to positron emitting radiopharmaceuticals in nuclear medicine. Pharmacopeia Committee of the Society of Nuclear Medicine. J Nucl Med. 1998;39:2190–2.
Silberstein EB, Ryan J. Prevalence of adverse reactions in nuclear medicine: Pharmacopeia Committee of the Society of Nuclear Medicine. J Nucl Med. 1996;37:185–92.
Schreuder N, Koopman D, Jager PL, Kosterink JGW, van Puijenbroek EP. Adverse events of diagnostic radiopharmaceuticals: a systematic review. Sem Nucl Med. 2019;49:382–410.
Hazell L, Shakir SA. Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2006;29:385–96.
Pinto SR, Santos LFC, Dos Reis SRR, Bastos MK, Gomes VDS, Vieira TO, et al. Adverse reactions to radiopharmaceuticals: a survey based on clinical cases using criteria of systematic review. Ther Innov Regul Sci. 2018;52:109–13.
Santos-Oliveira R, Machado M. Pitfalls with radiopharmaceuticals. Am J Med Sci. 2011;342:50–3.
Rolfes L, van Hunsel F, Wilkes S, van Grootheest K, van Puijenbroek E. Adverse drug reaction reports of patients and healthcare professionals-differences in reported information. Pharmacoepidemiol Drug Saf. 2015;24:152–8.
Foster JM, Van Der Molen T, Caeser M, Hannaford P. The use of questionnaires for measuring patient-reported side effects of drugs: its importance and methodological challenges. Pharmacoepidemiol Drug Saf. 2008;17:278–96.
Jarernsiripornkul N, Kakaew W, Loalukkana W, Krska J. Adverse drug reaction monitoring: comparing doctor and patient reporting for new drugs. Pharmacoepidemiol Drug Saf. 2009;18:240–5.
De Vries ST, Mol PGM, De Zeeuw D, Haaijer-Ruskamp FM, Denig P. Development and initial validation of a patient-reported adverse drug event questionnaire. Drug Saf. 2013;36:765–77.
Jarernsiripornkul N, Chaipichit N, Pratipanawatr T, Uchaipichat V, Krska J. Initial development and testing of an instrument for patient self-assessment of adverse drug reactions. Pharmacoepidemiol Drug Saf. 2016;25:54–63.
Duarte-Silva D, Figueiras A, Herdeiro MT, Teixeira Rodrigues A, Silva Branco F, Polónia J, Figueiredo IV. PERSYVE-Design and validation of a questionnaire about adverse effects of antihypertensive drugs. Pharm Pract (Granada). 2014;12:598.
Dos Santos Almeida R, Mamede M, Santos-Oliveira R. Pharmacovigilance of radiopharmaceuticals used for prostate and breast cancer in Brazil. Advers Drug React Bull. 2013;283:1091–4.
Giesen D, Meertens V, Vis-visschers R, Beukenhorst D. Questionnaire development. The Hague: Statistics Netherlands; 2012.
Sixma H, Hendriks M, Boer D, Delnoij D. Handboek CQI Ontwikkeling: richtlijnen en voorschriften voor de ontwikkeling van een CQI meetinstrument. 2nd ed. Utrecht: NIVEL; 2008; pp. 1–61. https://nivel.nl/sites/default/files/bestanden/Handboek-CQI-Ontwikkeling.pdf. Accessed 20 Aug 2019.
UNESCO. ISCED. International Standard Classification of Education 2011. Montreal: UNESCO Institute for Statistics; 2012.
EuroQol research foundation. EQ-5D Instruments. https://euroqol.org/. Accessed 04 July 2019.
The EuroQol Group. EuroQol-a new facility for the measurement of health related quality of life. Health Policy (New York). 1990;16:199–208.
Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239–45.
Lynn MR. Determination and quantification of content validity. Nurs Res. 1986;35:382–5.
Willis GB. Cognitive interviewing: a tool for improving questionnaire design. 1st ed. Thousand Oaks: Sage Publications Inc.; 2005.
Willis GB, Artino AR Jr. What do our respondents think we’re asking? Using cognitive interviewing to improve medical education surveys. J Grad Med Educ. 2013;5:353–6.
Willis GB, Schechter S, Whitaker K. A comparison of cognitive interviewing, expert review, and behavior coding: what do they tell us? In: Proceedings of the Section on Survey Research Methods. Alexandria: American Statistical Association; 1999. pp. 28–37.
de Rada VD, Domínguez-Álvarez JA. Response quality of self-administered questionnaires: a comparison between paper and web questionnaires. Soc Sci Comput Rev. 2014;32:256–69.
De Researchmanager. https://my-researchmanager.com/en/home-2/. Accessed 04 Jan 2019.
Kline P. The handbook of psychological testing. 2nd ed. Florence: Taylor & Frances; 1993. p. 10.
Norquist JM, Girman C, Fehnel S, DeMuro-Mercon C, Santanello N. Choice of recall period for patient-reported outcome (PRO) measures: criteria for consideration. Qual Life Res. 2012;21:1013–20.
Stull DE, Leidy NK, Parasuraman B, et al. Optimal recall periods for patient-reported outcomes: challenges and potential solutions. Curr Med Res Opin. 2009;25:929–42.
Dalziel K, Li J, Scott A, Clarke P. Accuracy of patient recall for self-reported doctor visits: is shorter recall better? Health Econ. 2018;27:1684–98.
MedDRA Browser (2016 version 2.0). https://www.meddra.org/. Accessed 27 Apr 2018.
Revicki DA, Gnanasakthy A, Weinfurt K. Documenting the rationale and psychometric characteristics of patient reported outcomes for labeling and promotional claims: the PRO Evidence Dossier. Qual Life Res. 2007;16:717–23.
Boynton PM, Greenhalgh T. Selecting, designing, and developing your questionnaire. BMJ. 2004;328:1312–5.
Blair J, Conrad FG. Sample size for cognitive interview pretesting. Public Opin Q. 2011;75:636–58.
Johnson T, Fendrich M. Modeling sources of self-report bias in a survey of drug use epidemiology. Ann Epidemiol. 2005;15:381–9.
CBS. Educational attainment amongst population aged 15–74 years. 2018. https://longreads.cbs.nl/trends18-eng/society/figures/education/. Accessed 13 Jan 2019.
van Gelder MM, Bretveld RW, Roeleveld N. Web-based questionnaires: the future in epidemiology? Am J Epidemiol. 2010;172:1292–8.
Acknowledgements
The authors would like to thank Rowena S. Oemrawsingh-Audhoe and Rike van Eekeren-Buiten for their advice during validation of the questionnaire. The authors would like to thank the EuroQol group for permitting the use of the EQ-5D instrument. Acknowledgement statement: The MedDRA® trademark is owned by the International Federation of Pharmaceutical Manufacturers and Associations on behalf of ICH.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. All authors provided valuable input during the development and validation of the questionnaire. Data collection and analysis was performed by Nanno Schreuder, Quincy de Hoog, and Eugène van Puijenbroek. The first draft of the manuscript was written by Nanno Schreuder and Quincy de Hoog, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Funding
No financial support was received for the conduct of this study or preparation of this manuscript.
Conflict of interest
Nanno Schreuder is employed by GE Healthcare. Quincy de Hoog, Sieta T. de Vries, Pieter L. Jager, Jos G.W. Kosterink, and Eugène van Puijenbroek have no conflicts of interest that are directly relevant to the content of this study.
Ethical Standards
Ethical exemption was obtained in writing from the Medical Ethics Committee of the Isala Hospital, in Zwolle in The Netherlands (reference number 16.08138), as this study did not require formal approval, according to Dutch law.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schreuder, N., de Hoog, Q., de Vries, S.T. et al. Patient-Reported Adverse Events of Radiopharmaceuticals: Development and Validation of a Questionnaire. Drug Saf 43, 319–328 (2020). https://doi.org/10.1007/s40264-019-00895-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-019-00895-2