
Abstract
Mitophagy, or mitochondrial autophagy, plays an important role in mitochondrial quality control for the selective removal of damaged or unwanted mitochondria. Several molecules, including Parkin, p62 and the mitophagy receptors ATG32, NIX/BNIP3 and FUNDC1, were found to participate selective mitophagy. One critical question is how mitochondrial damage-related signals are sensed and transduced to activate mitophagy. It is emerging that mitophagy is highly regulated by reversible protein phosphorylation. Several kinases were found to be involved in selective mitophagy. Pink1 can phosphorylate Parkin to facilitate the subsequent activation of mitophagy. Casein kinase 2 was found to phosphorylate ATG32 in yeast to promote mitophagy. In contrast, Src kinase phosphorylates FUNDC1 to prevent its interaction with LC3, and the dephosphorylation of FUNDC1 is correlated with the activation of mitophagy in mammalian cells in response to hypoxia. Here, we focus on recent advances in our understanding of the signaling events that activate mitophagy and the implications of these events in diseases. We further suggest the possibility that the phosphorylation status of mitophagy receptors may serve as a biochemical marker of this critical process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria play pivotal roles in the production of cellular ATP and metabolites required for normal cellular activities essential for cell survival and programmed cell death (both apoptosis and programmed necrosis) [1–4]. Moreover, mitochondria produce superoxide as an inevitable byproduct during electron transfer along the mitochondrial respiratory chain [5–8]. In addition, mitochondria are the center for iron metabolism for the synthesis of both the heme and iron–sulfur cluster, which are two classes of iron-containing molecules [7, 9]. To fulfill such diverse or even opposing functions, mitochondrial quality must be tightly monitored to avoid harmful effects from mitochondria and to maintain the health of the cell [10–14]. The accumulation of dysfunctional mitochondria is the characteristic of different types of diseases, including heart failure, Alzheimer’s disease, Parkinson’s disease and cancers [15–18]. One of the reasons could be defective mitochondrial quality control. A better understanding of the molecular mechanism of mitochondrial quality control holds promise in the fight against these incurable diseases.
Mitophagy is regarded as the major mechanism of mitochondrial quality control. Early studies observed the engulfment of mitochondria in glucagon-stimulated hepatocytes by electron microscopy [19, 20]. Lemasters et al. [21∙∙] more specifically described the selective autophagy of mitochondria as mitophagy. The authors went on to suggest that mitochondrially derived reactive oxygen species (ROS) can initiate mitophagy, likely via the mechanism of mitochondrial permeability transition (mPT) [21∙∙]. The opening of mPT pores renders mitochondria permeable to all solutes with a molecular mass up to 1,500 Da [22]. This permeability will lead to mitochondrial depolarization and reverse operation of the mitochondrial ATPase [23, 24]. Various pathological conditions, such as ischemia and reperfusion, often induce the loss of mitochondrial membrane potential (Δψ) and the opening of mPT pores, mechanisms that may be involved in ROS-induced ROS release or the so-called vicious cycle [25–27]. The exact mechanism explaining how mPT triggers mitophagy has yet to be defined.
A breakthrough in our understanding of the mechanisms of mitophagy came from Youle’s seminal findings [28∙∙]. His group found that in response to a loss of mitochondrial membrane potential, Parkin can translocate onto mitochondria for the selective removal of mitochondria that have lost membrane potential. In healthy mitochondria, phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) is constitutively imported, most likely via the TIM/TOM complex, to the inner membrane, where the kinase is cleaved by several proteases and ultimately proteolytically degraded [28∙∙]. When induced by CCCP or other uncouplers, PINK1 is stabilized on the outer mitochondrial membrane (OMM) where it can selectively recruit Parkin to depolarized mitochondria [29]. Parkin, as a potent E3 ligase, can polyubiquitinate numerous mitochondrial substrates that will ultimately lead to mitophagy [30∙, 31–34]. In particular, the ubiquitination of the mitofusins (Mfn1 and Mfn2), large GTPases that mediate mitochondrial fusion, will lead to their degradation and mitochondrial fission [30∙, 32]. The segregated mitochondria will undergo autolysosome-dependent degradation [35]. Mizushima and colleagues found that structures containing upstream autophagic proteins, including the ULK1 complex, can associate with depolarized mitochondria, even in the absence of LC3 [36∙]. Atg9A is recruited to autophagosome formation sites independent of the ULK1 complex [36∙]. LC3 is then recruited for the efficient incorporation of damaged mitochondria into the autophagosome at a later stage [36∙].
Receptors in Mitophagy
Since the first description of mitophagy, it was suggested that damaged mitochondria may crosstalk with autophagy machinery [37]. In other words, a mitochondrial surface protein may act as a cargo receptor that interacts with autophagy machinery to form selective mitophagy machinery [37]. An early study by Camougrand’s group identified an OMM protein, Uth1p, required for the autophagic degradation of excess mitochondria [38, 39]. Furthermore, ancient ubiquitous protein 1 (also known as Ptc6), a phosphatase 2C (PP2C) superfamily member, can efficiently eliminate overworked mitochondria in stationary-phase yeast cells [40].
In search of a specific mitophagy receptor, both Ohsumi’s and Klionsky’s groups [41∙∙, 42∙∙] have found that Atg32 is a mitophagy receptor in yeast. Atg32 is a 60-kDa protein with an intra-mitochondrial domain at its C terminus and the tetrapeptide sequence WQAI in its cytoplasmic domain, which is critical for the protein’s interaction with Atg8 [41∙∙, 42∙∙]. Mitochondrial proteins containing the tetrapeptide sequence W(Y)XXL, which is present in several other Atg8- or LC3-binding partners, are defined as mitophagy receptors mediating selective autophagy [41∙∙, 42∙∙]. Atg32 also binds to Atg11, which is a scaffold protein important for cargo selection [43] and autophagosomal formation [41∙∙, 42∙∙]. Unfortunately, there are no orthologs of Atg32, Uth1p and Aup1p in the mammalian system [37].
What are the mitochondrial molecules that mediated mitophagy in the mammalian system? Previous studies have suggested that NIX/BNIP3L is involved in the autophagic degradation of mitochondria in reticulocytes, a process essential for red blood cell maturation [44∙∙]. Genetic ablation of Nix retards mitochondrial clearance in maturing murine reticulocytes [45, 46∙∙]. Research from Ivan Dikic and colleagues [46∙∙] has subsequently shown that NIX also has a putative LC3-interacting region (LIR) that interacts with LC3 and defines NIX as a mitophagy receptor. BNIP3 also contains a typical LIR motif and functions as a mitophagy receptor [47, 48∙].
We have found that FUNDC1, an OMM protein, has the typical LC3-binding motif Y(18)xxL and have identified this protein as a new mitophagy receptor [49∙∙]. In response to hypoxia, FUNDC1 interacts with LC3 through its LIR, and mutation of the LIR impairs its interaction with LC3 and the subsequent induction of mitophagy. Knockdown of endogenous FUNDC1 significantly prevents hypoxia-induced mitophagy, which can be rescued by the expression of wild-type FUNDC1 but not LC3 interaction-deficient FUNDC1 mutants [49∙∙].
Both NIX/BNIP3 and FUNDC1 are involved in hypoxia-induced mitophagy. However, their mechanisms of action are different. Nix and BNIP3 are transcriptionally upregulated through HIF or FOXO3 [50, 51], whereas the mRNA level of FUNDC1 decreases under hypoxic conditions [49∙∙]. Nix and BNIP3 are localized to the ER and are involved in regulating apoptotic or programmed necrosis by affecting mitochondrial respiration or ROS production [51, 52], whereas FUNDC1 is exclusively localized on the OMM. NIX shows much weaker binding to LC3B, whereas the conversion of LC3B is correlated with elevated levels of autophagic vesicles in response to autophagy-inducing stress [49∙∙].
In addition to these receptors, p62 has also been implicated in selective mitophagy [31]. Early studies have shown that p62 functions as an adaptor, or scaffold protein, that participates in the regulation of the NF-kappa B pathway through its interactions with atypical PKC (aPKC), RIP1 kinase or TRAF6 [53–56]. Recently, p62 was reported to act as a cargo receptor for ubiquitinated protein aggregates intended for selective autophagy [57–60]. It was found that p62 translocates onto mitochondria via interaction with ubiquitin, which is significantly increased in response to mitochondrial stresses. Through this interaction, p62 could theoretically recruit LC3 and autophagosomes to facilitate subsequent selective autophagy [61∙, 62]. Whether ubiquitin/p62/LC3 complexing is sufficient for Parkin-mediated mitophagy requires further investigation.
Kinases and Phosphatases in Mitophagy
Mitophagy is a highly regulated and complex process essential for the mitochondrial turnover and quality control. A key question is how mitochondrial signals are sensed and then transduced to activate the autophagy machinery. Recently, post-translational modification, such as protein phosphorylation, was found to regulate the mitophagic process [63, 64]. ATG32 is phosphorylated in response to oxidative stress that promotes its interaction with ATG11 and LC3 [65, 66]. In particular, Ser-114 and Ser-119 of Atg32 are phosphorylated, which promotes the Atg11-Atg32 interaction. However, phosphorylation of these sites is not required for Atg32-Atg8 interaction [65]. Likewise, phosphorylation of Ser-17 and Ser-24 of Bnip3 also promotes its binding to LC3-B and GATE-16 [48∙] to facilitate subsequent mitophagy. In contrast, dephosphorylation activates FUNDC1-mediated mitophagy in mammalian systems [49∙∙]. We found that under normal (unstressed) conditions, FUNDC1 is phosphorylated, inhibiting its interaction with LC3 and subsequent mitophagy. Under hypoxic conditions, FUNDC1 is dephosphorylated, which will enhance its interaction with LC3 and subsequent mitophagy [49∙∙].
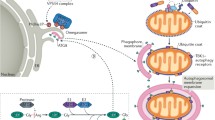
What are the kinases and phosphatases that mediate this reversible phosphorylation? A recent report using kinase screening showed that casein kinase 2 (CK2) phosphorylates the OMM protein Atg32 at Ser-114 and Ser-119 in vitro to promote its interaction with the cytosolic adaptor protein Atg11 in yeast, thus resulting in recruitment into vacuoles and mitophagy [67∙∙] Furthermore, CK2 inhibition selectively prevents Atg32-Atg11 interaction-dependent mitophagy without affecting the Cvt pathway, pexophagy and macroautophagy [67∙∙]. The adaptor protein Atg11 preferentially interacts via Atg32 with phosphorylated Ser-114 and relocates the mitochondrion to pre-autophagosomal complexes and phagophore assembly sites. At last, isolation membranes are produced, and mitochondria are specifically enveloped by autophagosomes. Therefore, Atg32 phosphorylation may represent mitophagy recognition and initiation signaling (see Fig. 1).

Phosphorylation regulates selective mitochondrial autophagy. There are PINK1/Parkin- dependent and receptor-mediated mitophagy pathways. In the PINK1/Parkin pathway, upon the loss of membrane potential in damaged mitochondria, PINK1 is stabilized and accumulated at the outer membrane to recruit Parkin, an E3 ligase to mitochondria. Parkin can be phosphorylated by PINK1 at Ser-65, and the activated Parkin mediates hyperubiquitination of the mitochondrial outer membrane proteins. P62 and other autophagy molecules are also recruited toward damaged mitochondria. Although its role in the PINK1/Parkin pathway remains controversial, P62 can also be phosphorylated by mTORC1 at Ser-351 and CK2 at Ser-403. ATG32, NIX/Bnip3 and FUNDC1 were identified as mitophagy receptors. In yeast, upon the oxidative stress, mitochondrial outer membrane protein Atg32 is phosphorylated at Ser-114 and Ser-119 by CK2, which is presumably activated by the MAPK signaling pathway, Hog1. This phosphorylation mediates the Atg32-Atg11 interaction and leads to the recruitment of mitochondria to PAS, and subsequently mitochondria surrounded by the phagophore membrane. In mammalian cells, mitochondrial outer membrane protein FUNDC1 is normally phosphorylated at Tyr-18 by Src kinase. Under hypoxic conditions, Src kinase is inactivated, and FUNDC1 becomes dephosphorylated. The dephosphorylated FUNDC1 will promote its interaction with LC3 for subsequent mitophagy. Also, under metabolic stress conditions Bnip3 protein can be phosphorylated by an unidentified kinase at Ser-17 and Ser-24. During red blood maturation, NIX protein mediates mitochondria removal via the mitophagy mechanism, and it is yet to be determined whether any kinase is involved
We found that FUNDC1-mediated mitophagy is inhibited by its phosphorylation at the Tyr-18 position in the LIR motif by Src kinase under normal physiological conditions. Upon hypoxia stimulation, Src is inactivated and degraded, and FUNDC1 becomes dephosphorylated, resulting in an increased interaction between FUNDC1 and LC3-II, leading to the selective incorporation and autophagic removal of the mitochondrion [49∙∙]. We propose that dephosphorylation of the Tyr-18 position in the LIR motif may cause interface exposure of the FUNDC1 protein, thus promoting its interaction with LC3 (our unpublished observations), although the exact molecular mechanisms need further investigation (see Fig. 1).
Interestingly, PINK1 kinase can phosphorylate Parkin for its mitophagic activity. Specifically, PINK1 was found to phosphorylate the Ubl domain of Parkin at Ser-65 and to enhance the activity of ubiquitin ligase in in vitro assays [68∙, 69]. Mutation of Ser-65 to Ala dramatically inhibits the recruitment of Parkin onto the mitochondria with depolarized potential, indicating that phosphorylation of Ser-65 primes the translocation of Parkin [68∙]. The exact mechanism of how PINK1 phosphorylation activates Parkin’s activities and translocation remains obscure. Other tyrosine protein kinases, such as c-Abl, diminish activity of Parkin ubiquitination both in vitro and in the cultured cells [70, 71]. Inactivation of Parkin is also implicated in oxidative stress conditions [72]. In addition, Parkin activation in the depolarized mitochondria induces proteasome-mediated degradation of Parkin, indicating that Parkin auto-inhibition might prevent ubiquitin-dependent Parkin degradation [73, 74]. Recently, Chen et al. [75] also found that Pink1 phosphorylated Mfn2 and promoted its Parkin-mediated ubiquitination, which functions as a mitochondrial receptor for Parkin and is required for quality control of cardiac mitochondria (see Fig. 1).
P62 is considered to act as a cargo receptor in selective autophagy (including mitophagy), and it was also found to be phosphorylated at multiple sites [76]. Specific phosphorylation of p62 at Ser-403 (S403) by CK2 in its ubiquitin-associated (UBA) domain increases the affinity between UBA and polyubiquitin chains [76]. This event will enhance p62′s targeting to polyubiquitinated proteins and promote autophagosomal formation. Additionally, mTOR was found to phosphorylates p62 at Ser-351 [77], which increase its binding to Keap1, an adaptor of the Cul3-ubiquitin E3 ligases, and sequesters Keap1 on its cargos such as mitochondria.
Sensing the Signals in Mitophagy
As a vital organelle with diverse cellular functions, mitochondria are highly sensitive to the cellular bioenergetic status, oxygen tension, ROS (including NO) levels, and other cellular and environmental cues. It is thus not surprising that a loss of mitochondrial membrane potential, an increase in cellular ROS [78] (either derived from the cytoplasmic or inside mitochondria), hypoxia [49∙∙], a disturbance of Ca2+ signaling, impaired bioenergetic and biogenesis of mitochondria [79], defects in mitochondrial protein import or export [79], mtDNA damages [80], a perturbation of mitochondrial protein quality control system and an accumulation of protein aggregates in mitochondria [81] are all able to trigger mitophagy. Given the critical role of reversible phosphorylation in mitophagy, it is reasonable to suggest that kinases or phosphatases are likely to play a role in sensing the mitochondrial stress signals responsible for the initiation of mitophagy.
Early studies have clearly shown that ROS can activate mitophagy [21∙∙]. It was reported that N-acetylcysteine (NAC), a compound that increases the cellular GSH pool, prevents mitophagy induction [82, 83], suggesting that the mitochondrial redox status or the ROS production level is one of the factors contributing to mitophagy. How do ROS signals lead to the activation of mitophagy? Although ROSs are short-lived molecules by nature, they can induce oxidative modification of ATGs and related pathways. Under oxidative conditions, Atg4 is oxidized and inactive, which allows Atg8 to bind to lipid and initiates autophagy, revealing that the modulation of the action of Atg4 on Atg8 can be controlled by H2O2 [84]. This phenomenon does not explain the selective effects of mitophagy. One possibility is that ROS production or the activation of the vicious cycle could lead to the opening of permeability transition pores and a loss of mitochondrial membrane potential. The loss of mitochondrial membrane potential may activate the PINK1/Parkin pathway of mitophagy. Indeed, it was shown that an acute burst of ROS within mitochondria was induced by a mitochondrial-targeted photosensitizer and that mitochondrial KillerRed (mtKR) resulted in a loss of membrane potential and subsequent activation of PARK2-dependent mitophagy [85]. ROS is implicated in NIX- and FCCP/Parkin-induced mitophagy [86]. The antioxidant NAC can effectively decrease the mitophagy induced by CCCP. ROS and the mitophagy receptor NIX are important in the induction and initiation of mitophagy by enhancing the translocation of Parkin onto the damaged mitochondria [86]. In the yeast system, CK2/ATG32 pathways can be regulated by MAPK Hog1 pathways, which can be modulated by oxidative stress and thus the downstream mitophagic activities [87]. In mammalian systems, we found that hypoxia can inactivate Src kinase to activate FUNDC1-mediated mitophagy [49∙∙]. NAC can also prevent hypoxia-induced mitophagy, suggesting that ROSs are involved (unpublished observations).
Additionally, studies have found that selective mitophagy induced by 6-OHDA, rotenone and staurosporine in neuron cells involve the externalization of cardiolipin, which binds LC3 to mediate mitophagy [88]. The Parkinson’s toxins result in a delayed phase of mitochondrial superoxide production that causes localized activation of ERK/MAPK at the mitochondria. Furthermore, mitochondrial localization of ERK is sufficient to induce mitophagy, even in the absence of mitochondrial injury [89, 90].
Mitophagy and Diseases
An accumulation of dysfunctional mitochondria and increased oxidative stress are characteristic of several diseases, including neurodegenerative diseases, cardiomyopathy and cancers [91–94]. Defective mitophagy is thus implicated in these diseases, although the molecular details remain to be defined. Due to space limitations, we only highlight a few diseases pertinent to mitophagy signaling and discuss how mitophagy contributes to these diseases.
PINK1/Parkin-Mediated Mitophagy and Parkinson’s Disease
Mutations in Parkin and PINK1 have been shown to be responsible for a small subset of autosomal-recessive Parkinson’s disease cases [95]. Mice deficient in Parkin had an abnormal mitochondrial respiratory chain [96]. Flies with Parkin knockout had pathology in muscle, with dysfunctional mitochondria and pronounced apoptosis [97, 98]. These results suggest that Parkin mutation that causes the defective of mitophagy indeed contributes to the disease. It is well documented that Parkin mediates the mitophagy in a PINK1-dependent manner [68∙, 99]. PINK1 mutations result in decreased Δψ and dysfunction of mitochondria. Although flies with PINK1 knockout are viable, they have a motor deficit and a decreased lifespan and are hypofertile or sterile [100]. In particular, these flies have abnormal mitochondrial morphology, decreased mitochondrial mass, lower ATP levels, flight muscle dysfunction and reduced dopaminergic neurons. Recent data suggest that PINK1 interacts with a mitochondrial phosphatase PGAM5, although they may not be reciprocal substrates [101]. A deficiency in PGAM5 inhibits the mitochondrial degeneration induced by PINK1 inactivation in Drosophila, suggesting that PINK1/Parkin- mediated mitophagy could be modulated by PGAM5 [102].
Parkin is a multifunctional protein that plays diverse roles in mitochondrial biogenesis, mitophagy, mitochondrial dynamics and transport through its regulation of PARIS (controlling the PGC-1alpha-NRF-1 pathway) [103], MFN1/2 [30∙, 75] and Drp-1 [[104]. Much of the work on PINK1/Parkin-mediated mitophagy has been performed in immortalized cell lines overexpressing high levels of Parkin [105]. The roles of endogenous Parkin and PINK1 that contribute to mitophagy in neurons in animal systems require further clarification. Identification of the upstream signaling mechanisms that regulate Parkin/PINK1-dependent mitophagy will help to explain the nature of the insults affecting mitophagy and mitochondrial functions.
Src Kinase/FUNDC1 Axis in Cancer
As early as the 1920s, Otto Warburg proposed a hypothesis to explain why tumors exhibited a metabolic energy change from aerobic respiration to glycolysis [106∙]. Defective mitochondria could be responsible for the Warburg effect, a widely accepted concept among cancer biologists [107, 108]. It is possible that damaged mitochondria progressively accumulate during cancer development [109, 110]. Impaired mitochondrial metabolism may provide a certain advantage in the increased glycolysis and lactate production contributing to the Warburg effect [111, 112]. Additionally, mitochondria are a major source of ROS production in cells, which may affect genomic instability via retrograde signaling [113].
Similar to the proposed dual roles of autophagy in cancers, mitophagy could be a double-edged sword in the development of cancer [16, 109]. During the early stage of cancer development, mitophagy functions to effectively remove damaged mitochondria to maintain the well-being of the cells and to prevent the occurrence of cancers [16]. During the late stage of cancer development, the hypoxic conditions inside the tumor will activate mitophagy, which can digest unwanted mitochondria to sustain the survival of the cancer [114, 115].
Src kinase is well known for its function in cancer development [116]. It is interesting to note that one of this kinase’s functions is to prevent mitophagy, as we demonstrated previously [49∙∙]. This function will lead to the accumulation of damaged mitochondria, which promotes the occurrence of cancer [117]. Additionally, hypoxia within cancer during the stage of cancer development can activate mitophagy, which will benefit cancer growth [110]. Further studies are underway to test how Src kinase modulates mitophagy in cancers.
NIX/BNIP3-Dependent Mitophagy and Heart Disease
Mitochondrial dysfunction is also implicated in the pathogenesis of several heart diseases during aging [118]. Given the postmitotic nature of cardiomyocytes, the efficient removal of dysfunctional mitochondria by mitophagy is critical for the maintenance of normal cellular functions and homeostasis [119]. Indeed, cardiac-specific ablation of both Nix and BNIP3 revealed normal cardiac structure and function at the age of 8 weeks in these mice [120]. However, by 30 weeks, these mice had developed massive cardiac abnormality. EM analysis revealed an abnormality in mitophagy and autophagic vesicle formation in senescent hearts deficient in NIX and/or BNIP3 [52]. As both NIX and BNIP3 also function in programmed cell death by affecting mPT and mitochondrial respiration, definitive proof connecting defective mitophagy and the resulting mitochondrial abnormality requires further experimental analysis. Additionally, the signaling events in mitophagy and myocardial injury warrant further investigations. For example, ischemia and reperfusion, which often incur side effects in the clinic, can trigger the opening of mPT pores, decreasing the mitochondrial membrane potential and massively increasing ROS levels [120]. These events may elicit a complex signaling pathway that activates mitophagy as a protective response or programmed cell death when the stresses are severe and persistent. It would be interesting to understand how these signals impinge upon NIX/BNIP3-dependent mitophagy. This information is important because the protective cardioprotective pathways relying on mitophagy, along with pharmacological drugs, might prevent heart damages in pathogenic heart conditions.
Detection of Mitophagy
Because the accumulation of damaged mitochondria correlates with the aging-related diseases, overall mitochondrial quality or mitophagic activities can serve as a prognostic indicator of disease risk. Additionally, advancing mitophagy research requires reliable and quantitative analysis of mitophagy. Like in autophagy, transmission electron microscopy (TEM) remains the best approach to detect mitophagy [121]. The engulfment of mitochondria by double-membrane autophagosomes can be visualized as a unique mitochondrial structure or a high electron density of mitochondria. Immuno-EM for specific mitochondrial markers, such as Tom20 or VDAC, and other mitochondrial proteins is important for confirming mitophagy. EM can be problematic in quantitative studies because of the limited cell numbers/sections that can be examined. Additionally, mitophagy is highly dynamic, so it is difficult to the capture typical engulfment of mitochondria within the autophagosomal membrane. Data from EM studies are highly subjective, and unbiased selection of samples is extremely important.
Other analyses include Western blotting of mitochondrial proteins from mitochondrial outer and inner membrane proteins, such as Tom 20 and Tim23, together with mitochondrial matrix proteins such as MnSOD or HSP60; measurements of the amount of mitochondrial DNA; and the colocalization of LC-3 and other autophagic markers with mitochondria [49∙∙]. It should be acknowledged that mitochondrial proteins can be degraded by other mechanisms of protein quality control, such as the AAA-ATPase inside mitochondria and proteasomal degradation of outer membrane proteins [122]. A decrease in mitochondrial DNA number is not specific to mitophagy, so caution needs to be exercised when interpreting these data. These analyses and a combination of these different measurements may provide a good indication of mitophagy.
Given the recent progress discussed above, we suggest using the reversible phosphorylation of mitophagy receptors as a biochemical marker for mitophagy. Phosphorylation of ATG32 at Ser-114 sites is clearly closely associated with mitophagy in yeast [65]. We have shown that de-phosphorylation of FUNDC1 resembles the activation of mitophagy in mammalian systems in response to hypoxia [49∙∙]. Further work is underway to examine whether dephosphorylation of FUNDC1 correlates with the activation of mitophagy in vivo and under pathophysiological conditions. One advantage is that both ATG32 and FUNDC1 are exclusively localized on mitochondria, while Parkin, p62 and BNIP3 are also phosphorylated in correlation with mitophagic activity [37]. Measurement of their phosphorylation statuses could be very useful. Profiling the distinct phosphorylation of mitophagy mediators, together with other morphological and biochemical analysis, may yield better and more reliable approaches to study mitophagy.
Conclusions
Mitophagy is an exquisitely regulated process comprising the segregation of to-be-removed mitochondria from the mitochondrial network and the crosstalk between mitochondria and the autophagy machinery. It is likely that mitophagy is highly regulated by reversible phosphorylation and is well coordinated with other mitochondrial events, such as mitochondrial ATP generation, ROS metabolism, mitochondrial dynamics and programmed cell death. Several kinases are found to play an essential role in selective mitophagy, namely PINK1 in the Parkin-dependent pathway, Hog1/CK2 in the ATG32 and Src kinase in the FUNDC1 and ERK2 though unknown targets [49∙∙, 68∙, 75, 122]. Other kinases and phosphatases are likely to be involved and are yet undiscovered. Future studies should aim to understand how these cues activate mitophagy via distinct pathways in distinct cellular contexts and how mitophagy is coordinated with other mitochondrial and cellular functions.
Defective of mitophagy may result in the accumulation of dysfunctional mitochondria, which is widely observed in aging-related diseases. Deciphering the causal roles of mitophagy in these diseases remains a challenge that requires useful model systems and reliable and quantitative measurements of mitophagy in vivo. A better understanding of these issues will not only improve our knowledge regarding general mitochondrial homeostasis but also reveal new avenues for treatments of diseases involving dysfunctional mitochondria.
References
Papers of particular interest have been highlighted as: ∙ Of importance ∙∙ Of major importance
Wallace DC et al (1998) Mitochondrial biology, degenerative diseases and aging. Biofactors 7(3):187–190
Galluzzi L et al (2012) Mitochondrial control of cellular life, stress, and death. Circ Res 111(9):1198–1207
Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412(6842):95–99
Wallace DC (1999) Mitochondrial diseases in man and mouse. Science 283(5407):1482–1488
Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91(23):10771–10778
Jezek P, Hlavata L (2005) Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol 37(12):2478–2503
Bartosz G (2009) Reactive oxygen species: destroyers or messengers? Biochem Pharmacol 77(8):1303–1315
Nohl H, Gille L, Staniek K (2005) Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol 69(5):719–723
Cheng VW et al (2006) The iron-sulfur clusters in Escherichia coli succinate dehydrogenase direct electron flow. J Biol Chem 281(37):27662–27668
Karbowski M, Youle RJ (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 10(8):870–880
Chan DC (2006) Dissecting mitochondrial fusion. Dev Cell 11(5):592–594
Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22(12):1577–1590
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12(1):9–14
Schon EA, Przedborski S (2011) Mitochondria: the next (neurode) generation. Neuron 70(6):1033–1053
Palikaras K, Tavernarakis N (2012) Mitophagy in neurodegeneration and aging. Front Genet 3:297
Taylor R, Goldman SJ (2011) Mitophagy and disease: new avenues for pharmacological intervention. Curr Pharm Des 17(20):2056–2073
DiMauro S, Schon EA (2008) Mitochondrial disorders in the nervous system. Annu Rev Neurosci 31:91–123
Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125(7):1241–1252
Lawrence BP, Brown WJ (1992) Autophagic vacuoles rapidly fuse with pre-existing lysosomes in cultured hepatocytes. J Cell Sci 102(Pt 3):515–526
Deter RL, De Duve C (1967) Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33(2):437–449
∙∙ Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8(1):3–5. Lemasters et al. firstly introduced mitophagy, the selective autophagy of mitochondria. They also proposed that reactive oxygen species (ROS) derived from mitochondria can initiate mitophagy, possibly through mitochondrial permeability transition (mPT)
Bradham CA et al (1998) The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Mol Cell Biol 18(11):6353–6364
Zima AV et al (2013) Effects of mitochondrial uncoupling on Ca(2+) signaling during excitation-contraction coupling in atrial myocytes. Am J Physiol Heart Circ Physiol 304(7):H983–H993
Qian T, Herman B, Lemasters JJ (1999) The mitochondrial permeability transition mediates both necrotic and apoptotic death of hepatocytes exposed to Br-A23187. Toxicol Appl Pharmacol 154(2):117–125
Petrosillo G et al (2003) Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J 17(6):714–716
Honda HM, Korge P, Weiss JN (2005) Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci 1047:248–258
Shiva S et al (2007) Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 204(9):2089–2102
∙∙ Narendra D et al. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803. Youle’s group discovered Parkin is selectively recruited to dysfunctional mitochondria with low membrane potential in mammalian cells. After recruitment, Parkin mediates the engulfment of mitochondria by autophagosomes and the selective elimination of impaired mitochondria
Vives-Bauza C et al (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107(1):378–383
∙ Gegg ME et al (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19(24):4861–4870. Parkin was found to ubiquitinate several mitochondrial proteins, including mitofusin 1 and mitofusin 2 following CCCP treatment
Geisler S (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12(2):119–131 (VDAC1 was found to be a target for Parkin-mediated Lys 27 polyubiquitylation and mitophagy.)
Tanaka A et al (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191(7):1367–1380
Van Humbeeck C et al (2011) Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 31(28):10249–10261
Ding WX et al (2012) Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem 287(50):42379–42388
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20(1):31–42
∙ Itakura E et al (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 125(Pt 6):1488–1499. Upstream autophagic proteins, including the ULK1, Atg9A, are recruited toward mitochondria for autophagosome formation during Parkin/PINK1-mediated mitophagy
Feng D et al (2013) Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res 319(12):1697–1705
Camougrand N et al (2004) Uth1p: a yeast mitochondrial protein at the crossroads of stress, degradation and cell death. FEMS Yeast Res 5(2):133–140
Kissova I et al (2004) Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 279(37):39068–39074
Gonzalez A et al (2013) Ptc6 Is required for proper rapamycin-induced down-regulation of the genes coding for ribosomal and rRNA processing proteins in S. cerevisiae. PLoS One 8(5):e64470
∙∙ Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17(1):98–109. Atg32 acts as a mitophagy-specific receptor and regulates selective degradation of mitochondria in yeast
∙∙ Okamoto K, Kondo-Okamoto N, Ohsumi Y (2009) Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17(1):87–97. Atg32 acts as a mitophagy-specific receptor and regulates selective degradation of mitochondria in yeast
Yorimitsu T, Klionsky DJ (2005) Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell 16(4):1593–1605
∙∙ Sandoval H et al. (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454(7201):232–235. Nix-dependent loss of membrane potential is important for targeting the mitochondria into autophagosomes for clearance during erythroid maturation, and interference with this function impairs erythroid maturation and results in anaemia
Schwarten M et al (2009) Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5(5):690–698
∙∙ Novak I et al. (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11(1):45–51. Nix functions as an autophagy receptor, which mediates mitochondrial clearance after mitochondrial damage and during erythrocyte differentiation
Hanna RA et al (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287(23):19094–19104
∙ Zhu Y et al. (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 288(2):1099–1113. Phosphorylation of serine residues 17 and 24 flanking the Bnip3 LIR promotes binding to specific Atg8 members LC3B and GATE-16, the phosphorylation state of the Bnip3 signals either the induction of apoptosis or pro-survival mitophagy
∙∙ Liu L et al. (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14(2):177–185. FUNDC1 has a typical LC3-binding motif Y(18)xxL and functions as a new mitophagy receptor. Upon hypoxia stimulation, Src is inactivated and FUNDC1 is dephosphorylated, thus enhancing interaction between FUNDC1 and LC3-II for the selective mitophagy
Chinnadurai G, Vijayalingam S, Gibson SB (2008) BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene 27(Suppl 1):S114–S127
Sowter HM et al (2001) HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61(18):6669–6673
Dorn GW 2nd, Kirshenbaum LA (2008) Cardiac reanimation: targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene 27 Suppl 1:S158–167
Moscat J, Diaz-Meco MT (2009) To aggregate or not to aggregate? A new role for p62. EMBO Rep 10(8):804
Mathew R et al (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137(6):1062–1075
Sanz L et al (2000) The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J 19(7):1576–1586
Yu HB et al (2009) Sequestosome-1/p62 is the key intracellular target of innate defense regulator peptide. J Biol Chem 284(52):36007–36011
Kirkin V et al (2009) A role for ubiquitin in selective autophagy. Mol Cell 34(3):259–269
Kirkin V et al (2009) NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5(5):732–733
Lamark T et al (2009) NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8(13):1986–1990
Kraft C, Peter M, Hofmann K (2010) Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12(9):836–841
∙ Narendra D et al. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6(8):1090–1106. VDAC1 and VDAC3 are dispensable for the recruitment of p62, mitochondrial clustering and mitophagy. They also suggest that proteins other than p62 are likely required for mitophagy downstream of Parkin substrates other than VDAC1
Huang C et al (2011) Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 6(6):e20975
Egan DF et al (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331(6016):456–461
Mao K et al (2011) Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol 193(4):755–767
Aoki Y et al (2011) Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell 22(17):3206–3217
Farre JC et al (2013) Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep 14(5):441–449
∙∙ Kanki T et al. (2013) Casein kinase 2 is essential for mitophagy. EMBO Rep 14(9):788–794. Casein kinase 2 (CK2) was found to phosphorylate the OMM protein Atg32 at Ser-114 and Ser-119 in vitro to promote its interaction with the cytosolic adaptor protein Atg11 in yeast, thus leading to recruitment into vacuoles and mitophagy
∙ Shiba-Fukushima K et al. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2:1002. PINK1 kinase phosphorylates Parkin at Ser-65 of the Ubl domain in Parkin and enhances the activity of ubiquitin ligase in vitro
Vincow ES et al (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci USA 110(16):6400–6405
Ko HS et al (2010) Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc Natl Acad Sci USA 107(38):16691–16696
Imam SZ et al (2011) Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson’s disease. J Neurosci 31(1):157–163
Winklhofer KF et al (2003) Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem 278(47):47199–47208
Rakovic A et al (2013) Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem 288(4):2223–2237
Heo JM, Rutter J (2011) Ubiquitin-dependent mitochondrial protein degradation. Int J Biochem Cell Biol 43(10):1422–1426
Chen Y, Dorn GW 2nd (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340(6131):471–475
Matsumoto G et al (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 44(2):279–289
Ichimura Y et al (2013) Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 51(5):618–631
Byrne AM, Lemasters JJ, Nieminen AL (1999) Contribution of increased mitochondrial free Ca2+ to the mitochondrial permeability transition induced by tert-butylhydroperoxide in rat hepatocytes. Hepatology 29(5):1523–1531
Priault M et al (2005) Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ 12(12):1613–1621
Kim I, Lemasters JJ (2011) Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal 14(10):1919–1928
Perlmutter DH (2002) Liver injury in alpha1-antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J Clin Invest 110(11):1579–1583
Kissova IB, Camougrand N (2009) Glutathione participates in the regulation of mitophagy in yeast. Autophagy 5(6):872–873
Deffieu M et al (2009) Glutathione participates in the regulation of mitophagy in yeast. J Biol Chem 284(22):14828–14837
Scherz-Shouval R et al (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760
Wang Y et al (2012) ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 8(10):1462–1476
Ding WX et al (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285(36):27879–27890
Prick T et al (2006) In yeast, loss of Hog1 leads to osmosensitivity of autophagy. Biochem J 394(Pt 1):153–161
Chu CT et al (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15(10):1197–1205
Kulich SM et al (2007) 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic Biol Med 43(3):372–383
Dagda RK et al (2008) Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy 4(6):770–782
Di Carlo M et al (2012) Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Radic Res 46(11):1327–1338
Keating DJ (2008) Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 104(2):298–305
Trushina E, McMurray CT (2007) Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145(4):1233–1248
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795
Gandhi S et al (2006) PINK1 protein in normal human brain and Parkinson’s disease. Brain 129(Pt 7):1720–1731
Palacino JJ et al (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279(18):18614–18622
Greene JC et al (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100(7):4078–4083
Pesah Y et al (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131(9):2183–2194
Gegg ME, Schapira AH (2011) PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: implications for Parkinson disease pathogenesis. Autophagy 7(2):243–245
Clark IE et al (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441(7097):1162–1166
Sekine S et al (2012) Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem 287(41):34635–34645
Imai Y et al (2010) The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet 6(12):e1001229
Castillo-Quan JI (2011) Parkin’ control: regulation of PGC-1alpha through PARIS in Parkinson’s disease. Dis Model Mech 4(4):427–429
Wang H et al (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286(13):11649–11658
Grenier K, McLelland GL, Fon EA (2013) Parkin- and PINK1-dependent mitophagy in Neurons: will the real pathway please stand up? Front Neurol 4:100
∙ Warburg O (1925) Iron, the oxygen-carrier of respiration-ferment. Science 61(1588):575–582. Otto Warburg proposed a hypothesis to explain why tumors exhibited a metabolic energy change from aerobic respiration to glycolysis
Shaw RJ (2006) Glucose metabolism and cancer. Curr Opin Cell Biol 18(6):598–608
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033
White E, DiPaola RS (2009) The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 15(17):5308–5316
Singh KK (2006) Mitochondria damage checkpoint, aging, and cancer. Ann N Y Acad Sci 1067:182–190
Pelicano H et al (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25(34):4633–4646
Kim JW, Dang CV (2006) Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 66(18):8927–8930
Lu J, Sharma LK, Bai Y (2009) Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res 19(7):802–815
Aredia F, Scovassi AI (2013) Manipulation of autophagy in cancer cells: an innovative strategy to fight drug resistance. Future Med Chem 5(9):1009–1021
Martinet W, De Meyer GR (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 104(3):304–317
Finn RS (2008) Targeting Src in breast cancer. Ann Oncol 19(8):1379–1386
Rosenfeldt MT, Ryan KM (2011) The multiple roles of autophagy in cancer. Carcinogenesis 32(7):955–963
Chinnery PF et al (2002) Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet 360(9342):1323–1325
Dutta D et al (2012) Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res 110(8):1125–1138
Diwan A et al (2007) Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117(10):2825–2833
Klionsky DJ et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8(4):445–544
Wang K et al (2013) Proteolytic processing of Atg32 by the mitochondrial i-AAA protease Yme1 regulates mitophagy. Autophagy 9(11) [Epub ahead of print]
Acknowledgments
We are grateful to all laboratory members for the useful discussion. This research was supported by the 973 Program Project (no. 2011CB910903 to Quan Chen and no. 2010CB91220 to Yushan Zhu) from the MOST, the Natural Science Foundation of China (81130045, 31271529, 31301130) and China postdoctoral grant (2013M541041).
Compliance with Ethics Guidelines
Conflict of Interest
Weilin Zhang, Hao Wu, Lei Liu, Yushan Zhu and Quan Chen declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, W., Wu, H., Liu, L. et al. Phosphorylation Events in Selective Mitophagy: Possible Biochemical Markers?. Curr Pathobiol Rep 1, 273–282 (2013). https://doi.org/10.1007/s40139-013-0033-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-013-0033-8