
Abstract
Introduction
Chronic low back pain (CLBP) is the leading cause of years lived with disability globally. The role of restorative neurostimulation in the treatment of patients with refractory mechanical CLBP and multifidus muscle dysfunction has been established in one randomized controlled trial (RCT) and several clinical studies that demonstrated both safety and clinical benefit. This post-market trial provides a direct comparison to optimized medical management to test the hypothesis that the addition of restorative neurostimulation to current care paradigms results in significant improvements in back pain-related disability.
Methods and Analysis
This trial will include people who have reported significant levels of back pain and back pain-related disability with symptoms that have persisted for longer than 6 months prior to enrollment and resulted in pain on most days in the 12 months prior to enrollment. Eligible patients will be randomized to either optimal medical management or optimal medical management plus ReActiv8® restorative neurostimulation therapy. Patient-reported outcomes will be collected at regular intervals out to the 1-year primary endpoint, at which time the patients in the control arm will be offered implantation with the ReActiv8 system. Assessment of each group will continue for an additional year.
Ethics and Dissemination
The RESTORE trial follows the principles of the Declaration of Helsinki. The WCG IRB acts as the Central Institutional Review Board (IRB) for most sites and some sites will receive local IRB approval prior to enrollment of patients. Each IRB assessed the protocol and related documentation. The protocol complies with Good Clinical Practice (GCP). All patients provide written informed consent to participate in the trial.
Protocol Version. Version C, 07 Sep 2022.
ClinicalTrials.gov registration number. NCT04803214 registered March 17, 2021.
Plain Language Summary
Restorative neurostimulation is a treatment for intractable CLBP associated with dysfunction of the multifidus muscle, which normally provides functional stability to the lumbar spine. To date, ReActiv8® (Mainstay Medical) is the only neurostimulator specifically developed and approved for this indication. Electrical stimulation of the muscle’s nerve overrides the dysfunction and reactivates it. Several prior studies demonstrated that the most of participants experienced clinically substantial and durable symptom relief compared to baseline. This protocol describes a second RCT in which all participants are on individualized optimal medical management and half of them are randomly selected to be implanted with a ReActiv8 system to receive restorative neurostimulation. The purpose of this design is to measure if there is any clinical benefit of restorative neurostimulation over individualized optimal medical management alone over the course of a full year.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Patients who suffer from intractable CLBP associated with multifidus muscle dysfunction despite receiving individualized optimal medical management (OMM), have a poor prognosis. The ReActiv8 restorative neurostimulation system is intended to address this unmet clinical need. The safety and clinical benefit of restorative neurostimulation was established by several earlier studies leading to approval in the US, Europe, and Australia. The purpose of this study is to test whether restorative neurostimulation leads to superior patient outcomes over individualized OMM. |
What might be learned from this study? |
The study may show that patients treated with restorative neurostimulation experience symptom relief superior to those receiving individualized optimal medical management alone. |
Introduction
Worldwide, low back pain is the most common pain condition and the leading cause of years lived with disability [1]. While acute low back pain is common and improves spontaneously in almost all cases within several weeks, chronic low back pain (CLBP), typically defined as low back pain lasting longer than 3 months, is associated with substantial economic costs, including work absenteeism, lost productivity as well as direct and indirect medical costs. In the United States, these costs are estimated to be as high as $296 billion annually [2,3,4]. CLBP can be subdivided into neuropathic and somatosensory causes, which have different etiologies as well as management strategies. Although neuropathic CLBP typically does not respond to non-opioid medications, it is often well treated with spinal surgery and neuromodulation including spinal cord and dorsal root ganglion stimulation [5, 6]. In contrast, mechanical CLBP, which comprises predominantly nociceptive mechanical pain resulting from tissue injury and inflammation, has fewer effective treatment options.
A variety of treatment strategies have been investigated for mechanical CLBP, including non-pharmacologic therapies such as physical therapy, medications (including opioids and non-opioids), minimally invasive interventions, and spine surgery. Conservative management typically consists of maximizing the use of non-opioid medications and physical/exercise therapies, which have been associated with small-to-moderate effects on pain [7]. Mechanical CLBP is also a leading reason for chronic opioid use, despite limited evidence for its efficacy and increasing evidence of potential harm [8,9,10,11]. Minimally invasive interventional therapies such as nerve blocks, facet joint injections, and radio-frequency ablations of the medial branches may provide relief for patients with mechanical CLBP. However, such relief is typically transient and therapies repeated [12,13,14,15]. Faced with the limited management options, patients with chronic low back pain may pursue spine surgery despite the limited long-term benefits, huge economic burden [16], and the increased potential of poor outcomes [17]. Therefore, patients with nociceptive chronic low back pain have no long-term treatment options beyond the limited success of conservative management.
Restorative Neurostimulation
The ReActiv8 restorative neurostimulation system is a device that treats intractable mechanical CLBP by incorporating principles of motor stimulation to overcome motor control impairment of the multifidus muscle. By delivering electrical stimulation to the medial branches of the dorsal rami spinal nerves, ReActiv8 overrides underlying inhibition and elicits contractions of the deep lumbar multifidus muscles. The ReActiv8 system is indicated for bilateral stimulation of the L2 medial branch of the dorsal ramus as it crosses the L3 transverse process as an aid in the management of intractable CLBP associated with multifidus muscle dysfunction.
Prior Clinical Studies
Feasibility of restorative neurostimulation for CLBP was demonstrated almost a decade ago [18, 19], followed by several international multicenter observational studies and one randomized controlled trial (RCT) under Investigational Device Exemption (IDE), which supported the safety and clinical benefit claims and Premarket Approval (PMA) in the USA [20,21,22,23,24,25]. The outcomes of both the randomized phase [20] and longitudinal follow-up [20,21,22] have been discussed in depth in prior publications.
Objectives
The primary objective of the RESTORE trial is to compare the effectiveness of ReActiv8 restorative neurostimulation to optimal medical management (OMM) for the treatment of intractable chronic low back pain (CLBP) associated with multifidus dysfunction. The hypothesis of this trial is that ReActiv8 therapy will be superior to OMM alone in the relief of mechanical nociceptive low back pain-related disability at the 1-year follow-up visit.
Methods and Analysis
Trial Setting
RESTORE is a multi-center, open-label RCT, performed at up to 30 clinical sites in the United States. This trial protocol is produced according to the applicable Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) reporting guidelines [26].
Patient and Public Involvement
Patients are not involved in planning of research questions, outcome measures, or design of the trial.
Eligibility Criteria
Candidates with CLBP will be assessed for eligibility using the trial-specific inclusion/exclusion criteria detailed in Tables 1 and 2. To verify eligibility, medical records and imaging will be reviewed by a three-member panel of independent medical experts prior to randomization.
The treatment of mechanical CLBP does not follow a well-defined care pathway but is typically individually optimized over multiple consultations with the patient’s physician. Despite treatment optimization, many patients may still experience high levels of residual pain, disability, and or treatment side effects. These patients are candidates for this trial, provided they meet the eligibility criteria.
Optimal Medical Management
The Optimal Medical Management treatment plan is individualized based on the patient’s needs and known responsiveness to therapies tried previously or in current use. It is documented on a standardized worksheet before randomization and should consider non-investigational pharmacologic agents (e.g., non-steroidal anti-inflammatory drugs, muscle relaxants, duloxetine, or opioids) and/or non-pharmacologic or psychosocial therapies (e.g., spinal manipulation, exercise program, and cognitive behavioral therapy) as appropriate. If the physician decides that there is a relevant therapy that has yet to be tried, the patient is not to be included in the trial until the effect of the new therapy has been observed. At this point, the patient may be enrolled provided the inclusion and exclusion criteria are met.
Any therapy changes during the study should be managed through the study investigator. If other therapies (e.g., medications, physical therapy) are being managed by another provider, it will be important for the study investigator to be in contact with the patient’s provider throughout the study.
Interventions
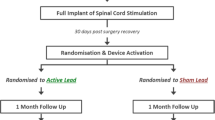
Participants will be randomized to receive the ReActiv8 neurostimulation system (treatment) or OMM (control). At physician discretion, all patients continue to receive OMM per the predetermined individual treatment plan and patients randomized to the treatment arm will receive the implantable ReActiv8 restorative neurostimulation system. The technique for implantation has been described elsewhere [20]. Patients in the treatment group will be instructed to deliver two 30-min stimulation sessions per day through the 1-year primary endpoint assessment visit. Thereafter, patients in the treatment group will be advised to continue treatment at the same level, but will be permitted to reduce the amount of stimulation as desired. Patients in the control group (OMM alone) may elect to receive the ReActiv8 system at that time.
Patients in both arms will return to the clinic at regular intervals (Table 3) to collect data and adjust treatments as needed, however no specific strategies will be employed to improve adherence to OMM in either group. Patients in the treatment group will be encouraged to comply with the twice-daily delivery of stimulation and their compliance with this regime will be reviewed at follow-up visits (the implanted pulse generator records device use).
All patients will be followed for 2 years, at which point they will exit the trial.
Outcomes
The primary endpoint will be a comparison of the mean change in Oswestry Disability Index (ODI) between the Treatment and Control groups at 1-year post randomization. The ODI is a disease-specific assessment of the disabling effects of back pain that includes one item on pain and nine items on activities of daily living (personal care, lifting, walking, sitting, standing, sleeping, sex life, social life, and traveling) [27]. The ODI is reported as a score from 0 to 100. Patients with ODI between ≥ 30 and ≤ 60, i.e., those with moderate and severe disability are included.
Secondary endpoints at the 1-year visit include between-group comparison of:
-
1.
The change from baseline in “average low back pain in the last 24 h” measured using the 11-point Numeric Rating Scale (NRS).
-
2.
The change from baseline in quality-of-life measured using the EQ-5D-5L utility score.
Tertiary endpoints at the 1-year visit include between-group comparison of:
-
1.
Percent pain relief
-
2.
Subject global impression of change
-
3.
Treatment satisfaction
-
4.
Proportion of patients with a ≥ 15-point ODI improvement and/or a ≥ 50% low back pain NRS improvement and no worsening in either measure.
-
5.
The mean change from baseline in leg pain NRS
The supporting efficacy analyses at the 1-year visit include between-group comparison of:
-
Proportion of patients with a ≥ 15-point ODI improvement.
-
Cumulative proportion of responders (a comparison of ranks of the proportion of patients across all possible ODI thresholds).
-
Proportion of patients with a ≥ 50% low back pain NRS improvement.
-
Cumulative proportion of responders (a comparison of ranks of the proportion of patients across all possible NRS thresholds).
Further efficacy analyses include:
-
All outcome measures at the 2-year visit compared to baseline.
-
Health economic outcome measures at the 1- and 2-year visits compared to baseline.
-
Activity monitoring through the 2-year visit compared to baseline in a subset of patients.
Participant Timeline
All patients will be followed at regular intervals for 2 years, at which point they will be exited from the trial. The trial visit schedule is provided in Table 3. Patient enrollment started on July 16, 2021 and patients continue to be enrolled. The trial procedures schedule is provided in Tables 4 and 5. Recruitment is expected to be completed in 2023 and the primary endpoint reached in 2024.
Recruitment and Allocation
A minimum of 204 evaluable patients is required to sufficiently power the primary endpoint. To allow for attrition, approximately 230 patients will be randomized at up to 30 clinical sites within the US. Patients will be recruited from each clinic’s referral base. To account for screen failures prior to randomization, approximately 400 patients may be enrolled. Power calculations for this trial rely on the following assumptions: minimum power of 80%, type I error rate of 5%, assumed mean change in treatment group of 18.2, assumed mean change in control group of 12.2, and pooled standard deviation of 15. The assumed mean changes and standard deviation are based on a previous trial of the ReActiv8 system.
Patients will be randomized to continuing OMM (control arm) or ReActiv8 (treatment arm) in a ratio of 1:1 at enrolment. Randomization will be performed according to a random permuted block design stratified by clinical site. The assignment is provided electronically according to the random permuted block design for each clinical site. Assignment to the treatment or control arm will be performed by the investigators according to the randomization.
Adverse Events and Assessment Process
Reportable adverse events (AEs) are those related to the device, procedure, stimulation, or other therapies utilized to treat LBP, and all serious adverse events (SAEs), whether related or not. All reportable AEs will be documented and reported from the time of informed consent through the end of the trial with summary statistics presented for observed rates. No formal statistical hypotheses will be tested in the safety assessment.
The investigator must determine whether the event was related to the device, stimulation, procedure, and/or other therapies. The categories used for relatedness are listed in Table 6.
Data Collection Methods
Data will be collected and stored in an electronic database, which shall have written procedures and document requirements. Security, reliability, and data consistency will be maintained throughout the trial. Database access will be restricted to staff with appropriate training as designated by the investigator. Questionnaires will be completed by patients electronically or on paper and subsequently transferred to the database by designated study site personnel.
Data Management
The sponsor will be responsible for collection of the data required for this trial in accordance with Health Insurance Portability Accountability Act (HIPAA) and GCP. The sponsor will use an electronic database, which shall have written procedures and document requirements. Patient questionnaires will be completed electronically or on paper CRFs, which are then transferred to the database by the designated study site personnel.
Statistical Methods
Analyses will be conducted using SAS version 9.3 or later (SAS Institute Inc., Cary, NC, USA). Continuous variables will be summarized with means and standard deviations or as medians and interquartile ranges. Categorical variables will be summarized with the number and proportion of patients in each category. Binary outcomes will be presented as proportions with corresponding 95% confidence limits. Statistical analyses for all outcomes will compare treatment and control groups for the average change at 1-year follow-up compared with baseline using two-sided two-sample t tests for difference in mean changes. Analyses will be conducted with the null hypothesis representing no difference across treatment and control groups in each primary and secondary outcome, with alternative hypotheses representing significant differences across these groups at the p < 0.05 level.
Strengths and Limitations
The main limitation of the study is that it is unblinded. While blinding and sham therapy were considered during the design phase, it was decided that blinding is now impossible due to widespread availability of information describing the therapy. Unlike during the ReActiv8-B trial, the device is currently commercially available and there is substantial patient facing educational material available to improve patient expectations and understanding of the therapy. A significant strength is the extended duration of the randomized phase of this trial. The observation of the accrual of therapeutic benefit over baseline after crossover in the ReActiv8 B trial suggests that a larger effect size may be achieved with 1 year of therapy compared to 120 days.
Data Monitoring
The Advisory Committee will provide oversight for the trial. This includes physician advisors independent of the trial as well as select advisors. The database of trial data will be housed with a database management company. Data will be analyzed by an independent statistician. The trial sponsor will ensure proper monitoring of the trial. Appropriately trained personnel will perform trial monitoring at clinical sites to ensure the trial is conducted in accordance with the protocol, the signed Clinical Study Agreement, and IRB requirements. Trial safety and integrity will be periodically monitored by physician advisors.
Ethics and Dissemination
Research Ethics Approval
The RESTORE Trial follows the principles of the Declaration of Helsinki. The WCG IRB acts as the Central IRB (RN# 20211219) for most sites and some sites have or will receive local IRB approval prior to enrollment of patients. Each IRB committee assessed the protocol and related documentation. The protocol complies with GCP. All patients provide written informed consent to participate in the trial.
Protocol Amendments
The protocol complies with GCP Protocol amendments are recorded in a Quality Management system in accordance with BS EN ISO13485:2016 + A11:2021. In case of major amendments, for example, changes to the consent form, they are submitted for approval and training deployed and documented at each of the sites.
Consent
All patients provide written informed consent to participate in the trial.
Confidentiality
Confidentiality will be maintained at all times throughout the trial and all data shall be secured against unauthorized access. Data entered in the database are de-identified and only authorized personnel and their designees will have access to patient data. Confidentiality will be preserved in reports and publications of results. All patients’ health information will be kept confidential in accordance with all applicable laws and regulations.
Access to Data
Only members of the research team who need to contact trial patients, enter data or perform data quality control have access to identifiable patient information.
Data are de-identified upon entry into the database and only authorized and trained personnel will have access to these data for analysis.
Dissemination Policy
One-year results from this trial will be published in a peer-reviewed journal and further manuscripts examining primary and secondary outcomes will be planned. Authorship is based on International Committee of Medical Journal Editors 2018 Recommendations.
Scientific Relevance and Broader Impact
This trial provides evidence for the effectiveness of ReActiv8 versus OMM. Importantly the duration of follow-up highlights the impact of the restorative mechanism of action.
References
Rubin DI. Epidemiology and risk factors for spine pain. Neurol Clin. 2007;25:353–71.
Woolf Bruce Pfleger A. Burden of major musculoskeletal conditions. Bull World Health Organ. 2003;81:646–56.
Mehra M, Hill K, Nicholl D, Schadrack J. The burden of chronic low back pain with and without a neuropathic component: a healthcare resource use and cost analysis. J Med Econ. 2012;15:245–52.
Katz JN. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. J Bone Joint Surg Am. 2006;88(Suppl 2):21–4.
Kapural L, Peterson E, Provenzano DA, Staats P. Clinical evidence for spinal cord stimulation for failed back surgery syndrome (FBSS): systematic review. Spine. 2017;42(Suppl 14):S61–6.
Liem L, Russo M, Huygen FJPM, Van Buyten JP, Smet I, Verrills P, et al. One-year outcomes of spinal cord stimulation of the dorsal root ganglion in the treatment of chronic neuropathic pain. Neuromodulation. 2015;18:41–9.
Chou R, Huffman LH. Nonpharmacologic therapies for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med. 2007;147:492–504.
Jennifer F, Waljee CMB. Opioid prescribing for low back pain what is the role of payers? JAMA Netw Open. 2018;1: e180236.
Lin DH, Jones CM, Compton WM, Heyward J, Losby JL, Murimi IB, et al. Prescription Drug coverage for treatment of low back pain among US Medicaid, Medicare Advantage, and commercial insurers. JAMA Netw Open. 2018;1: e180235.
The effectiveness and risks of long-term opioid treatment of chronic pain [Internet]. Rockville, MD: Agency for Healthcare Research and Quality. [cited 2021 Oct 7]. Available from https://archive.ahrq.gov/research/findings/evidence-based-reports/opoidstp.html.
Busse JW, Wang L, Kamaleldin M, Craigie S, Riva JJ, Montoya L, et al. Opioids for chronic noncancer pain: a systematic review and meta-analysis. JAMA. 2018;320:2448–60.
Chou R, Hashimoto R, Friedly J, Fu R, Dana T, Sullivan S, et al. Pain management injection therapies for low back pain. Rockville, MD: Agency for Healthcare Research and Quality US; 2015.
Nelemans P. Injection therapy for subacute and chronic low‐back pain. Cochrane Database Syst Rev [Internet]. John Wiley & Sons, Ltd; 2008. Available from https://doi.org/10.1002/14651858.CD001824.pub3.
Cohen SP, Doshi TL, Constantinescu OC, Zhao Z, Kurihara C, Larkin TM, et al. Effectiveness of lumbar facet joint blocks and predictive value before radiofrequency denervation. Anesthesiol Ovid Technol (Wolters Kluwer Health). 2018;129:517–35.
Juch JNS, Maas ET, Ostelo RWJG, George Groeneweg J, Kallewaard JW, Koes BW, et al. Effect of radiofrequency denervation on pain intensity among patients with chronic lowback pain the mint randomized clinical trials. JAMA J Am Med Assoc. 2017;318:68–81.
Deyo RA, Weinstein JN. Low back pain. N Engl J Med. 2001;344:363–70.
Hebert JJ, Fritz JM, Koppenhaver SL, Thackeray A, Kjaer P. Predictors of clinical outcome following lumbar disc surgery: the value of historical, physical examination, and muscle function variables. Eur Spine J. 2016;25:310–7.
Deckers K, De Smedt K, Van Buyten JP, Smet I, Eldabe S, Gulve A, et al. Chronic low back pain: restoration of dynamic stability. Neuromodulation. 2015;18:478–86.
Russo M, Deckers K, Eldabe S, Kiesel K, Gilligan C, Vieceli J, et al. Muscle control and non-specific chronic low back pain. Neuromodulation. 2018;21:1–9.
Gilligan C, Volschenk W, Russo M, Green M, Gilmore C, Mehta V, et al. An implantable restorative-neurostimulator for refractory mechanical chronic low back pain: a randomized sham-controlled clinical trial. Pain. 2021;162:2486–98.
Gilligan C, Volschenk W, Russo M, Green M, Gilmore C, Mehta V, et al. Long-term outcomes of restorative neurostimulation in patients with refractory chronic low back pain secondary to multifidus dysfunction: two-year results of the ReActiv8-B pivotal trial. Neuromodulation Technol Neural Interface. 2023;26:87–97.
Gilligan C, Volschenk W, Russo M, Green M, Gilmore C, Mehta V, et al. Three-year durability of restorative neurostimulation effectiveness in patients with chronic low back pain and multifidus muscle dysfunction. Neuromodulation. 2022. https://doi.org/10.1016/j.neurom.2022.08.457.
Deckers K, De Smedt K, Mitchell B, Vivian D, Russo M, Georgius P, et al. New therapy for refractory chronic mechanical low back pain—restorative neurostimulation to activate the lumbar multifidus: one year results of a prospective multicenter clinical trial. Neuromodulation. 2018;21:48–55.
Mitchell B, Deckers K, Smedt KD, Russo M, Georgius P, Green M, et al. Durability of the therapeutic effect of restorative neurostimulation for refractory chronic low back pain. Neuromodulation. 2021;24:1024–32.
Thomson S, Chawla R, Love-Jones S, Sharma M, Vajramani G, Williams A, et al. Restorative neurostimulation for chronic mechanical low back pain: results from a prospective multi-centre longitudinal cohort. Pain Ther. 2021;10:1451–65. https://doi.org/10.1007/s40122-021-00307-3.
Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158:200–7.
Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain-United States, 2016. JAMA. 2016;315:1624–45.
Acknowledgements
We thank Greg Maislin, MS, MA, and Teresa Yurik for their statistical advice and data analysis support in designing the trial and Jan Pieter Heemels MS and Ben Goss PhD for reviewing drafts of the manuscript. We also thank the physicians and their research and clinical staff at each site undertaking the trial. In particular, we would also like to thank the patients who will participate in this trial.
Funding
Funding to support this trial was provided by Mainstay Medical, Inc. The Rapid Service Fee was paid by Mainstay Medical Inc.
Editorial Assistance
was provided by Ben Goss PhD (Mainstay Medical).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
CG, DB, and LG designed the trial and wrote the protocol, all authors contributed to the preparation of this manuscript and reviewed the final version prior to submission. RY and PM prepared the manuscript NM CG and FS reviewed and edited the manuscript. FS is the overall trial principal investigator, CG and NM are the trial medical advisors.
Compliance with Ethics Guidelines
The RESTORE trial follows the principles of the Declaration of Helsinki. The WCG IRB acts as the Central IRB (RN# 20211219) for most sites and some sites have or will receive local IRB approval prior to enrollment of patients. Each IRB committee assessed the protocol and related documentation. The protocol complies with Good Clinical Practice (GCP). All patients provide written informed consent to participate in the trial.
Disclosures
Dr. Gilligan Mainstay Medical pays partial salary cost directly to his institution and holds options in Mainstay Medical. Ms Burnside: is an employee of Mainstay Medical and holds options and equity. Ms Grant: has a consultancy (clinical research services) agreement with Mainstay Medical and holds stock options. Dr. Yong and Dr. Mullins have no competing interests. Dr. Schwab is the principal investigator for the RESTORE Trial has a consultancy agreement with Mainstay Medical. Dr. Mekhail functions as independent medical monitor of the RESTORE trial sponsored by Mainstay Medical and has a consultancy agreement with Mainstay Medical.
Data Availability
Not applicable to this article as no datasets were generated or analyzed in the preparation of this protocol. Data generated from this trial will be made available upon reasonable request to the sponsor.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gilligan, C., Burnside, D., Grant, L. et al. ReActiv8 Stimulation Therapy vs. Optimal Medical Management: A Randomized Controlled Trial for the Treatment of Intractable Mechanical Chronic Low Back Pain (RESTORE Trial Protocol). Pain Ther 12, 607–620 (2023). https://doi.org/10.1007/s40122-023-00475-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-023-00475-4