
Abstract
Introduction
In the phase 3 TACKLE study, outpatient treatment with AZD7442 (tixagevimab/cilgavimab) was well tolerated and significantly reduced progression to severe disease or death through day 29 in adults with mild-to-moderate coronavirus disease 2019 (COVID-19) at the primary analysis. Here, we report data from the final analysis of the TACKLE study, performed after approximately 15 months’ follow-up.
Methods
Eligible participants were randomized 1:1 and dosed within 7 days of symptom onset with 600 mg intramuscular AZD7442 (n = 456; 300 mg tixagevimab/300 mg cilgavimab) or placebo (n = 454).
Results
Severe COVID-19 or death through day 29 occurred in 4.4% and 8.8% of participants who received AZD7442 or placebo, a relative risk reduction (RRR) of 50.4% [95% confidence interval (CI) 14.4, 71.3; p = 0.0096]; among participants dosed within 5 days of symptom onset, the RRR was 66.9% (95% CI 31.1, 84.1; p = 0.002). Death from any cause or hospitalization for COVID-19 complications or sequelae through day 169 occurred in 5.0% of participants receiving AZD7442 versus 9.7% receiving placebo, an RRR of 49.2% (95% CI 14.7, 69.8; p = 0.009). Adverse events occurred in 55.5% and 55.9% of participants who received AZD7442 or placebo, respectively, and were mostly mild or moderate in severity. Serious adverse events occurred in 10.2% and 14.4% of participants who received AZD7442 or placebo, respectively, and deaths occurred in 1.8% of participants in both groups. Serum concentration–time profiles recorded over 457 days were similar for AZD7442, tixagevimab, and cilgavimab, and were consistent with the extended half-life reported for AZD7442 (approx. 90 days).
Conclusions
AZD7442 reduced the risk of progression to severe COVID-19, hospitalization, and death, was well tolerated through 15 months, and exhibited predictable pharmacokinetics in outpatients with mild-to-moderate COVID-19. These data support the long-term safety of using long-acting monoclonal antibodies to treat COVID-19.
Trial Registration
Clinicaltrials.gov, NCT04723394. (https://clinicaltrials.gov/study/NCT04723394.
Plain Language Summary
The body’s immune system produces proteins called antibodies that specifically target foreign substances such as viruses. AZD7442 is a combination of two antibodies (called tixagevimab and cilgavimab) that bind to the severe acute respiratory syndrome coronavirus 2 virus spike protein, preventing it from causing coronavirus disease 2019 (COVID-19). AZD7442 was designed to be “long-acting” and therefore provide prolonged protection against COVID-19 lasting several months from a single dose. It was tested in a clinical trial (TACKLE) to see if it could prevent people who had recently developed symptoms of COVID-19 from getting sicker, being hospitalized, or dying. Around 900 adults took part in this clinical trial. Half of this group were treated with a dose of AZD7442, given as two injections. The other half received a placebo (injections that look like the AZD7442 injections but contain no medicine). The effect of AZD7442 treatment against COVID-19 was monitored over 6 months, and safety was monitored over 15 months. Around the same percentage of participants in the trial reported side effects with AZD7442 and placebo, suggesting there were no safety issues with AZD7442. AZD7442 treatment reduced the risk of participants getting severe COVID-19 or dying from COVID-19 by approximately half, compared with the placebo group. Participants receiving AZD7442 also had fewer hospitalizations due to COVID-19 complications, compared with the placebo group. These results showed the long-term safety of using long-acting antibodies such as AZD7442 as a treatment for COVID-19.
AbstractSection Graphical Abstract
Similar content being viewed by others


Why carry out this study? |
There is an ongoing need for effective coronavirus disease 2019 (COVID-19) treatments to prevent severe disease and reduce the risk of hospitalization and death in at-risk individuals. |
What did the study ask? |
The TACKLE study evaluated the efficacy and safety of a single intramuscular injection of 600 mg AZD7442 (tixagevimab/cilgavimab) in outpatients with mild-to-moderate COVID-19 administered within 7 days of symptom onset. |
What was learned from this study? |
The final 15-month analysis of TACKLE confirmed the efficacy of AZD7442 in reducing progression to severe COVID-19 or death through day 29 and death from any cause or hospitalization for COVID-19 complications or sequelae through day 169. |
The safety profile of AZD7442 over 15 months of follow-up was comparable with placebo and consistent with that observed in prior reports of shorter duration. |
These long-term follow-up data confirm the findings of the TACKLE primary and key secondary analyses and suggest that intramuscular injection of long-acting therapeutic monoclonal antibodies can be used to treat outpatients with mild-to-moderate COVID-19. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.25055054 .
Introduction
AZD7442 is a combination of two long-acting monoclonal antibodies (mAbs), tixagevimab and cilgavimab, derived from B cells isolated from individuals with prior severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection [1]. The fragment crystallizable (Fc) regions of the progenitor mAbs were enhanced with YTE (amino acid substitutions M252Y/S254T/T256E) and TM (triple modification; amino acid substitutions L234F/L235E/P331S) modifications to extend their serum half-lives and reduce Fc effector functions, respectively [2,3,4,5]. AZD7442 has been authorized for the treatment of coronavirus disease 2019 (COVID-19) in the European Union, Japan, Canada, and Australia [6,7,8,9], although several Omicron subvariants have since demonstrated reduced susceptibility to neutralization by tixagevimab and cilgavimab in vitro [10,11,12]. Following emergence of resistant variants, clinical use of AZD7442 has been restricted [13]. However, long-term safety and efficacy data for AZD7442 may assist the development of new therapeutic mAbs with YTE and TM modifications.
The phase 3 TACKLE study (clinicaltrials.gov identifier NCT04723394) demonstrated the safety and efficacy of AZD7442 versus placebo as an outpatient treatment for adults with mild-to-moderate COVID-19 [14, 15]. Here, we report data from the final analysis of the TACKLE study, including final evaluations of the primary and key secondary efficacy endpoints, longer-term safety evaluated over 15 months, and pharmacokinetics.
Methods
Trial Overview
TACKLE was a phase 3, double-blind, placebo-controlled, multicenter study, full details of which have previously been reported [14]. Briefly, eligible participants were adults aged ≥ 18 years who had not received a COVID-19 vaccine and who had laboratory-confirmed SARS-CoV-2 infection determined by reverse transcription-polymerase chain reaction (RT-PCR) or antigen test from any respiratory tract specimen collected within 3 days of enrollment and had a World Health Organization (WHO) Clinical Progression Scale score > 1 and < 4. Participants were randomized 1:1 to receive 600 mg AZD7442 (consecutive intramuscular injections of 300 mg tixagevimab and 300 mg cilgavimab) or placebo within 7 days of symptom onset. The final analysis was performed when those participants who had not withdrawn from the study had completed follow-up through day 457 (approximately 15 months).
Ethical Approval
The study adhered to Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable laws and regulations. The protocol was reviewed and approved by an institutional review board or ethics committee at the respective study sites (Table S1). All participants provided informed, written consent.
Randomization and Masking
Participants were randomly assigned in a 1:1 ratio to either AZD7442 or placebo via a central interactive response system, with stratification by time from symptom onset (≤ 5 or > 5 days) and risk of progression to severe COVID-19 (low or high risk; with high risk including age ≥ 65 years, immunocompromised, and comorbidities such as cancer and chronic diseases). Participants, investigators, and sponsor staff who were involved in treatment, clinical evaluation, and monitoring of the participants were masked to the randomly assigned study treatments.
Endpoints
The primary efficacy endpoint was a composite of severe COVID-19 or death from any cause through day 29, with severe COVID-19 defined as a minimum of pneumonia or hypoxemia together with a WHO Clinical Progression Scale score of ≥ 5. The primary analysis was conducted 30 days after 43 primary endpoint events had occurred. As the primary analysis was conducted whilst the study was ongoing, data continued to accumulate as participants completed protocol visits. Evaluation of the primary efficacy endpoint was repeated at the final analysis (using methods as previously described [14]) to include participants whose day 29 visit came after the primary data cutoff. Also repeated at the final analysis (using methods as previously described [14]) were evaluations of the first supportive estimand of the primary efficacy endpoint, which included participants dosed within 5 days of symptom onset, the key secondary efficacy endpoint, which was a composite of death from any cause or hospitalization for COVID-19 complications or sequelae through day 169, and the levels of SARS-CoV-2 RNA detected in nasal swabs up to day 29.
Safety was evaluated on the basis of incidences of adverse events (AEs), serious AEs (SAEs), and AEs of special interest (AESIs). AESIs included anaphylaxis and any other serious hypersensitivity reactions, injection site reactions, and cardiovascular and thrombotic events.
Single-dose pharmacokinetics of AZD7442 were assessed over the course of the study using methods as previously described [14]. Incidence and serum titers of treatment-emergent antidrug antibodies (TE-ADA) in participants receiving AZD4772, and the impact of TE-ADAs on the pharmacokinetics of AZD7442, were also evaluated (methods as previously described [14]). ADA positivity was defined by ADA titers ≥ 160 for tixagevimab or ≥ 80 for cilgavimab. ADAs were defined as treatment-emergent if they occurred post-dose in participants who were ADA negative at baseline, or if they occurred in participants who were ADA positive at baseline but experienced an increase in ADA titers of ≥ 4-fold over the course of the study.
Protocol Amendments
Amendments to the original study protocol included the redefinition of an AESI to include cardiovascular and thrombotic events, as noted above. A cardiovascular event adjudication committee was commissioned to independently validate the diagnosis of all cardiovascular events, including cardiac ischemic, cardiac failure, cerebrovascular, and thromboembolic AESIs, as well as all fatal AEs (to determine if they were cardiovascular deaths). The protocol amendments also clarified that reinfection/recurrence of COVID-19 or new, confirmed asymptomatic SARS-CoV-2 infections were to be captured as AEs or SAEs and not considered to be the original disease, and that prolonged COVID-19 symptoms lasting approximately 3 months were to be captured as an AE of post-acute COVID-19 symptoms (long COVID-19).
Statistical Analyses
The primary efficacy endpoint was evaluated using the modified full analysis set (mFAS), comprising all participants administered study drug within 7 days of symptom onset and not hospitalized at baseline (day 1) for isolation purposes (hospitalization was allowed for isolation purposes in Japan and Russia as a result of local requirements). The first supportive estimand of the primary efficacy endpoint was evaluated in an early intervention subgroup of participants from the mFAS who were dosed within 5 days of symptom onset. The key secondary endpoint and the change in SARS-CoV-2 RNA from baseline were evaluated in the mFAS. Safety was evaluated using the safety analysis set, comprising all participants who received study drug regardless of baseline hospitalization status. The single-dose pharmacokinetics of AZD7442 were evaluated using the pharmacokinetics analysis set, comprising all participants who received AZD7442 and had at least one quantifiable serum pharmacokinetic observation post-dose. TE-ADAs and their impact on the pharmacokinetics of AZD7442 were evaluated using the AZD7442 ADA set, comprising all participants in the safety analysis set who received AZD7442 and were assessed for ADAs at baseline and at least one post-baseline visit.
The relative risk reduction (RRR) in the primary efficacy endpoint between the AZD7442 and placebo groups was assessed using a Cochran–Mantel–Haenszel test, stratified by the randomization stratification factors. The key secondary efficacy endpoint was assessed by the same methods. Kaplan–Meier curves were used to summarize the time to primary endpoint events. A Cox proportional hazards model used to obtain a hazard ratio (HR) and 95% confidence intervals (CIs), with the stratification factors included as covariates, and a stratified log-rank test was used to assess differences between groups.
The changes from baseline in SARS-CoV-2 RNA levels were compared between groups at day 3 and day 6 using a mixed model for repeated measures. These results were confirmed by calculating the time-weighted average change in log10 SARS-CoV-2 RNA from baseline to day 6 and day 29 from an analysis of covariance model, including terms for log10 baseline value, treatment, randomization stratification factors, and log10 baseline by treatment interaction.
Results
Trial Population
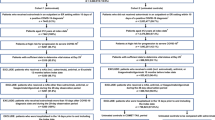
Of 910 randomized participants, 452 and 451 received AZD7442 or placebo, respectively. In total, 797 (87.6%) participants completed the study and 113 (12.4%) discontinued (Fig. S1). Median (range) follow-up was 458.5 (6–571) days in the AZD7442 group and 458.0 (6–533) days in the placebo group.
Baseline characteristics have been reported previously and were generally similar between groups [14]. Briefly, mean age was 46.1 (standard deviation 15.2) years, 12.8% of participants were aged ≥ 65 years, 50.4% were female, and 88.7% had one or more comorbidities considered risk factors for progression to severe COVID-19 or death, such as obesity (43.0%) or smoking (40.3%). Most participants were recruited in Latin America (42.2%) and Europe (41.9%), with 11.5% from the USA and 4.4% from Asia. Participants reported their race as White (61.9%), American Indian or Alaska Native (including Mexican participants who identified as Native American) (23.8%), Asian (5.6%), and Black or African American (4.0%), and 51.8% of participants described their ethnicity as Hispanic or Latino.
Efficacy
Primary Efficacy Endpoint: Severe COVID-19 or Death from Any Cause Through Day 29
In the primary analysis, AZD7442 reduced the risk of progression to severe COVID-19 or death by 50.5% versus placebo (p = 0.0096) in participants dosed within 7 days of symptom onset and by 66.9% (p = 0.0017) in participants dosed within 5 days of symptom onset [14]. The final analysis of the primary efficacy endpoint included three additional participants in the AZD7442 group and four additional participants in the placebo group compared with the primary analysis. Progression to severe COVID-19 or death occurred in 4.4% of participants in the AZD7442 group and 8.8% in the placebo group when dosed within 7 days of symptom onset, an RRR of 50.4% (95% CI 14.4, 71.3; p = 0.0096). When dosed within 5 days of symptom onset, severe COVID-19 or death occurred in 3.5% of participants in the AZD7442 group and 10.7% in the placebo group, an RRR of 66.9% (95% CI 31.1, 84.1; p = 0.002). This result is supported by Kaplan–Meier analysis of the primary efficacy endpoint, which shows separation of the curves for AZD7442 and placebo occurring from day 5 (Fig. S2).
Key Secondary Efficacy Endpoint: Death from Any Cause or Hospitalizations for COVID-19 Complications or Sequelae Through Day 169
With respect to the key secondary endpoint, AZD7442 administered within 7 days of symptom onset reduced the risk of death from any cause or hospitalization for COVID-19 complications or sequelae through day 169 by 49.1% versus placebo (p = 0.009) in the key secondary analysis. The final analysis of the key secondary endpoint included five additional participants in the AZD7442 group and four additional participants in the placebo group compared with the key secondary analysis. Death from any cause or hospitalization for COVID-19 complications or sequelae through day 169 occurred in 5.0% of participants in the AZD7442 group versus 9.7% in the placebo group, an RRR of 49.2% (95% CI 14.7, 69.8; p = 0.009).
Secondary Efficacy Endpoint: SARS-CoV-2 RNA Detected in Nasal Swabs up to Day 29
Treatment with AZD7442 resulted in numerically greater reductions in viral load (log10 SARS-CoV-2 RNA) versus placebo at day 3 (least squares mean difference −0.27; 95% CI −0.63, 0.10) and day 6 (−0.47; 95% CI −0.75, −0.20) (Fig. 1). These results were confirmed by time-weighted analysis of viral load between baseline and day 6 (least squares mean difference −0.21; 95% CI −0.31, −0.12) and day 29 (−0.18; 95% CI −0.29, −0.07).

Least squares mean change from baseline in viral load (log10 viral RNA from nasal swab) over time
Safety
AEs occurred in 251 (55.5%) participants in the AZD7442 group and 252 (55.9%) in the placebo group (Table 1). The most common AEs in both groups were COVID-19, post-acute COVID-19 syndrome, and COVID-19 pneumonia (Table S2). Post-acute COVID-19 syndrome occurred in 38 (8.4%) participants in the AZD7442 group and 29 (6.4%) in the placebo group. In the AZD7442 group, all events of post-acute COVID-19 syndrome were mild or moderate in severity, none were assessed as being related to AZD7442, and the majority occurred in participants with chronic comorbidities such as diabetes, obesity, or other cardiovascular risk factors. Reinfection with COVID-19 within 6 months occurred in one (0.2%) participant in the AZD7442 group and two (0.4%) in the placebo group (all other COVID-19 AEs were judged to be sequelae from the original event and not recurrent infections). Most AEs were mild or moderate in severity. Grade 3–4 AEs occurred in 33 (7.3%) participants in the AZD7442 group and 51 (11.3%) in the placebo group. The most common grade 3–4 AE in both groups was COVID-19 pneumonia. SAEs occurred in 46 (10.2%) participants in the AZD7442 group and 65 (14.4%) in the placebo group (Table 1). The most common SAE in both groups was COVID-19 pneumonia (Table S3). AESIs occurred in 19 (4.2%) participants in the AZD7442 group and 17 (3.8%) in the placebo group (Table 1). The most common AESI in both groups was injection site pain (Table S4). Cardiac disorder AESIs occurred in two (0.4%) participants in the AZD7442 group and one (0.2%) in the placebo group. The cardiovascular events adjudication committee confirmed the diagnosis for two events (one event of myocardial ischemia in a participant who received AZD7442, and one event of acute myocardial infarction in a participant who received placebo). Eight (1.8%) participants died in each of the AZD7442 and placebo groups (Table 1). The most common fatal AE in each group was COVID-19 pneumonia.
Pharmacokinetics and ADAs
Serum concentration–time profiles recorded for AZD7442, tixagevimab, and cilgavimab were similar over 457 days, and reflected the long-acting duration of the antibodies (Fig. 2a). Incidence and serum titers of TE-ADAs in participants receiving AZD7442 are summarized in Table S5. Geometric mean serum concentrations of AZD7442 were similar among participants with TE-ADAs versus those without TE-ADAs, indicating that ADAs had no apparent effect on the pharmacokinetics of AZD7442 (Fig. 2b).

Serum concentrations of a AZD7442, tixagevimab, and cilgavimab over time, and b of AZD7442 by TE-ADA status. ADA antidrug antibody, CV% percent geometric coefficient of variation, SD standard deviation, TE-ADA treatment-emergent ADA. aDay 0 = day 1 pre-dose; day 1 post-dose (n = 410), geometric mean not calculated; day 3, n = 157; day 6, n = 414; day 15, n = 164. bDay 0 (baseline), geometric mean not calculated
Discussion
Tixagevimab and cilgavimab were the first therapeutic mAbs containing YTE and TM modifications to be studied in large, human populations. The TACKLE study was therefore designed to follow participants for up to 15 months, a timeframe spanning five serum half-lives of AZD7442 (approximately 90 days [2, 16]), so that important long-term safety data could be collected.
In the repeated evaluations of the primary efficacy endpoint and its first supportive estimand, AZD7442 significantly reduced the risk of severe COVID-19 or death through day 29 by 50.4% in participants dosed within 7 days of symptom onset and by 66.9% in participants dosed within 5 days of symptom onset. In the repeated evaluation of the key secondary endpoint, AZD7442 significantly reduced the risk of death from any cause or hospitalization for COVID-19 complications or sequelae through day 169 by 49.2%. These results are consistent with those of the primary analysis, which reported risk reductions of 50.5% for the primary efficacy endpoint and 66.9% for its first supportive estimand [14], and 49.1% for the key secondary endpoint [15]. The efficacy of AZD7442 is also supported by nasal swabs showing greater reductions in SARS-CoV-2 viral RNA compared with placebo, an observation that is also consistent with the findings of the primary analysis [14].
In this final analysis of the TACKLE study, performed after approximately 15 months of follow-up, the safety profile of the single intramuscular dose of 600 mg AZD7442 was found to be consistent with the primary and key secondary analyses [14, 15] and reports from other studies of AZD7442 [17, 18], with no new safety signals observed. SAEs, AESIs, and deaths occurred with similar frequency in the AZD7442 and placebo groups, and no SAEs in the AZD7442 group were assessed as being related to the study drug. Cardiac disorder AESIs also occurred in similar frequencies between the groups (0.4% with AZD7442 and 0.2% with placebo) over 15 months of follow-up.
AZD7442 exhibited predictable pharmacokinetics. Following a single 600 mg intramuscular dose, serum concentration–time profiles of tixagevimab and cilgavimab recorded over 457 days were similar and consistent with the extended half-lives reported for both mAbs (approximately 90 days) [2, 16]. ADA titers among participants with TE-ADAs were similar between the AZD7442 and placebo groups, and ADAs to AZD7442 did not increase in magnitude over time. Moreover, TE-ADAs were found to have no clinically relevant impact on AZD7442 pharmacokinetics.
Limitations of this study include the small number of participants aged ≥ 65 years, although this age group was prioritized for vaccination during the COVID-19 pandemic and vaccinated individuals were ineligible to participate in this study. Furthermore, a large proportion of participants were from Latin America, with a relatively high enrollment from Mexico, whereas few participants were Black or African American or Asian. However, results of population pharmacokinetic modeling including other large global studies of AZD7442 as well as studies specifically performed in Japan and China have not indicated any clinically relevant differences in pharmacokinetics due to race and no differences in AZD7442 pharmacokinetics between treatment and prevention settings [19, 20]. The primary efficacy analysis was performed prior to the emergence of the Omicron variant, although follow-up for this study did overlap with the emergence of some subvariants.
Conclusions
This final analysis of the TACKLE study confirms the long-term safety and efficacy of AZD7442 as an outpatient treatment for mild-to-moderate COVID-19. Overall, data from the TACKLE study [14, 15] together with data from other clinical and real-world studies of AZD7442 [17, 18, 21,22,23,24] confirm that therapeutic mAbs with extended half-lives can prevent progression to severe COVID-19, hospitalization, and death after a single intramuscular dose in general populations and immunocompromised individuals. These data also provide proof of concept for the development of new YTE/TM-modified mAbs that can neutralize Omicron subvariants and provide vital protection for vulnerable individuals, such as immunocompromised individuals, who respond suboptimally to COVID-19 vaccines [25, 26].
Data Availability
Requests for data underlying the findings described in this manuscript will be considered in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
References
Zost SJ, Gilchuk P, Case JB, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584(7821):443–9.
Loo YM, McTamney PM, Arends RH, et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in nonhuman primates and has an extended half-life in humans. Sci Transl Med. 2022;14(635):eabl8124.
Robbie GJ, Criste R, Dall’acqua WF, et al. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57(12):6147–53.
Yu XQ, Robbie GJ, Wu Y, et al. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-Staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob Agents Chemother. 2017;61(1):e01020–e1116.
Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008;64:700–4.
European Medicines Agency. Evusheld: EPAR - Product information. 2022. https://www.ema.europa.eu/en/documents/product-information/evusheld-epar-product-information_en.pdf. Accessed 8 January 2024.
AstraZeneca. Evusheld long-acting antibody combination approved for prevention and treatment of COVID-19 in Japan. 2022. https://www.astrazeneca.com/media-centre/press-releases/2022/evusheld-approved-for-covid-19-in-japan.html. Accessed 8 January 2024.
AstraZeneca Canada Inc. EVUSHELD. Tixagevimab and cilgavimab injection. Product monograph including patient medication information. 2022. https://covid-vaccine.canada.ca/info/pdf/evusheld-pm-en.pdf. Accessed 8 January 2024.
Australian Government Department of Health and Aged Care. Therapeutic Goods Administration. Evusheld. 2022. https://www.tga.gov.au/resources/auspmd/evusheld-0. Accessed 8 January 2024.
Imai M, Ito M, Kiso M, et al. Efficacy of antiviral agents against Omicron subvariants BQ.1.1 and XBB. N Eng J Med. 2023;388(1):89–91.
Arora P, Kempf A, Nehlmeier I, et al. Omicron sublineage BQ 1 1 resistance to monoclonal antibodies. Lancet Infect Dis. 2022;23(1):S1473-3099.
Wang Q, Li Z, Ho J, et al. Resistance of SARS-CoV-2 omicron subvariant BA 4.6 to antibody neutralisation. Lancet Infect Dis. 2022;22(12):1666–8.
United States Food and Drug Administration. FDA announces Evusheld is not currently authorized for emergency use in the U.S. 2023. https://www.fda.gov/drugs/drug-safety-and-availability/fda-announces-evusheld-not-currently-authorized-emergency-use-us. Accessed 8 January 2024.
Montgomery H, Hobbs FDR, Padilla F, et al. Efficacy and safety of intramuscular administration of tixagevimab-cilgavimab for early outpatient treatment of COVID-19 (TACKLE): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2022;10(10):985–96.
Hobbs FDR, Montgomery H, Padilla F, et al. Outpatient treatment with AZD7442 (tixagevimab/cilgavimab) prevented COVID-19 hospitalizations over 6 months and reduced symptom progression in the TACKLE randomized trial. Infect Dis Ther. 2023;12(9):2269–87.
Forte-Soto P, Albayaty M, Brooks D, et al. Safety, tolerability and pharmacokinetics of half-life extended severe acute respiratorysyndrome coronavirus 2 neutralizing monoclonal antibodies AZD7442 (tixagevimab-cilgavimab) in healthy adults. J Infect Dis. 2023;227(10):1153–63.
Levin MJ, Ustianowski A, De Wit S, et al. Intramuscular AZD7442 (tixagevimab-cilgavimab) for prevention of Covid-19. N Engl J Med. 2022;386(23):2188–200.
ACTIV-3–Therapeutics for Inpatients with COVID-19 (TICO) Study Group. Tixagevimab-cilgavimab for treatment of patients hospitalised with COVID-19: a randomised, double-blind, phase 3 trial. Lancet Respir Med. 2022;10(10):P972–84.
Clegg LE, Stepanov O, Schmidt S, et al. Consistency of AZD7442 (cilgavimab/tixagevimab) pharmacokinetics across prophylaxis and treatment and adult and pediatric participants: application of population pharmacokinetics to enable rapid decision-making during the COVID-19 pandemic. Open Forum Infect Dis. 2023. https://doi.org/10.1093/ofid/ofad500.464.
Okada H, Ishikawa K, Itoh Y, et al. Safety, tolerability, and pharmacokinetics of half-life extended SARS-CoV-2-neutralizing monoclonal antibodies AZD7442 (tixagevimab/cilgavimab) in healthy Japanese adults. J Infect Chemother. 2023;29:1061–7.
Akinosoglou K, Rigopoulos EA, Kaiafa G, et al. Tixagevimab/cilgavimab in SARS-CoV-2 prophylaxis and therapy: a comprehensive review of clinical experience. Viruses. 2022;15(1):118.
Elias LB, Jaber A, Manzano M, Leekoff M, Sylvester A, Tremblay MA. Real-world efficacy of COVID-19 pre-exposure prophylaxis with tixagevimab/cilgavimab in people with multiple sclerosis. Vaccines (Basel). 2023;11(12):1855.
Al Jurdi A, Morena L, Cote M, Bethea E, Azzi J, Riella LV. Tixagevimab/cilgavimab pre-exposure prophylaxis is associated with lower breakthrough infection risk in vaccinated solid organ transplant recipients during the omicron wave. Am J Transplant. 2022;22(12):3130–6.
Angelico R, Romano F, Coppola L, et al. Effects of anti-COVID-19 vaccination and pre-exposure prophylaxis with tixagevimab-cilgavimab in kidney and liver transplant recipients. Medicina (Kaunas). 2023;59(12):2101.
Evans RA, Dube S, Lu Y, et al. Impact of COVID-19 on immunocompromised populations during the Omicron era: insights from the observational population-based INFORM study. Lancet Reg Health-Eur. 2023;35: 100747.
Ketkar A, Willey V, Pollack M, et al. Assessing the risk and costs of COVID-19 in immunocompromised populations in a large United States commercial insurance health plan: the EPOCH-US Study. Curr Med Res Opin. 2023;39(8):1–16.
Acknowledgements
The authors thank the participants and their families, and all investigators involved in this study.
Medical Writing Assistance
Medical writing support was provided by Matthew Young, DPhil, and Linda Brown, BSc (Hons), and editorial support was provided by Jess Galbraith, BSc, all of Core (a division of Prime), London, UK, supported by AstraZeneca according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/full/10.7326/M22-1460).
Funding
The TACKLE study and the journal’s Rapid Service fee for this manuscript were funded by AstraZeneca.
Author information
Authors and Affiliations
Contributions
AstraZeneca was involved in the study design, collection, analysis, and interpretation of the study data, as well as data checking of information provided in the manuscript. Marius Albulescu and Karen Near of AstraZeneca were involved in the study design. Ultimate responsibility for the opinions, conclusions, and data interpretations lies with the authors. All authors were involved in the drafting and critical revision of the manuscript. All authors approved the final version of the manuscript. F.D. Richard Hobbs, Hugh Montgomery, Francisco Padilla, and Jesus Abraham Simón-Campos were study investigators. Douglas Arbetter performed the statistical analyses. All authors contributed to the interpretation of study data.
Corresponding author
Ethics declarations
Conflict of Interest
F.D. Richard Hobbs has received funding from AstraZeneca to cover meeting attendances and operationalization of TACKLE in the UK as the UK Principal Investigator. He has received funding from UK Research and Innovation and the National Institute for Health and Care Research (NIHR) for national Urgent Public Heath COVID-19 trials and directorship of the NIHR Applied Research Collaboration, Oxford Thames Valley. Hugh Montgomery has received funding from the UK NIHR BRC at University College London Hospitals. He has received consultation fees from AstraZeneca and has been a consultant for Millfield Medical Ltd. During the development of a new Closed Circuit Continuous Positive Airway Pressure machine. Francisco Padilla has received personal fees and grants from Amgen, AstraZeneca, Boehringer Ingelheim, Ferrer, Kowa, Medix, Merck, Merck Sharp and Dohme, Novartis, Pfizer, Sanofi, Servier, and Silanes. Jesus Abraham Simón-Campos has served on advisory boards for Pfizer, AstraZeneca, Roche, Regeneron, Atea, and Eli Lilly, is a speaker for Pfizer, AstraZeneca, Roche, and Regeneron, and has received funding from AstraZeneca, Roche, and Pfizer to cover meeting attendances. Douglas Arbetter, Seth Seegobin, Alexandre Kiazand, Katie Streicher, Taylor S. Cohen, and Mark T. Esser are employees of, and may hold stock and/or stock options in, AstraZeneca. Nuria Martinez-Alier was an employee of AstraZeneca at the time of this analysis. Dr Martinez Alier’s current affiliation is: Department of Paediatric Infectious Diseases and Immunology, Evelina London Children’s Hospital, Guy’s and St Thomas’ NHS Foundation Trust, London, UK.
Ethical Approval
The study adhered to Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable laws and regulations. The protocol was reviewed and approved by an institutional review board or ethics committee at the respective study sites (Table S5). All participants provided informed, written consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Nuria Martinez-Alier’s affiliation was correct at the time of this analysis.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hobbs, F.D.R., Montgomery, H., Padilla, F. et al. Safety, Efficacy and Pharmacokinetics of AZD7442 (Tixagevimab/Cilgavimab) for Treatment of Mild-to-Moderate COVID-19: 15-Month Final Analysis of the TACKLE Trial. Infect Dis Ther 13, 521–533 (2024). https://doi.org/10.1007/s40121-024-00931-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-024-00931-4