
Abstract
Introduction
AZD7442 (tixagevimab/cilgavimab) comprises neutralising monoclonal antibodies (mAbs) that bind to distinct non-overlapping epitopes on the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein. Viral evolution during mAb therapy can select for variants with reduced neutralisation susceptibility. We examined treatment-emergent SARS-CoV-2 variants during TACKLE (NCT04723394), a phase 3 study of AZD7442 for early outpatient treatment of coronavirus disease 2019 (COVID-19).
Methods
Non-hospitalised adults with mild-to-moderate COVID-19 were randomised and dosed ≤ 7 days from symptom onset with AZD7442 (n = 452) or placebo (n = 451). Next-generation sequencing of the spike gene was performed on SARS-CoV-2 reverse-transcription polymerase chain reaction-positive nasopharyngeal swabs at baseline and study days 3, 6, and 15 post dosing. SARS-CoV-2 lineages were assigned using spike nucleotide sequences. Amino acid substitutions were analysed at allele fractions (AF; % of sequence reads represented by substitution) ≥ 25% and 3% to 25%. In vitro susceptibility to tixagevimab, cilgavimab, and AZD7442 was evaluated for all identified treatment-emergent variants using a pseudotyped microneutralisation assay.
Results
Longitudinal spike sequences were available for 461 participants (AZD7442, n = 235; placebo, n = 226) and showed that treatment-emergent variants at any time were rare, with 5 (2.1%) AZD7442 participants presenting ≥ 1 substitution in tixagevimab/cilgavimab binding sites at AF ≥ 25%. At AF 3% to 25%, treatment-emergent variants were observed in 15 (6.4%) AZD7442 and 12 (5.3%) placebo participants. All treatment-emergent variants showed in vitro susceptibility to AZD7442.
Conclusion
These data indicate that AZD7442 creates a high genetic barrier for resistance and is a feasible option for COVID-19 treatment.
Similar content being viewed by others

Why carry out this study? |
Emergence of novel viral variants has been shown to occur following mono- and combination therapy with monoclonal antibodies (mAbs) for coronavirus disease 2019 (COVID-19). |
Emergent mutations in antibody epitopes that reduce binding affinity and inhibit neutralising activity suggest that immune pressure from mAbs drives development of escape mutations; furthermore, studies have shown greater viral loads in individuals with treatment-emergent mutations. |
In this study we wanted to understand the scope and effects of treatment-emergent mutations in participants from the TACKLE phase 3, randomised clinical trial who received ≤ 7 days of either placebo or AZD7442, a COVID-19 antibody therapy designed to provide a high threshold of protection against escape mutations. |
What was learned from this study? |
Treatment-emergent variants with AZD7442 were rare when measured up to 15 days post dosing, and all treatment-emergent variants showed in vitro susceptibility to AZD7442. |
While treatment-emergent variants can reduce neutralising activity of some therapies, our data indicate that AZD7442 creates a high genetic barrier for resistance, making it a feasible option for COVID-19 treatment. |
Introduction
The development and deployment of vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has had a significant impact on the global burden of coronavirus disease 2019 (COVID-19) [1]. However, individuals considered clinically vulnerable to SARS-CoV-2 infection and severe COVID-19 outcomes, including those with comorbidities or immunosuppression, and older adults, may benefit from additional protection [2,3,4,5,6]. To this end, anti-SARS-CoV-2 spike protein monoclonal antibody (mAb) therapies have been widely used in pandemic responses to provide protection to immunocompromised individuals and individuals who cannot be vaccinated as pre-exposure prophylaxes, and to limit disease progression [7,8,9].
RNA viruses, such as SARS-CoV-2, are prone to rapid evolution due to inherently high rates of mutation and recombination [10]. Owing to ongoing intra-host viral mutations, viral variants with decreased susceptibility to mAb neutralisation may emerge, and be selected for, during COVID-19 treatment [9, 11,12,13]. AZD7442 is a combination of two fully human SARS-CoV-2-neutralising mAbs—tixagevimab (AZD8895) and cilgavimab (AZD1061)—derived from potent antibodies isolated from B cells of individuals with prior SARS-CoV-2 infection [14]. Tixagevimab and cilgavimab were selected to be used in combination to provide a high threshold of protection against virus mutational escape from mAb neutralisation. Tixagevimab and cilgavimab carry a triple modification (L234F/L235E/P331S; TM modification) that reduces fragment crystallizable effector function and the risk of antibody-dependent disease enhancement, and a YTE modification (M257Y/S259T/T261E) that extends the in vivo half-life of each mAb [15]. Following administration, the AZD7442 component mAbs bind to distinct epitopes on the receptor-binding domain of the SARS-CoV-2 spike protein, and potently neutralise the virus by preventing interaction with the host human angiotensin-converting enzyme 2 receptor [15].
In the phase 3, randomised, double-blind, placebo-controlled TACKLE study (NCT04723394) a single intramuscular dose of AZD7442 was shown to protect against severe outcomes from COVID-19 in a population of unvaccinated outpatients comprising predominantly (approx. 90%) individuals at high risk of severe disease [8]. The risk of COVID-19 progression was found to be reduced in outpatients who received AZD7442 versus placebo; the relative risk reduction was 50.5% and absolute risk reduction 4.5% for severe COVID-19 or death [8]. Here, we examine the emergence of viral variants following AZD7442 treatment during TACKLE.
Methods
TACKLE Study Design
TACKLE (NCT04723394) was a phase 3, randomised, double-blind, placebo-controlled study, conducted across 95 sites in Europe, Japan, Latin America, and the USA, designed to assess the safety and efficacy of a single dose of AZD7442 for the prevention of severe outcomes from COVID-19, including death. Full details of the study, including primary and key secondary outcomes, were reported previously [8]. In short, between January and July 2021, this study enrolled unvaccinated, non-hospitalised adults (≥ 18 years) with mild-to-moderate COVID-19 confirmed by laboratory reverse-transcription polymerase chain reaction (RT-PCR) or antigen test on a sample collected ≤ 3 days prior to enrolment. Participants were randomised and dosed ≤ 7 days from symptom onset with a single 600-mg dose of AZD7442 (two consecutive 3-mL intramuscular injections, one each of 300 mg tixagevimab and 300 mg cilgavimab) or matched saline placebo (two consecutive 3-mL intramuscular injections). Randomisation in a 1:1 ratio was stratified, using centralised blocked randomisation, by time from symptom onset (≤ 5 vs. > 5 days), and risk of progression to severe COVID-19 (high vs. low risk; high risk included those aged ≥ 65 years, immunocompromised individuals, and those with comorbidities, such as cancer and chronic diseases). Per protocol, cases of severe COVID-19 were diagnosed by the treating principal investigator and defined as pneumonia (fever, cough, tachypnoea or dyspnoea, and lung infiltrates) or hypoxaemia (saturation of arterial blood with oxygen < 90% in room air, severe respiratory distress, or both), and a World Health Organization (WHO) Clinical Progression Scale score of 5 or more [16]. Participants, investigators, and the sponsor were blind to treatment-group assignments.
Ethical Approval
The study was conducted in accordance with the Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonization Good Clinical Practice guidelines, and all applicable laws and regulations. The protocol, protocol amendments, and all other relevant documentation were reviewed and approved by an institutional review board or ethics committee (Supplementary Material Table S1) and are available with the previous report [8]. An independent Data Safety Management Board provided oversight throughout the study to ensure the safe and ethical conduct of the study. All participants provided written informed consent.
Objective
We examined treatment-emergent viral variants following AZD7442 treatment as an exploratory objective of the TACKLE study. With longitudinal viral genotypic analysis, we explored the relationship between treatment-emergent viral variants and in vitro changes in viral susceptibility to AZD7442, viral load, and prevalence of severe COVID-19 outcomes, including death.
Sample Collection and Handling
Nasopharyngeal (NP) swabs were taken at study visits occurring at baseline, and 3, 6, and 15 days after administration of AZD7442 or placebo. Immediately after collection, all swabs were placed in transport medium for storage at − 70 °C. Samples then were shipped to the centralised testing laboratory on dry ice where they were stored at − 70 °C until viral nucleic acid extraction.
Genotypic Analysis
Nucleic Acid Extraction, Amplification, and Sequencing
Viral nucleic acids were extracted from NP swabs on a Kingfisher Flex instrument using the MagMax™ Viral/Pathogen Nucleic Acid Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA). A validated protocol for next-generation sequencing (NGS) of the SARS-CoV-2 spike gene was employed on SARS-CoV-2 RT-PCR-positive swabs to identify viral mutations and assign SARS-CoV-2 lineages at Monogram Biosciences (South San Francisco, CA, USA). Briefly, the full-length S gene (amino acids 1–1274) was amplified and sequenced in a GenoSure SARS-CoV-2 spike 2 × 150 base pair MiSeq sequencing (v2 chemistry; Illumina, San Diego, CA, USA) NGS assay. Sequence files were analysed to determine frequency of amino acid polymorphisms (consensus; reported at allele fraction [AF] ≥ 25%) and minor variants (minimum coverage > 1000×; reported at AF 3% to 25%).
SARS-CoV-2 Lineage Designation
For each sample, a consensus SARS-CoV-2 spike nucleotide sequence was derived from NGS representing variants at AF ≥ 25% with International Union of Pure and Applied Chemistry-based codes. A spike-only version of Pangolin COVID-19 lineage assigner (Hedgehog; https://github.com/aineniamh/hedgehog, version 1.0.7) was used to classify SARS-CoV-2 spike sequences to current Pango lineages or sets of lineages [17]. Variant of concern (VOC) and variant of interest (VOI) classification followed nomenclature as defined by the WHO during the study period [18, 19].
Treatment-Emergent Substitution Analysis
NGS-derived consensus SARS-CoV-2 spike nucleotide sequences were aligned to Wuhan-Hu-1/2019 SARS-CoV-2 reference sequence (GenBank accession number NC_045512) using Geneious (version 2023.0.4; Dotmatics, Boston, MA, USA). Longitudinal sequence quality control was performed by computing intra-participant Hamming’s distances (R version 4.2.0 and DescTools package version 0.99.47), and participants with at least one pairwise Hamming’s distance greater than the mean + 3 standard deviations of the intra-participant distribution (i.e. 12.21 substitutions) were retained in the baseline analysis but were removed from the longitudinal analysis, as they may have represented sample mislabelling/misassignment.
Sanger sequencing, a common method of minor variant detection, has a traditionally accepted capacity to detect minor variants at > 25% AF [20,21,22]. The technique we used in this study, NGS, has been predicted to accurately detect minor sequence variants at a 1% AF [20]. However, it has also been suggested that variants in the 1–3% AF range can only be accurately determined if the obtained sequence data are of high quality and when the variants are confirmed by a secondary method [23]. Therefore, our treatment-emergent substitution analysis was performed by intra-participant comparison of baseline to post-baseline amino acid sequences, and by computing amino acid substitutions (i.e. amino acid replacements, and in-frame insertions and deletions) at AF ≥ 25% and AF 3% to 25% in the binding sites of tixagevimab (positions 455–456, 458, 475–480, 483–489, and 493) and cilgavimab (positions 345–346, 439–441, 443–447, 449–450, 452, 484, 490, 492–494, and 499) [14]. The notation employed for treatment-emergent substitutions is of the following format: amino acid residue(s) present at baseline plus residue position, followed by amino acid residue(s) observed at the post-baseline time point.
In Vitro Microneutralisation of SARS-CoV-2 Spike Protein Pseudotyped Lentivirus
Spike protein sequences representing the major SARS-CoV-2 lineages identified at baseline, and single substitutions in the spike protein identified at baseline and treatment-emergent analyses were engineered into SARS-CoV-2 Wuhan-Hu-1/2019 + D614G spike protein pseudotyped lentiviruses and assessed for in vitro susceptibility to AZD7442 and its component mAbs via a microneutralisation assay. Generation of spike protein pseudotyped lentivirus and pseudovirus microneutralisation assays were performed as previously reported, with several modifications [24, 25]. Inserts encoding for residues 1–1254 of the Wuhan-Hu-1/2019 + D614G SARS-CoV-2 spike protein with single substitutions or of major viral variants (Supplementary Material Table S2) were designed through codon optimisation and were incorporated in the pCAGG-Sdl19 plasmid. SARS-CoV-2 spike pseudotyped lentiviral particles were generated using a third-generation human immunodeficiency virus (HIV)-based lentiviral vector system expressing luciferase. Pseudotyped lentiviral particles were produced, purified, and titrated to assess AZD7442 susceptibility in the microneutralisation assay.
In vitro susceptibility of SARS-CoV-2 variants to AZD7442 and its component mAbs was assessed using one of three versions of the microneutralisation assay (i.e. research-grade, qualified version 1.0 [pVNTv1.0], and qualified version 2.0 [pVNTv2.0]). For all three versions of this assay, serial dilutions of mAbs were prepared in a 384-well microtiter plate and pre-incubated with pseudovirus for 30 min at 37 °C. Following incubation, AD293-ACE2-ARCB cells stably expressing ACE2 (for research-grade and pVNTv1.0 assay versions) or HEK-Blue-ACE2/TMPRSS2 cells stably expressing ACE2/TMPRSS2 (for pVNTv2.0 assay version; InVivogen, San Diego, CA, USA) were added to the wells and the plates were incubated for 48 h at 37 °C. Relative quantitation of infectivity was determined by measuring the luminescence of the expressed luciferase activity as relative luminescence units on an EnVision 2105 Multimode Plate Reader (Perkin Elmer, Waltham, MA, USA) using the Steady-Glo™ Luciferase Assay System (Promega, Madison, WI, USA) for research-grade and pVNTv1.0 assay version or Bright-Glo™ Luciferase Assay System for pVNTv2.0 assay version, according to the manufacturer’s recommendations. Percent inhibition was calculated by normalisation to the virus-only control for all assays. For the research-grade assay, half-maximal inhibitory concentration (IC50) values were determined by non-linear regression using Prism software (GraphPad, version 9.0.0). The average IC50 value for each antibody was determined from a minimum of two independent experiments. The corresponding fold change IC50 against each variant was determined for each mAb individually and together (tixagevimab and cilgavimab) compared with the reference SARS-CoV-2 Wuhan-Hu-1/2019 + D614G spike protein pseudovirus. Reference IC50 was measured within each run of the research-grade assay. The assay methodology underwent a qualification based on specificity, intermediate precision, inter-assay precision, and intra-assay precision to support regulatory submission [26]. For assays performed using the qualified pseudovirus microneutralisation assay (pVNTv1.0 and pVNTv2.0), IC50 values were subsequently determined by fitting a four-parameter logistic model to the replicate relative luminescence signal obtained across a dilution of mAb concentrations and back-calculating the concentrations that give 50% inhibition between the virus only and the no virus control (X50) using ordinary least squares in RStudio, version 4.0.2 (PBC, Boston, MA, USA). The average reference IC50, established during qualification, was utilised for all qualified assay runs for fold-change calculations.
Quantitative PCR to Measure SARS-CoV-2 Viral Load
NP swab samples acquired at baseline and study day 6 that were assessed as positive in the Cobas® SARS-CoV-2 RT-PCR assay (Roche Diagnostics International AG, Rotkreuz, Switzerland) were further evaluated by quantitative RT-PCR to measure viral load using the Centers for Disease Control and Prevention’s 2019-nCoV Real-Time RT-PCR Diagnostic Panel (Centers for Disease Control and Prevention, Atlanta GA, USA). The testing was performed at Covance Central Laboratory Services (Indianapolis, IN, USA). The validation included precision, sensitivity, accuracy, and linearity. The lower limit of quantitation for the viral load assay was 3.348 log10 copies/mL (2228 copies/mL).
Statistical Analysis
Statistical analysis of viral shedding was conducted by first calculating the log-transformation of the reduction in viral shedding at study day 6 versus baseline. The reduction was then fit to a robust linear model (R version 4.1.3). A one-sided t test was used to compare the mean reduction in viral shedding between participants who exhibited treatment-emergent substitutions and participants who had no detected substitutions.
Results
Participants
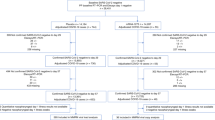
A total of 1014 participants were enrolled between 28 January 2021 and 22 July 2021, with 910 randomly assigned to receive AZD7442 (n = 456) or placebo (n = 454), as described previously [8] (Fig. 1). Approximately half (n = 396; 50.5%) of all participants included in the genotypic analyses were female, the majority (n = 652; 87.5%) were aged < 65 years (mean age [standard deviation] 45.8 [15.2] years), and baseline sociodemographic characteristics of participants were balanced between the two groups (Table 1).

Participant disposition. A severe COVID-19 event was defined as pneumonia (fever, cough, tachypnoea or dyspnoea, and lung infiltrates) or hypoxaemia (saturation of arterial blood with oxygen < 90% in room air, severe respiratory distress, or both), and a WHO Clinical Progression Scale score of 5 or more. COVID-19 coronavirus disease 2019, QC quality control, WHO World Health Organization
Genotypic Analysis
Baseline Prevalence of SARS-CoV-2 Variants
Of 452 participants who received AZD7442 and 451 who received placebo, baseline spike sequences were available for analysis from 745 (82.5%) study participants (AZD7442, n = 380; placebo, n = 365; Fig. 1). Across all participants with samples available at baseline, 483/745 (64.8%) sequences corresponded to VOC or VOI based on WHO classification during the study period [18, 19]. Prevalence of SARS-CoV-2 variants was balanced between treatment groups and was in concordance with SARS-CoV-2 circulating at geographical locations at the time of recruitment [27], with Alpha (38.0%), B.1.1.519 (18.7%), Gamma (12.2%), and Delta (9.8%) accounting for the majority of overall cases (Table 2).
At baseline, 11/34 positions in the binding sites of tixagevimab and cilgavimab showed the presence of amino acids that differed from the ancestral Wuhan-Hu-1 sequence at an AF ≥ 25%, with frequencies ranging from 0.1% to 29.3% in the full analysis set (Supplementary Material Tables S3 and S4). The most common amino acid differences were T478K (218 participants; 29.3%) in the tixagevimab binding site, E484K (135; 18.1%) in the tixagevimab and cilgavimab sites, and L452R (83; 11.1%) in the cilgavimab site, which represent lineage-defining signatures of the major SARS-CoV-2 variants detected in the study. All tested variants and single amino acid differences showed in vitro susceptibility to AZD7442, with a less than fivefold change versus Wuhan-Hu-1 + D614G reference (Table 2; Supplementary Material Tables S3 and S4). Overall, 417/745 (56.0%) participants carried at least one amino acid difference to the Wuhan-Hu-1 reference sequence in the binding sites of tixagevimab and cilgavimab at an AF ≥ 25%, with 117/745 (15.7%) participants carrying at least two amino acid differences (Supplementary Material Table S5).
Treatment-Emergent SARS-CoV-2 Substitution Analysis
The analysis of treatment-emergent substitutions was conducted for each participant by comparing baseline sequences to any post-baseline sequence. A baseline spike sequence and at least one follow-up sequencing dataset were available for treatment-emergent substitution analysis of 235/452 (52.0%) and 226/451 (50.1%) participants in the AZD7442 and placebo groups, respectively. This excluded 14 participants (n = 8 in the AZD7442 treatment group and n = 6 in the placebo group) who did not pass longitudinal sequence quality control (Supplementary Material Table S6). The sociodemographic characteristics and the distribution of SARS-CoV-2 variants in the longitudinal dataset were similar to the ones in the overall dataset (Tables 1, 2; Supplementary Material Tables S7 and S8).
Treatment-emergent substitutions were uncommon, with 5 (2.1%) participants who received AZD7442 having treatment-emergent substitutions in the tixagevimab/cilgavimab binding site at an AF ≥ 25%; none at an AF ≥ 25% were identified in the placebo group (Fig. 2; Table 3; Supplementary Material Fig. S1). Fifteen (6.4%) participants in the AZD7442 group and 12 (5.3%) who received placebo had treatment-emergent substitutions in the tixagevimab/cilgavimab binding site at an AF 3% to 25% (Fig. 3; Supplementary Material Table S9). Across these individuals, 25 unique treatment-emergent substitutions at an AF 3% to 25% were detected (Supplementary Material Table S9). The majority of treatment-emergent substitutions were detected at study day 6 following treatment with AZD7442 or placebo (Figs. 2, 3).

Treatment-emergent substitutions in tixagevimab or cilgavimab binding sites according to SARS-CoV-2 lineage at baseline (AF ≥ 25%). For each time point, the number of sequences assessed is shown. Only subjects with an emergent mutation at any time point are longitudinally tracked and only sequences carrying treatment-emergent substitutions in tixagevimab or cilgavimab binding sites at an AF ≥ 25% are shown. Blue lines depict longitudinal assessment from individual participants. Black ovals depict sequences that had no detectable substitution in the tixagevimab or cilgavimab binding sites compared to the participant baseline sequence. The participant infected with Gamma variant had an event of severe COVID-19 presenting with pneumonia and hypoxaemia, with a maximum WHO Clinical Progression Scale score of 6. All other participants with treatment-emergent substitutions in tixagevimab or cilgavimab binding sites had no severe COVID-19 events. Substitution notation is of the following format: amino acid residue(s) present at baseline plus residue position, followed by amino acid residue(s) observed at the post-baseline time point. B.1.1_6: PANGO lineages B.1.1.222 or B.1.1.322. AF allele fraction, COVID-19 coronavirus disease 2019, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, WHO World Health Organization

Treatment-emergent substitutions in tixagevimab or cilgavimab binding sites according to SARS-CoV-2 lineage at baseline (AF 3 to 25%). For each time point, the number of sequences assessed is shown. Only subjects with an emergent mutation at any time point are longitudinally tracked and only sequences carrying treatment-emergent substitutions in tixagevimab or cilgavimab binding sites at an AF 3% to 25% are shown. Blue lines depict longitudinal assessment from individual participants. Black ovals depict sequences that had no detectable substitution in the tixagevimab or cilgavimab binding sites compared to the baseline sequence. The participant infected with Alpha variant with study day 6 G446G/V and C488C/F substitutions, and the participant infected with Alpha variant with study day 6 S477N/S substitution both had an event of severe COVID-19 presenting with pneumonia and hypoxaemia, with a maximum WHO Clinical Progression Scale score of 5. Substitution notation is of the following format: amino acid residue(s) present at baseline plus residue position, followed by amino acid residue(s) observed at the post-baseline time point. AF allele fraction, COVID-19 coronavirus disease 2019, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, WHO World Health Organization
Treatment-emergent substitutions with an AF ≥ 25% arising in participants in the AZD7442 group were tested in vitro for susceptibility to AZD7442 (Table 4). Four of the five variants remained susceptible to AZD7442 with a less than fivefold change of in vitro susceptibility to AZD7442 compared to the reference Wuhan-Hu-1 + D614G. Lentiviral pseudovirus particles produced for the remaining one variant did not achieve high enough titer for in vitro susceptibility testing. Twenty-four out of 25 treatment-emergent substitutions detected with an AF 3% to 25% had a less than fivefold change of in vitro susceptibility to AZD7442 compared to the reference Wuhan-Hu-1 + D614G (Supplementary Material Table S9). Substitution F456F/S, in the tixagevimab binding site, which emerged at day 3 post dosing at AF 5.6% in one (0.4%) participant treated with AZD7442, showed a 23.2-fold change in in vitro susceptibility to AZD7442; however, this is consistent with AZD7442 being active against this substitution. Among AZD7442 recipients, treatment-emergent substitutions at the tixagevimab and cilgavimab binding sites did not appear to be associated with an increase in cases of severe COVID-19 events or death (Table 5). Severe COVID-19 or death occurred in 3/20 (15.0%) participants with and 11/215 (5.1%) participants without treatment-emergent substitutions. However, the low number of participants and the lack of study power to statistically infer differences between groups precludes our ability to draw firm conclusions.
SARS-CoV-2 viral loads in NP swabs at baseline and study day 6 were compared to determine if there was a difference in viral shedding between treatment groups. In participants in the longitudinal analysis subset who did not have treatment-emergent substitutions, reduction in viral shedding by study day 6 was significantly stronger for the AZD7442 group than the placebo group (P = 0.018). No formal analysis of the difference in viral shedding between treatment groups was undertaken for participants with treatment-emergent substitutions because of the small cohort numbers; however, the changes in viral shedding recorded in participants with and without treatment-emergent substitutions were comparable within treatment groups (Supplementary Material Fig. S2).
Discussion
Error-prone replication of the SARS-CoV-2 genome allows for the generation of viral mutants, which can be selected by the evolutionary pressure exerted by antiviral drugs [28]. On the basis of knowledge gained through clinical treatment of infections with other RNA viruses (e.g. HIV), the emergence of viral resistance under treatment is influenced by the balance between the selective pressure exerted by the antiviral drug and the capacity of the virus populations to generate a pool of viable resistant mutants [29,30,31]. The likelihood of resistance emergence is maximal when the drug levels at the site of action are low enough to allow for viral replication, thus allowing for continuing generation of viral mutants, while high enough to exert a selective pressure (Supplementary Material Fig. S3). In the current study, based on a clinical trial demonstrating protection from progression to severe COVID-19 or death in treated participants, the observed low levels of treatment-emergent substitutions are consistent with effective drug levels being achieved at the site of action.
We found a low incidence of SARS-CoV-2 variants bearing substitutions at tixagevimab/cilgavimab binding sites following treatment with AZD7442 in the TACKLE study, with frequencies comparable to those seen in the placebo group. None of the tested treatment-emergent substitutions with AF ≥ 25% showed reduced susceptibility to AZD7442. Only 1/25 treatment-emergent substitution detected at AF 3% to 25% in 1/235 participants showed greater than fivefold change in susceptibility to AZD7442. Importantly, the treatment-emergent substitutions observed in the tixagevimab/cilgavimab binding sites were not associated with any detectable decreases in susceptibility to the mAbs, severe COVID-19/death, or increases in viral shedding between study day 6 and baseline. Overall, this indicates that AZD7442 created a high genetic barrier to resistance during the study period (in the background of highly susceptible viral variants).
Data herein were obtained from participants of the TACKLE study who were enrolled between January and July 2021, with a follow-up for longitudinal assessment of treatment-emergent variants up to 15 days post dosing. Therefore, data were collected prior to and during a global SARS-CoV-2 Delta variant wave, and thus a limitation of this study is that infections were caused by circulating variants that are no longer classified as variants of concern/interest [18, 19]. At present, the Omicron variant and its sublineages account for the vast majority of sequenced infections [32]. Although treatment-emergent variants following AZD7442 treatment for Omicron could not be assessed in the present study, previous in vitro studies demonstrated that AZD7442 retains neutralising activity against BA.1, BA.2, BA.2.12.1, BA.3, BA.4, and BA.5 Omicron subvariants [33, 34]. However, recently circulating Omicron sublineages escape the strong neutralising potency of AZD7442 in vitro [25, 35, 36].
As novel variants emerge with resistance to available antiviral therapies, alternative targeted treatment options are required. Results from the TACKLE study suggest that new mAbs with a similar mechanism of action to AZD7442 could protect against severe outcomes associated with SARS-CoV-2 infection.
In the present study, examination of disease severity was limited by the rarity of treatment-emergent variants. As a result of the low numbers of treatment-emergent substitutions observed in the tixagevimab/cilgavimab binding sites, we cannot make firm conclusions on the impacts of such substitutions on the severity of COVID-19 or risk of death, nor could we conduct a formal analysis of the differences in viral shedding in participants with treatment-emergent substitutions according to treatment group. However, it was notable that the changes we observed in viral shedding from baseline to study day 6 in individuals with treatment-emergent substitutions were comparable to individuals without treatment-emergent substitutions in respective treatment groups.
Despite reduced activity against recent variants, AZD7442 has demonstrated that mAb approaches are a viable option for providing additional protection against severe outcomes of SARS-CoV-2 infection. Monitoring of emerging variants will remain an important part of clinical trial design and real-world monitoring for mAb treatments.
Data Availability
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://www.astrazenecaclinicaltrials.com/our-transparency-commitments/. Data for studies directly listed on Vivli can be requested through Vivli at www.vivli.org. Data for studies not listed on Vivli could be requested through Vivli at https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. AstraZeneca Vivli member page is also available outlining further details: https://vivli.org/ourmember/astrazeneca/.
References
Watson OJ, Barnsley G, Toor J, Hogan AB, Winskill P, Ghani AC. Global impact of the first year of COVID-19 vaccination: a mathematical modelling study. Lancet Infect Dis. 2022;22(9):1293–302. https://doi.org/10.1016/S1473-3099(22)00320-6.
Bonelli M, Mrak D, Tobudic S, et al. Additional heterologous versus homologous booster vaccination in immunosuppressed patients without SARS-CoV-2 antibody seroconversion after primary mRNA vaccination: a randomised controlled trial. Ann Rheum Dis. 2022;81(5):687–94. https://doi.org/10.1136/annrheumdis-2021-221558.
Manothummetha K, Chuleerarux N, Sanguankeo A, et al. Immunogenicity and risk factors associated with poor humoral immune response of SARS-CoV-2 vaccines in recipients of solid organ transplant: a systematic review and meta-analysis. JAMA Netw Open. 2022;5(4):e226822. https://doi.org/10.1001/jamanetworkopen.2022.6822.
Re D, Seitz-Polski B, Brglez V, et al. Humoral and cellular responses after a third dose of SARS-CoV-2 BNT162b2 vaccine in patients with lymphoid malignancies. Nat Commun. 2022;13(1):864. https://doi.org/10.1038/s41467-022-28578-0.
Marlet J, Gatault P, Maakaroun Z, et al. Antibody responses after a third dose of COVID-19 vaccine in kidney transplant recipients and patients treated for chronic lymphocytic leukemia. Vaccines. 2021;9(10):1055. https://doi.org/10.3390/vaccines9101055.
Bajaj V, Gadi N, Spihlman AP, Wu SC, Choi CH, Moulton VR. Aging, immunity, and COVID-19: how age influences the host immune response to coronavirus infections? Front Physiol. 2020;11:571416. https://doi.org/10.3389/fphys.2020.571416.
Levin MJ, Ustianowski A, De Wit S, et al. Intramuscular AZD7442 (tixagevimab-cilgavimab) for prevention of Covid-19. N Engl J Med. 2022;386(23):2188–200. https://doi.org/10.1056/NEJMoa2116620.
Montgomery H, Hobbs FDR, Padilla F, et al. Efficacy and safety of intramuscular administration of tixagevimab-cilgavimab for early outpatient treatment of COVID-19 (TACKLE): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2022;10(10):985–96. https://doi.org/10.1016/S2213-2600(22)00180-1.
Focosi D, Novazzi F, Baj A, et al. Sotrovimab-emergent resistance in SARS-CoV-2 Omicron: a series of three cases. J Clin Virol Plus. 2022;2(3):100097. https://doi.org/10.1016/j.jcvp.2022.100097.
Dolan PT, Whitfield ZJ, Andino R. Mechanisms and concepts in RNA virus population dynamics and evolution. Ann Rev Virol. 2018;5(1):69–92. https://doi.org/10.1146/annurev-virology-101416-041718.
Choudhary MC, Chew KW, Deo R, et al. Emergence of SARS-CoV-2 escape mutations during bamlanivimab therapy in a phase II randomized clinical trial. Nat Microbiol. 2022;7(11):1906–17. https://doi.org/10.1038/s41564-022-01254-1.
Rockett R, Basile K, Maddocks S, et al. Resistance mutations in SARS-CoV-2 delta variant after sotrovimab use. N Engl J Med. 2022;386(15):1477–9. https://doi.org/10.1056/NEJMc2120219.
Focosi D, McConnell S, Casadevall A, Cappello E, Valdiserra G, Tuccori M. Monoclonal antibody therapies against SARS-CoV-2. Lancet Infect Dis. 2022;22(11):e311–26. https://doi.org/10.1016/S1473-3099(22)00311-5.
Dong J, Zost SJ, Greaney AJ, et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat Microbiol. 2021;6(10):1233–44. https://doi.org/10.1038/s41564-021-00972-2.
Loo YM, McTamney PM, Arends RH, et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in nonhuman primates and has an extended half-life in humans. Sci Transl Med. 2022;14(635):eabl8124. https://doi.org/10.1126/scitranslmed.abl8124.
WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis. 2020;20(8):e192–e7. https://doi.org/10.1016/S1473-3099(20)30483-7.
O’Toole A, Pybus OG, Abram ME, Kelly EJ, Rambaut A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genomics. 2022;23(1):121. https://doi.org/10.1186/s12864-022-08358-2.
World Health Organization (WHO). Tracking SARS-CoV-2 variants. Updated 30 March 2023. https://www.who.int/activities/tracking-SARS-CoV-2-variants. Accessed 25 Jul 2023.
World Health Organization (WHO). Historical working definitions and primary actions for SARS-CoV-2 variants. Updated 15 March 2023. https://www.who.int/publications/m/item/historical-working-definitions-and-primary-actions-for-sars-cov-2-variants. Accessed 25 Jul 2023.
Zhou S, Swanstrom R. Fact and fiction about 1%: next generation sequencing and the detection of minor drug resistant variants in HIV-1 populations with and without unique molecular identifiers. Viruses. 2020;12(8):850. https://doi.org/10.3390/v12080850
Solomon D. Integrating molecular diagnostics with surgical neuropathology. Amsterdam: Elsevier; 2018. p. 71–89. https://doi.org/10.1016/B978-0-323-44941-0.00005-9.
Larder BA, Kohli A, Kellam P, Kemp SD, Kronick M, Henfrey RD. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature. 1993;365(6447):671–3. https://doi.org/10.1038/365671a0.
Petrackova A, Vasinek M, Sedlarikova L, et al. Standardization of sequencing coverage depth in NGS: recommendation for detection of clonal and subclonal mutations in cancer diagnostics. Front Oncol. 2019;9:851. https://doi.org/10.3389/fonc.2019.00851.
Zost SJ, Gilchuk P, Case JB, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584(7821):443–9. https://doi.org/10.1038/s41586-020-2548-6.
Roe TL, Brady T, Schuko N, et al. Molecular characterization of AZD7442 (tixagevimab-cilgavimab) neutralization of SARS-CoV-2 omicron subvariants. Microbiol Spectr. 2023;11(2):e0033323. https://doi.org/10.1128/spectrum.00333-23.
Tuffy K, Ahani B, Aksyuk A, et al. Breakthrough SARS-CoV-2 infections in the PROVENT prevention trial were not associated with AZD7442 (tixagevimab/cilgavimab) resistant variants. J Infect Dis. 2023. https://doi.org/10.1093/infdis/jiad210.
Our World in Data (University of Oxford). SARS-CoV-2 variants in analyzed sequences; 20 April 2023. https://ourworldindata.org/grapher/covid-variants-area?time=2021-01-04..2021-07-19&facet=entity&country=GBR~USA~ARG~BRA~CZE~DEU~HUN~ITA~JPN~MEX~PER~POL~RUS~ESP~UKR. Accessed 25 Jul 2023.
Focosi D, McConnell S, Sullivan DJ, Casadevall A. Analysis of SARS-CoV-2 mutations associated with resistance to therapeutic monoclonal antibodies that emerge after treatment. www.medrxiv.org. 2023. https://doi.org/10.1101/2023.03.02.23286677.
Motyan JA, Mahdi M, Hoffka G, Tozser J. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: lessons learned from HIV-1 protease. Int J Mol Sci. 2022;23(7):3507. https://doi.org/10.3390/ijms23073507.
Richman DD. The implications of drug resistance for strategies of combination antiviral chemotherapy. Antiviral Res. 1996;29(1):31–3. https://doi.org/10.1016/0166-3542(95)00911-6.
Back D, Gatti G, Fletcher C, et al. Therapeutic drug monitoring in HIV infection: current status and future directions. AIDS. 2002;16(Suppl 1):S5-37. https://doi.org/10.1097/00002030-200203001-00002.
World Health Organization (WHO). Statement on the update of WHO’s working definitions and tracking system for SARS-CoV-2 variants of concern and variants of interest. Updated 16 March 2023. https://www.who.int/news/item/16-03-2023-statement-on-the-update-of-who-s-working-definitions-and-tracking-system-for-sars-cov-2-variants-of-concern-and-variants-of-interest. Accessed 25 Jul 2023.
Cao Y, Yisimayi A, Jian F, et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022;608(7923):593–602. https://doi.org/10.1038/s41586-022-04980-y.
Tuekprakhon A, Nutalai R, Dijokaite-Guraliuc A, et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell. 2022;185(14):2422–33.e13. https://doi.org/10.1016/j.cell.2022.06.005.
Wang Q, Li Z, Ho J, et al. Resistance of SARS-CoV-2 omicron subvariant BA.4.6 to antibody neutralisation. Lancet Infect Dis. 2022;22(12):1666–8. https://doi.org/10.1016/S1473-3099(22)00694-6.
Arora P, Kempf A, Nehlmeier I, et al. Omicron sublineage BQ.1.1 resistance to monoclonal antibodies. Lancet Infect Dis. 2023;23(1):22–3. https://doi.org/10.1016/S1473-3099(22)00733-2.
Acknowledgements
We thank Kevin Tuffy, MSc, for generating structural models of the SARS-CoV-2 spike protein.
Medical Writing/Editorial Assistance
Medical writing support by Jon Moran, PhD, of Ashfield MedComms (Macclesfield, UK), an Inizio company, in accordance with Good Publication Practice (GPP) 2022 guidelines (www.ismpp.org/gpp-2022; Ann Intern Med. 2022;175(9):1298–304) was funded by AstraZeneca.
Funding
This analysis was funded by AstraZeneca. AZD7442 is being developed with support from the US Government, including federal funds from the Department of Health and Human Services, Administration for Strategic Preparedness and Response, Biomedical Advanced Research and Development Authority in partnership with the Department of Defense, and Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under contract no. W911QY-21-9-0001. AstraZeneca were responsible for the design of this study, acquisition, analysis, and interpretation of data. AstraZeneca funded payment of the journal’s Rapid Service Fee.
Author information
Authors and Affiliations
Contributions
Douglas Arbetter, Elizabeth J. Kelly, F. D. Richard Hobbs, Gustavo H. Kijak, and Katie Streicher conceptualised this study; F.D. Richard Hobbs contributed to funding acquisition; Amy Nguyen, Elizabeth J. Kelly, Hugh Montgomery, Nicolette Schuko, and Tyler Brady served as study investigators; Elizabeth J. Kelly, Fernando Chuecos, F.D. Richard Hobbs, Francisco Padilla, Katie Streicher, and Vancheswaran Gopalakrishnan supervised this study; Hugh Montgomery provided project administration; Amy Nguyen, Douglas Arbetter, Elizabeth J. Kelly, Gustavo H. Kijak, Nicolette Schuko, Tyler Brady, and Tiffany L. Roe developed the experimental methodology; Francisco Padilla, Gustavo H. Kijak, and Vancheswaran Gopalakrishnan contributed to data visualisation; Bahar Ahani, Gustavo H. Kijak, Nicolette Schuko, Tyler Brady, Tiffany L. Roe, and Vancheswaran Gopalakrishnan curated the data; Bahar Ahani, Douglas Arbetter, Gustavo H. Kijak, Jagadish Beloor, Tianhui Zhang, and Vancheswaran Gopalakrishnan conducted formal analyses and data generation; Francisco Padilla, Douglas Arbetter, and Vancheswaran Gopalakrishnan validated the data; and Gustavo H. Kijak, Hugh Montgomery, and Katie Streicher wrote an original draft of this work. All authors reviewed and provided feedback on the manuscript drafts, and approved the manuscript for submission. The manuscript was written under the direction of all authors by medical writers funded by the study sponsor.
Corresponding author
Ethics declarations
Conflict of Interest
Gustavo H. Kijak, Bahar Ahani, Douglas Arbetter, Jagadish Beloor, Tyler Brady, Amy Nguyen, and Katie Streicher are employees of AstraZeneca and owns stocks/shares. Vancheswaran Gopalakrishnan is an employee of AstraZeneca and owns stocks/shares, and is co-inventor on US patent PCT/US17/53,717 and on a provisional US patent WO2020106983A1. Fernando Chuecos, Tiffany L. Roe and Nicolette Schuko are employees of AstraZeneca. Tianhui Zhang was an employee of AstraZeneca and owns stocks/shares, and is currently employed at Eli Lilly and Company (Indianapolis, Indiana, USA). F. D. Richard Hobbs acknowledges part support as Director of the NIHR Applied Research Collaboration (ARC) Oxford Thames Valley, and Theme Lead of the NIHR OUH BRC, and has also received occasional fees or expenses for speaking or consultancy from AstraZeneca, Boehringer Ingelheim, Bayer, Bristol Myers Squibb/Pfizer, and Novartis. Francisco Padilla has received fees for conferences and research from Amgen, AstraZeneca, Ferrer, Lilly, Novartis, Servier, and Silanes. Elizabeth J. Kelly was an employee of AstraZeneca and owns stocks/shares, and is now employed at Sanofi (Swiftwater, PA, USA). Hugh Montgomery has received fees for consulting and advisory committees from AstraZeneca.
Ethical Approval
The study was conducted in accordance with the Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonization Good Clinical Practice guidelines, and all applicable laws and regulations. The protocol, protocol amendments, and all other relevant documentation were reviewed and approved by an institutional review board or ethics committee (see Supplementary Material Table S1 for ethics approval committee information) and are available with the previous report [8]. An independent Data Safety Management Board provided oversight throughout the study to ensure the safe and ethical conduct of the study. All participants provided written informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Prior Presentation: Part of this work was presented in an abstract submitted to IDWeek, 19–23 October, 2022, Washington, DC, USA: Kijak GH, Ahani B, Arbetter D, et al. 1160. Treatment-Emergent Viral Variants in the Phase 3 TACKLE Trial Investigating Efficacy and Safety of AZD7442 (Tixagevimab/Cilgavimab) for the Treatment of Mild to Moderate COVID-19 in Adults. Open Forum Infectious Diseases. 2022;9(supplement_2):ofac492.997. https://doi.org/10.1093/ofid/ofac492.9970.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kijak, G.H., Ahani, B., Arbetter, D. et al. Analysis of SARS-CoV-2 Emergent Variants Following AZD7442 (Tixagevimab/Cilgavimab) for Early Outpatient Treatment of COVID-19 (TACKLE Trial). Infect Dis Ther 12, 2691–2707 (2023). https://doi.org/10.1007/s40121-023-00882-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00882-2