
Abstract
Introduction
Regdanvimab, a neutralising monoclonal antibody (mAb) against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), received approval for the treatment of coronavirus disease 2019 (COVID-19) in South Korea in 2021. The Ministry of Food and Drug Safety in South Korea mandate that new medications be re-examined for safety and effectiveness post-approval in at least 3000 individuals. This post-marketing surveillance (PMS) study was used to evaluate the safety and effectiveness of regdanvimab in real-world clinical care.
Methods
This prospective, multicentre, phase 4 PMS study was conducted between February 2021 and March 2022 in South Korea. Eligible patients were aged ≥ 18 years with confirmed mild COVID-19 at high risk of disease progression or moderate COVID-19. Patients were hospitalised and treated with regdanvimab (40 mg/kg, day 1) and then monitored until discharge, with a follow-up call on day 28. Adverse events (AEs) were documented, and the COVID-19 disease progression rate was used to measure effectiveness.
Results
Of the 3123 patients with COVID-19 infection identified, 3036 were eligible for inclusion. Approximately 80% and 5% of the eligible patients were diagnosed with COVID-19 during the delta- and omicron-dominant periods, respectively. Median (range) age was 57 (18–95) years, and 50.6% of patients were male. COVID-19 severity was assessed before treatment, and high-risk mild and moderate COVID-19 was diagnosed in 1030 (33.9%) and 2006 (66.1%) patients, respectively. AEs and adverse drug reactions (ADRs) were experienced by 684 (22.5%) and 363 (12.0%) patients, respectively. The most common ADR was increased liver function test (n = 62, 2.0%). Nine (0.3%) patients discontinued regdanvimab due to ADRs. Overall, 378 (12.5%) patients experienced disease progression after regdanvimab infusion, with extended hospitalisation/re-admission (n = 300, 9.9%) as the most common reason. Supplemental oxygen was required by 282 (9.3%) patients. Ten (0.3%) patients required intensive care monitoring and 3 (0.1%) died due to COVID-19.
Conclusion
This large-scale PMS study demonstrated that regdanvimab was effective against COVID-19 progression and had an acceptable safety profile when used in real-world clinical practice.
Similar content being viewed by others

Why carry out this study? |
Regdanvimab is a neutralising monoclonal antibody against SARS-CoV-2 that received approval for the treatment of COVID-19 in South Korea in 2021. |
This post-marketing surveillance (PMS) study was conducted in accordance with regulations issued by the Ministry of Food and Drug Safety in South Korea to obtain post-approval safety and effectiveness data in at least 3000 individuals. |
The study aimed to evaluate the safety and effectiveness of regdanvimab in routine clinical care for the treatment of COVID-19. |
What was learned from the study? |
Regdanvimab was well tolerated, and no safety concerns were raised. Overall, 12.5% of treated patients experienced disease progression, and the mortality rate was low (0.1%). |
These real-world data from over 3000 patients support the tolerability and effectiveness of regdanvimab in clinical practice for the treatment of high-risk mild or moderate COVID-19. |
Introduction
Since the World Health Organization (WHO) declared a coronavirus disease 2019 (COVID-19) pandemic on 11 March 2020 [1,2,3] and up to the time of writing (16 July 2023), there have been more than 767 million confirmed cases of COVID-19 and over 6.9 million deaths [4]. The rapid response from the scientific community resulted in the unprecedented approval of vaccines and new antiviral therapies, including monoclonal antibodies (mAbs), within a year [5,6,7]. In the early phase of the pandemic, mAb treatment contributed to preventing a worsening of the disease and reducing hospitalisation and mortality rates [5, 8,9,10]. In line with research on virus mutations, other antibodies were quickly developed to respond to virus mutations [8, 11]. In particular, mAb treatment is used for early response to disease and is a vital option for some patients, including immunocompromised individuals, as it provides a prompt immune response [5, 7, 12, 13].
Regdanvimab is a potent neutralising mAb that binds to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike-protein receptor binding domain (RBD) for the angiotensin-converting enzyme 2 receptor and disrupts viral cell entry [14]. Regdanvimab has high binding affinity for the RBD and neutralising potential as compared with other mAbs [14,15,16,17,18,19]. In a phase 2/3 randomised, placebo-controlled, double-blind study conducted in patients with mild-to-moderate COVID-19, regdanvimab reduced disease progression and clinical recovery time compared with placebo, and its acceptable safety profile was confirmed [20, 21]. Following these results, regdanvimab has received approval or emergency authorisation use for the treatment of COVID-19 [22,23,24]. To expedite use, conditional approval was provided by the Ministry of Food and Drug Safety (MFDS) in South Korea in February 2021 for the treatment of adult patients with mild symptoms of COVID-19 at high risk of progression (over the age of 60 or at least one underlying medical condition; cardiovascular disease, chronic respiratory disease, diabetes, hypertension), or adult patients with moderate symptoms of COVID-19, on the condition of submitting phase 3 results and closely monitoring for any side effects [23]. Later in 2021, regdanvimab received full approval in South Korea (September 2021) for use in adult patients with mild symptoms of COVID-19 at high risk of progression (over the age of 50 or at least one underlying medical condition; obesity, cardiovascular disease, chronic lung disease, diabetes, chronic kidney disease, chronic liver disease, and patients on immunosuppressive agents) or adult patients with moderate symptoms of COVID-19 [24]. In November 2021, the European Union approved regdanvimab treatment for adults with COVID-19 who are at increased risk of progression to severe disease but do not require supplementary oxygen [22]. Overall, regdanvimab has received approval in 40 countries worldwide for the purpose of preventing progression to severe COVID-19. Several retrospective studies conducted in South Korea have provided further evidence for the safety and effectiveness of regdanvimab in patients with mild or mild-to-moderate COVID-19 [25,26,27,28,29,30,31,32].
Post-marketing surveillance (PMS) studies provide important information, after regulatory approval, on the safety and effectiveness of drugs, in a broad patient population attending clinical practice [33, 34]. This phase 4, PMS, multicentre study was conducted in accordance with regulations issued by the MFDS in South Korea whereby new drugs are re-examined for safety and effectiveness post-approval in 3000 or more individuals [35, 36]. The study aims to evaluate the safety and effectiveness of regdanvimab in routine clinical care.
Methods
Study Design
This was a prospective, open-label, observational, phase 4, PMS study of regdanvimab conducted at 17 study centres in South Korea (see Table S1 in the electronic supplementary material). Surveillance was planned over a 6-year period, but, due to the rapid spread of the disease and subsequent high patient enrolment, the necessary number of participants was reached after only 1 year, and the study was closed early.
Compliance with Ethics Guidelines
The study was conducted according to the ethical principles of the Declaration of Helsinki and in compliance with the International Council for Harmonisation Good Clinical Practice and applicable regulatory requirements. The study protocol was approved by the MFDS of the Republic of Korea. Per South Korean laws, additional approval from individual institutional review boards is not required for surveillance studies. All patients provided written informed consent.
Study Population
The study enrolled patients who received regdanvimab for the first time according to its approved indication in South Korea for the treatment of COVID-19. Eligible patients were aged ≥ 18 years with COVID-19 confirmed using reverse transcription-polymerase chain reaction (RT-PCR). Within this population, patients with mild COVID-19 at high risk of progression to severe disease or moderate COVID-19 could receive regdanvimab if they had developed COVID-19 symptoms within 7 days prior to regdanvimab administration, did not require supplemental oxygen, and had oxygen saturation (SpO2) > 94% on room air.
Study definitions of COVID-19 severity were based on guidelines from the Korea Disease Control and Prevention Agency (KDCA) [37]. Patients with mild COVID-19 were those who had clinical symptoms of COVID-19 without viral pneumonia and hypoxia. Patients with moderate COVID-19 were those diagnosed with COVID-19 who had pneumonia (fever, cough, dyspnoea, tachypnoea) but no symptoms of severe pneumonia.
Patients with disease at high risk of progression were defined as patients with ≥ 1 of the following risk factors: age > 50 years; body mass index (BMI) > 30 kg/m2; cardiovascular disease, including hypertension; chronic lung disease, including asthma; type 1 or type 2 diabetes mellitus; chronic kidney disease, including those on dialysis; chronic liver disease; or immunosuppressed status due to disease or treatment (e.g., cancer treatment, bone marrow or organ transplantation, immune deficiencies, HIV, sickle-cell anaemia, thalassaemia, and prolonged use of immune-weakening medications) based on the investigator’s assessment. Patients were excluded if they were prohibited from receiving regdanvimab as specified by the prescribing information [38], had received regdanvimab for purposes other than the approved indication, or if they were considered unfit for participation in this study as decided by the investigator.
Treatment
Regdanvimab 40 mg/kg was administered as a single intravenous infusion over 60 min (± 15 min) according to the prescribing information and as approved by the MFDS in Korea [22, 23].
Assessments
Patient medical history and COVID-19 treatment history were collected, including date of COVID-19 symptom onset, date of diagnosis, disease severity, and variant type, if available. Details of any concomitant medications, any abnormal changes in clinical laboratory tests which require safety reporting as determined by the investigator, and any occurrence of antibody-dependent enhancement after regdanvimab infusion were also recorded.
Patients received regdanvimab on day 1 and were isolated at the hospital for quarantine. Patients were monitored for up to 10 days at the start of the PMS, but this subsequently changed to up to 7 days from 26 January 2022 as dictated by the KDCA. The discharge decision was at the investigator’s discretion, taking into account the isolation period set by the KDCA and the patient’s condition. On day 28 (± 3 days), a follow-up telephone call was conducted to ascertain any additional information about drug safety and effectiveness.
Safety
Adverse events (AEs) and adverse drug reactions (ADRs), including serious AEs, serious ADRs, unexpected AEs, and unexpected ADRs, were documented. Only symptoms of an infusion-related reaction (IRR) grade 1–3 in intensity were considered as possibly expected. For each AE, the type, date of symptom onset/end, severity, seriousness, outcome, causal relationship to regdanvimab and other treatments, and actions taken and/or treatment received were recorded. AEs were recorded regardless of their causality or relationship to regdanvimab treatment. The causal relationship of AEs to regdanvimab treatment was characterised as ‘certain’, ‘probable/likely’, ‘possible’, ‘unlikely’, ‘conditional/unclassified’, or ‘unassessable/unclassifiable’. AEs were deemed to be ADRs (i.e. causally related to regdanvimab) if the relationship to regdanvimab was deemed by the investigator to be ‘certain’, ‘probable/likely’, ‘possible’, ‘conditional/unclassified’, or ‘unassessable/unclassifiable’. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA; version 25.0).
Effectiveness
Effectiveness was assessed based on the rate of COVID-19 disease progression, defined as the occurrence of any of the following criteria: requirement for supplementary oxygen or mechanical ventilation for ≥ 24 h due to SpO2 measuring ≤ 94% on room air because of SARS-CoV-2 infection; hospitalisation for ≥ 24 h longer than planned or re-hospitalisation for ≥ 24 h after discharge due to worsening symptoms of SARS-CoV-2 infection; admittance to intensive care unit (ICU) due to SARS-CoV-2 infection; death due to SARS-CoV-2 infection; requirement for remdesivir for the treatment of severe SARS-CoV-2 infection after regdanvimab administration (includes only cases where remdesivir administration started before 7 January 2022, when the approval of remdesivir in South Korea was extended from severe SARS-CoV-2 infection to also include patients with mild and moderate disease who were at high risk of progression to severe disease).
Effectiveness was analysed in the overall population and further characterised in the following patient subgroups: COVID-19 severity (mild or moderate) prior to regdanvimab infusion; SARS-CoV-2 variant (wild-type, alpha, delta, gamma, omicron, or unknown); patient risk status (≥ 1 high-risk factor or no high-risk factors); COVID-19 vaccination status (vaccinated or unvaccinated); time from symptom onset to regdanvimab infusion (≤ 3 or > 3 days); outbreak period (defined by dominant variant: pre-delta, delta, or omicron). In South Korea, during each outbreak period, the dominant variant was initially detected in more than 50% of patients and rapidly reached about 100% [39].
Statistical Analysis
The safety analysis set comprised all patients who received a partial or full dose of regdanvimab and in whom safety follow-up was completed after regdanvimab administration. The effectiveness analysis set comprised all patients in the safety analysis set who completed the effectiveness evaluation after regdanvimab administration. Patient demographics and baseline characteristics, incidence of AEs and ADRs, and rate of disease progression are reported using descriptive statistics. All statistical analyses were performed using SAS software, version 9.4 (SAS Institute Inc., Cary, North Carolina, USA).
Results
The PMS study was initiated on 5 February 2021. Enrolment of the last patient was on 25 February 2022, and the last visit was performed on 23 March 2022. Final data cutoff was 6 October 2022.
Patients
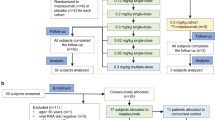
Patient disposition is presented in Fig. 1. In total, 3123 patients with COVID-19 confirmed by RT-PCR were identified across 17 centres in South Korea. Of these patients, 87 were excluded from the study due to violation of the inclusion criteria (n = 72), the patient not receiving regdanvimab (n = 9), loss to follow-up (n = 9), informed consent uncertainty (n = 7), violation of the exclusion criteria (n = 7), or duplicate entries (n = 3) (note that some patients were excluded for more than one reason). The remaining 3036 patients formed the safety analysis set and the effectiveness analysis set.

Patient disposition. aHigh-risk mild patients were defined as patients with ≥ 1 of the following risk factors: age > 50 years; BMI > 30 kg/m2; cardiovascular disease, including hypertension; chronic lung disease, including asthma; type 1 or type 2 diabetes mellitus; chronic kidney disease, including those on dialysis; chronic liver disease; or immunosuppressed status due to disease or treatment (e.g., cancer treatment, bone marrow or organ transplantation, immune deficiencies, HIV, sickle-cell anaemia, thalassaemia, and prolonged use of immune-weakening medications) based on the investigator’s assessment. bPatients could be counted multiple times if there was > 1 reason for exclusion. BMI body mass index, ICF informed consent form, RT-PCR reverse transcription-polymerase chain reaction
Patient demographics and baseline characteristics are presented in Table 1. The median (range) age was 57.0 (18–95) years. Overall, there was a comparable number of females (n = 1500; 49.4%) and males (n = 1536; 50.6%). A total of 46.9% patients received regdanvimab within 3 days of COVID-19 symptom onset, and 71.2% of enrolled patients were not vaccinated. There were 1030 (33.9%) patients classified as having mild COVID-19 and 2006 (66.1%) with moderate COVID-19 prior to regdanvimab infusion. The most common time of diagnosis among patients in the study was during the delta outbreak period (July 2021 to December 2021) (n = 2422; 79.8%). The variant of SARS-CoV-2 was unknown in most patients (n = 2258; 74.4%). Where known, the delta variant was the most commonly occurring variant (n = 516; 17.0%). There were 2294 (75.6%) patients classed as being in the high-risk group.
Regdanvimab Exposure
Among the 3036 patients in the safety analysis set, most (n = 2989, 98.5%) received regdanvimab in accordance with the approved dosage of 40 mg/kg. The median (range) dose administered was 40.0 mg/kg (4.8–56.7) (Table S2). In total, 9 patients (0.3%) permanently discontinued regdanvimab, all due to AEs. The median (range) administered dose in patients with permanent discontinuation of regdanvimab was 24.0 mg/kg (4.8–39.8).
Safety
Table 2 summarises safety findings. In the safety analysis set, 684 (22.5%) patients experienced an AE and 6 (0.2%) experienced a serious AE. One serious AE (cholangiocarcinoma) led to death (drug-related causality was deemed unlikely).
Altogether, 363 (12.0%) patients experienced an ADR. No notable difference was observed in ADR trends between outbreak periods. ADRs that occurred in ≥ 10 patients are reported in Table 3: the most commonly occurring ADRs were increased liver function tests (n = 62; 2.0%), nausea (n = 35; 1.2%), and diarrhoea (n = 26; 0.9%). Unexpected ADRs were reported in 293 (9.7%) patients. Two (0.1%) patients experienced serious ADRs (anaphylactic shock [grade 4] and cerebral infarction [grade 2]); both recovered. For cerebral infarction, the patient had an existing medical history of hypertension and hyperlipidaemia. The investigator assessed the causality as unclassified. A total of 9 patients permanently discontinued regdanvimab due to AEs which were evaluated as ADRs (3 cases of pyrexia, 3 cases of urticaria, and 1 each of anaphylactic shock, anxiety, and rash). No antibody-dependent enhancement events were reported.
Effectiveness
The rate of COVID-19 disease progression is summarised overall and by subgroup in Table 4. Overall, 378 (12.5%) patients in the effectiveness analysis set experienced disease progression after regdanvimab infusion. Extended hospitalisation period or hospital re-admission (lasting ≥ 24 h due to COVID-19) was the most commonly occurring reason for disease progression, occurring in 300 (9.9%) patients, followed by requirement for supplemental oxygen therapy (282 [9.3%] patients, 4 of whom [0.1%] required mechanical ventilation). Administration of remdesivir for the treatment of severe COVID-19, before remdesivir indication had been extended to patients with mild and moderate COVID-19 at high risk of progression to severe disease, was documented as the reason for disease progression in 242 (8.0%) patients. There were 10 (0.3%) patients who required ICU monitoring and 3 (0.1%) patients who died. Among 1030 patients who had mild COVID-19 before regdanvimab infusion, 68 (6.6%) experienced disease progression, compared with 310 (15.5%) of the 2006 patients who had moderate COVID-19 before regdanvimab infusion. Supplemental oxygen therapy was administered to 5.4% (56/1030) of patients with mild COVID-19 and 11.3% (226/2006) with moderate COVID-19. Among those who received oxygen therapy, 35.7% (20/56) of patients with mild COVID-19 and 65.0% (147/226) with moderate COVID-19 received regdanvimab > 3 days after symptom onset (Table S3).
The rate of disease progression by outbreak period was not substantially different between the pre-delta and delta subgroups (16.1% and 12.3%, respectively). Fewer progression events (3.4%) were observed during the omicron period; however, we note the small patient number in this group (Table 5). Similar disease progression rates were observed when using different criteria for disease progression. The rate of disease progression by SARS-CoV-2 variant was not significantly different between the alpha variant, delta variant, wild-type, and unknown variant subgroups (ranging from 12.1% to 16.7%, p = 0.755).
Significant differences were observed according to the presence or absence of high-risk factors, whether a vaccine was prescribed before administration of regdanvimab, and timing of administration. Among 2294 patients with ≥ 1 high-risk factor, 307 (13.4%) experienced disease progression, compared with 71 (9.6%) of the 742 patients without a high-risk factor (p = 0.006). Of the 875 patients vaccinated against COVID-19, 71 (8.1%) experienced disease progression, compared with 307 (14.2%) of the 2161 patients who were unvaccinated (p < 0.001). The proportion of patients experiencing progression was lower when regdanvimab was administered within 3 days of symptom onset (n = 149, 10.5%) compared with > 3 days after onset (n = 229, 14.2%; p = 0.002). Overall, in patients with mild COVID-19, no significant difference in disease progression was observed between patients who received regdanvimab within 3 days of symptom onset (7.0%) and patients who received regdanvimab later (6.1%). A higher proportion of patients with moderate COVID-19, who received regdanvimab 3 or more days after symptom onset, experienced disease progression (16.9%) compared with patients who received regdanvimab earlier (13.3%; p = 0.031) (Table 6).
Discussion
This phase 4 PMS study investigated regdanvimab in patients with high-risk mild or moderate COVID-19, confirmed by RT-PCR. Data from 3036 patients support the tolerability and effectiveness of regdanvimab in clinical practice. PMS studies are an integral component of the regulatory process and are mandated by the MFDS in Korea for any new approved drug product [34,35,36, 40]. Although randomised controlled trials are the gold standard for evaluating drug efficacy and safety, the strict eligibility criteria and restricted patient numbers can mean that factors such as rare ADRs and important drug–drug interactions are missed [34, 40]. PMS studies are especially important following the rapid development and approval of COVID-19 vaccines and therapeutics, and the additional data may help inform treatment decisions in the clinic [33, 41].
Analysis of regdanvimab treatment in this large real-world patient population did not reveal any new safety concerns. In total, AEs and ADRs were reported in 684 (22.5%) and 363 (12.0%) of patients, respectively. The most common ADR was increased liver function tests, observed in 62 (2.0%) of patients. Unexpected ADRs occurred in 293 (9.7%) of patients. Discontinuation of regdanvimab administration due to AE was in all cases related to IRR and was observed in only 9 patients (0.3%). IRRs are known to be commonly associated with mAb infusion [42]. In the present study, pruritus, urticaria, rash, and dizziness—AEs often related to IRRs—all occurred in < 1% of patients. Of the 3036 patients treated with regdanvimab, 2 reported serious ADR (anaphylactic shock and cerebral infarction), both of whom recovered.
The overall safety findings of this PMS study were consistent with those of previous controlled studies of CT-P59 and other therapeutic anti-SARS-CoV-2 antibodies [20, 21]. In the phase 3 study of regdanvimab (CT-P59), treatment-emergent AEs (TEAEs) were reported in 30.4% of patients. The most frequently reported treatment-related AEs in the regdanvimab group were liver enzyme increases and hypertriglyceridemia (both 1.1%). IRRs were reported for 0.6% patients treated with regdanvimab [20]. ‘Real-world’ evidence for other SARS-CoV-2 mAb therapies is limited, although evidence from clinical trials demonstrates comparable instances of TEAEs (up to 28.1%), with IRRs reported to be among the most frequent observations (up to 4.2%) [43, 44].
In general, similar to our PMS study, reports of serious adverse reactions following anti-SARS-CoV-2 mAb treatment in the clinic are few [45], and there are conflicting reports in the literature regarding the effect of mAb treatments for COVID-19 on cardiovascular events. Analysis of the US Food and Drug Administration Adverse Event Reporting System (FAERS) revealed an increase in hypertension events with casirivimab/imdevimab, bamlanivimab, bamlanivimab/etesevimab, and bebtelovimab, and ischaemic heart disease for casirivimab/imdevimab and bamlanivimab, but no association between cardiovascular events and treatment with sotrovimab or tixagevimab/cilgavimab [46]. In contrast, analysis of VigiBase, the safety database of the WHO, discovered an increased risk for arterial and venous thromboembolic events with tixagevimab/cilgavimab as compared with other anti-SARS-CoV-2 mAbs [47]. In our PMS study, no substantial new safety signals were identified. This is notable considering that cardiovascular disease was categorised in 1199 (39.5%) patients at baseline as one of the high-risk criteria, and only a small number (0.4%) of cardiac disorder ADRs were reported.
This PMS study suggests real-world effectiveness of regdanvimab for the treatment of mild-to-moderate COVID-19. A higher proportion of patients (12.5%) experienced disease progression compared with the 3.1% of patients who received regdanvimab in the phase 3 trial [20]. However, this should be considered in the context of the additional criteria used to define disease progression in the PMS study versus the phase 3 study. Progression for the PMS study included supplemental oxygen therapy, mechanical ventilation, extension of the hospitalisation period or re-admission after discharge, ICU monitoring, remdesivir administration, and death. In the phase 3 study, progression was defined as clinical symptoms requiring hospitalisation, oxygen therapy, or death. The proportions of patients who received regdanvimab and required supplemental oxygen therapy (9.3%) or hospital re-admission (9.9%) were higher in the PMS compared with the phase 3 trial (2.3% and 2.4%, respectively) [20]. According to treatment guidelines in South Korea, all SARS-CoV-2 confirmed positive cases are admitted to a residential treatment centre, and patients who progress to symptomatic COVID-19 are admitted to hospital for treatment and isolation. So, the PMS study was based on a hospitalised patient population where oxygen therapy can be given promptly and easily to patients who are in need, whereas the phase 3 study was conducted in ambulatory patients from 13 countries where differences in country-specific practices could have had an impact. In addition, extension of hospitalisation was also included as a progressive criterion in the PMS study, which might have been prolonged according to investigator’s judgement in addition to the aggravation of COVID-19 and could also be confounded by non-COVID-19 factors, such as comorbidities. Despite these differences, the mortality rates between the PMS study (0.1%) and phase 3 studies (0.2%) were similar [20].
Retrospective studies conducted in South Korea under similar conditions have reported a similar effectiveness of regdanvimab in preventing disease progression or reducing the need for supplemental oxygen compared with this PMS study [25, 26, 32]. In a retrospective cohort study with a primary endpoint of proportion of patients with mild-to-moderate COVID-19 who deteriorated with SpO2 ≤ 94% on room air up to day 28, 13.4% (17/127) progressed in the regdanvimab cohort compared with 39.5% (75/190) in the standard of care cohort [25]. A propensity-score-matched retrospective study reported that 7.1% (8/113) of patients with mild-to-moderate COVID-19 treated with regdanvimab progressed to severe/critical COVID-19 or died within 28 days of treatment compared with 16.1% (26/161) in the control group [26]. In another retrospective study, 8.1% (19/234) of patients with high-risk mild COVID-19 treated with regdanvimab required supplemental oxygen and 2.1% (5/234) progressed to severe disease compared with 18.4% (100/544) and 9.6% (52/544) of patients in the supportive care group, respectively [32]. Overall, the PMS study results align with those reported from a recent meta-analysis of randomised controlled and retrospective studies of regdanvimab. This meta-analysis found that morbidity (in terms of supplemental oxygen use and/or progression to severe disease) and mortality were improved for regdanvimab versus control [48].
The rate of COVID-19 disease progression was reduced if regdanvimab treatment was administered promptly, within 3 days of symptom onset. Notably, the subgroup analysis of patients who required oxygen therapy indicated that the median time from symptom onset to regdanvimab infusion exceeded 3 days in moderate patients. When considered in contrast with the generally early (≤ 3 days) administration of regdanvimab in patients with mild disease, this suggests that the higher rate of disease progression in moderate patients could be attributed to the delayed administration of regdanvimab following onset of symptoms. A similar observation was made in Japan following casirivimab/imdevimab administration: a sharp increase in disease progression was noted after day 5 of symptom onset [49]. Both studies support the early administration of mAb treatment for COVID-19.
The effectiveness of regdanvimab against the delta variant should be noted, especially as this variant has been associated with higher intrinsic severity and progression than other variants of SARS-CoV-2 [50]. However, these data should be interpreted with caution given the varying numbers of patients in each subgroup and the fact that for most (74.4%) patients the variant was unknown due to the sample survey method performed by the KDCA, although at the time of this study the delta variant was the predominant circulating variant in South Korea. Between July and December 2021, the delta variant accounted for almost 100% of COVID-19 cases in South Korea [39]. During the same period, a total of 2452 (81%) patients in this PMS study received regdanvimab, and therefore it is highly likely that many of the patients in the Unknown variant group were also infected with the delta variant. A retrospective study of over 700 patients with the delta variant treated with regdanvimab does support a clinical benefit for regdanvimab against this variant [51]. The proportion of patients requiring ICU monitoring after treatment with regdanvimab (0.3%) is lower than that observed in an analysis of a large real-world cohort of high-risk patients (n = 10,775) treated with bamlanivimab/etesevimab, casirivimab/imdevimab, or sotrovimab during the delta surge (1.0%, 1.0%, and 0.4%, respectively) [52]. A real-world effectiveness study in the United States of early mAb treatment for mild-to-moderate COVID-19 in over 2500 patients found lower rates of hospitalisation or death for a range of mAbs and presumed variants of SARS-CoV-2 (based on treatment date and circulating variants) compared with a propensity-score-matched non-treated control group (4.6% vs 7.6%, respectively) [53]. Only two patients in the PMS study were known to have been infected with the omicron variant, so it is difficult to make any conclusions with respect to the effectiveness of regdanvimab against omicron. In vitro studies have described a reduced sensitivity of neutralising antibodies, including regdanvimab, to omicron variants [54,55,56].
The emergence of further SARS-CoV-2 variants is to be expected, and the development of combination therapy strategies may be required to overcome possible resistance to mAbs [8]. Antibody cocktails have been used in the clinic, and more are in development, providing an important treatment option [8]. As described above, regdanvimab is not inferior to other mAbs in terms of safety and efficacy. Therefore, when considering single prescriptions or future antibody cocktails for treating COVID-19, regdanvimab can be included as an option.
This large phase 4 study in over 3000 patients implies that regdanvimab could be effective and tolerable in routine clinical practice. As all patients were prospectively observed in an inpatient setting, the possibility of under-reporting might be minimised. However, there are some limitations. Due to the open-label nature of the study, no comparison can be made to untreated patients. The study was conducted in a limited number of sites in South Korea only. In addition, the SARS-CoV-2 variant was unknown for the majority of patients. Overall, only 147 (5%) eligible patients in this study were diagnosed with COVID-19 during the omicron-dominant period.
Conclusion
Data reported herein are from the period after the approval of use of regdanvimab in South Korea, providing further insight into the safety and effectiveness of regdanvimab. The results of this phase 4 PMS study show that regdanvimab is effective, well tolerated, and has an acceptable safety profile in patients with high-risk mild or moderate COVID-19. These findings support those of earlier randomised controlled clinical trials and retrospective studies.
Data Availability
All data generated or analysed during this study are included in this published article/as supplementary information files.
References
Pollard CA, Morran MP, Nestor-Kalinoski AL. The COVID-19 pandemic: a global health crisis. Physiol Genomics. 2020;52:549–57.
Fernandes Q, Inchakalody VP, Merhi M, et al. Emerging COVID-19 variants and their impact on SARS-CoV-2 diagnosis, therapeutics and vaccines. Ann Med. 2022;54:524–40.
World Health Organization. Coronavirus disease (COVID-19) pandemic. 2023. https://www.who.int/europe/emergencies/situations/covid-19. Accessed 13 Apr 2023.
World Health Organization. WHO coronavirus (COVID-19) dashboard. 2023. https://covid19.who.int/. Accessed 10 Mar 2023.
Corti D, Purcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184:3086–108.
Forman R, Shah S, Jeurissen P, Jit M, Mossialos E. COVID-19 vaccine challenges: what have we learned so far and what remains to be done? Health Policy. 2021;125:553–67.
Liu M, Gan H, Liang Z, et al. Review of therapeutic mechanisms and applications based on SARS-CoV-2 neutralizing antibodies. Front Microbiol. 2023;14:1122868.
de Almeida Oliveira A, Praia Borges Freire D, Rodrigues de Andrade A, et al. The landscape of neutralizing monoclonal antibodies (nAbs) for treatment and prevention of COVID-19. J Pharm Innov. 2023. https://doi.org/10.1007/s12247-023-09713-w.1-19. (Epub Ahead of Print).
Wynia MK, Beaty LE, Bennett TD, et al. Real-world evidence of neutralizing monoclonal antibodies for preventing hospitalization and mortality in COVID-19 outpatients. Chest. 2023;163:1061–70.
Deng J, Heybati K, Ramaraju HB, Zhou F, Rayner D, Heybati S. Differential efficacy and safety of anti-SARS-CoV-2 antibody therapies for the management of COVID-19: a systematic review and network meta-analysis. Infection. 2023;51:21–35.
Seo JM, Kang B, Song R, et al. Preclinical assessment and randomized Phase I study of CT-P63, a broadly neutralizing antibody targeting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Emerg Microbes Infect. 2022;11:2315–25.
Chavda VP, Vuppu S, Mishra T, et al. Recent review of COVID-19 management: diagnosis, treatment and vaccination. Pharmacol Rep. 2022;74:1120–48.
Chinta S, Rodriguez-Guerra M, Shaban M, Pandey N, Jaquez-Duran M, Vittorio TJ. COVID-19 therapy and vaccination: a clinical narrative review. Drugs Context. 2023;12:2022-7–2.
Kim C, Ryu DK, Lee J, et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021;12:288.
Almagro JC, Mellado-Sanchez G, Pedraza-Escalona M, Perez-Tapia SM. Evolution of anti-SARS-CoV-2 therapeutic antibodies. Int J Mol Sci. 2022;23:9763.
Das NC, Chakraborty P, Bayry J, Mukherjee S. In silico analyses on the comparative potential of therapeutic human monoclonal antibodies against newly emerged SARS-CoV-2 variants bearing mutant spike protein. Front Immunol. 2021;12: 782506.
Ryu DK, Kang B, Noh H, et al. The in vitro and in vivo efficacy of CT-P59 against Gamma, Delta and its associated variants of SARS-CoV-2. Biochem Biophys Res Commun. 2021;578:91–6.
Ryu DK, Song R, Kim M, et al. Therapeutic effect of CT-P59 against SARS-CoV-2 South African variant. Biochem Biophys Res Commun. 2021;566:135–40.
Kumar S, Chandele A, Sharma A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021;17: e1009885.
Kim JY, Săndulescu O, Preotescu LL, et al. A randomized clinical trial of regdanvimab in high-risk patients with mild-to-moderate coronavirus disease 2019. Open Forum Infect Dis. 2022;9:ofac406.
Streinu-Cercel A, Săndulescu O, Preotescu LL, et al. Efficacy and safety of regdanvimab (CT-P59): a phase 2/3 randomized, double-blind, placebo-controlled trial in outpatients with mild-to-moderate coronavirus disease 2019. Open Forum Infect Dis. 2022;9:ofac053.
European Medicines Agency. Regkirona EPAR product information. 2021. https://www.ema.europa.eu/en/documents/product-information/regkirona-epar-product-information_en.pdf. Accessed 10 Mar 2023.
Ministry of Food and Drug Safety. MDFS grants marketing authorization for COVID-19 treatment, Regkirona Inj. 2020. https://www.mfds.go.kr/eng/brd/m_64/view.do?seq=49&srchFr=&srchTo=&srchWord=&srchTp=&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=1. Accessed 9 Mar 2023.
Syed YY. Regdanvimab: first approval. Drugs. 2021;81:2133–7.
Jang YR, Oh YJ, Kim JY. Clinical effectiveness of regdanvimab treatment for mild-to-moderate COVID-19: a retrospective cohort study. Curr Ther Res Clin Exp. 2022;96:100675.
Lee S, Lee SO, Lee JE, et al. Regdanvimab in patients with mild-to-moderate SARS-CoV-2 infection: a propensity score-matched retrospective cohort study. Int Immunopharmacol. 2022;106:108570.
Park S, Je NK, Kim DW, Park M, Heo J. Effectiveness and safety of regdanvimab in patients with mild-to-moderate COVID-19: a retrospective cohort study. J Korean Med Sci. 2022;37:e102.
Choi SJ, Park SW, Lee E. Effectiveness of regdanvimab at preventing the need for oxygen therapy in patients with mild-to-moderate COVID-19: a retrospective cohort study. Infect Chemother. 2022;54:91–101.
Kim T, Joo DH, Lee SW, Lee J, Lee SJ, Kang J. Real-world efficacy of regdanvimab on clinical outcomes in patients with mild to moderate COVID-19. J Clin Med. 2022;11:1412
Hong SI, Ryu BH, Hong KW, Bae IG, Cho OH. Real world experience with regdanvimab treatment of mild-to-moderate coronavirus disease-19 in a COVID-19 designated hospital of Korea. Infect Chemother. 2022;54:114–24.
Kwak YG, Song JE, Kang J, et al. Use of the monoclonal antibody regdanvimab to treat patients hospitalized with COVID-19: real-world data during the Delta variant predominance. Infect Chemother. 2022;54:781–6.
Lee JY, Lee JY, Ko JH, et al. Effectiveness of regdanvimab treatment in high-risk COVID-19 patients to prevent progression to severe disease. Front Immunol. 2021;12:772320.
Guo W, Pan B, Sakkiah S, et al. Informing selection of drugs for COVID-19 treatment through adverse events analysis. Sci Rep. 2021;11:14022.
Beaulieu-Jones BK, Finlayson SG, Yuan W, et al. Examining the use of real-world evidence in the regulatory process. Clin Pharmacol Ther. 2020;107:843–52.
Song H, Yim DS. Problems within the post-marketing surveillance system in Korea: time for a change. Transl Clin Pharmacol. 2016;24:63–5.
Ministry of Food and Drug Safety. Safety control after releasing medicinal products. 2021. https://www.mfds.go.kr/eng/brd/m_18/down.do?brd_id=eng0003&seq=71532&data_tp=A&file_seq=1. Accessed 13 Apr 2023.
Korea Disease Control and Prevention Agency. The response guidelines on COVID-19 for medical institution, version 1–2. 2020. https://www.kdca.go.kr/board/board.es?mid=a20507020000&bid=0019&act=view&list_no=711657. Accessed 3 Apr 2023.
Korean Ministry of Food and Drug Safety. Lekirona Inj. 960mg (regdanvimab) 2021. https://nedrug.mfds.go.kr/pbp/CCBBB01/getItemDetailCache?cacheSeq=202101124aupdateTs2023-03-25%2008:11:11.089797b. Accessed 1 June 2023. (Epub Ahead of Print).
Kim I, Park A, Lee H, et al. Status and characteristics of the SARS-CoV-2 variant outbreak in the Republic of Korea in January 2021. Public Health Wkly Rep. 2022;15:505–10.
Raj N, Fernandes S, Charyulu NR, Dubey A, G SR, Hebbar S. Postmarket surveillance: a review on key aspects and measures on the effective functioning in the context of the United Kingdom and Canada. Ther Adv Drug Saf. 2019;10:2042098619865413.
Younus MM, Al-Jumaili AA. An overview of COVID-19 vaccine safety and post-marketing surveillance systems. Innov Pharm. 2021;1210.24926/iip.v12i4.4294.
Doessegger L, Banholzer ML. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin Transl Immunol. 2015;4:e39.
Herman GA, O’Brien MP, Forleo-Neto E, et al. Efficacy and safety of a single dose of casirivimab and imdevimab for the prevention of COVID-19 over an 8-month period: a randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2022;22:1444–54.
Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325:632–44.
Goldin L, Elders T, Werhane L, Korwek K, Poland R, Guy J. Reactions and COVID-19 disease progression following SARS-CoV-2 monoclonal antibody infusion. Int J Infect Dis. 2021;112:73–5.
Zou J, Jing F. Cardiovascular adverse events associated with monoclonal antibody products in patients with COVID-19. Pharmaceuticals (Basel). 2022;15:1472.
Montastruc F, Lafaurie M, Flumian C, de Canecaude C. Increased reporting of venous and arterial thromboembolic events reported with tixagevimab-cilgavimab for coronavirus disease 2019. Clin Microbiol Infect. 2023;29:543.e1–3.
Yang M, Li A, Jiang L, Wang Y, Tran C, Ao G. Regdanvimab improves disease mortality and morbidity in patients with COVID-19: a meta-analysis. J Infect. 2022;85:e122–4.
Kadowaki T, Imajou S, Matsumoto N, Takao S, Yorifuji T. Timing of REGEN-COV administration and progression to severe COVID-19. J Infect Chemother. 2022;28:1459–63.
Varea-Jiménez E, Aznar Cano E, Vega-Piris L, et al. Comparative severity of COVID-19 cases caused by Alpha, Delta or Omicron SARS-CoV-2 variants and its association with vaccination. Enferm Infecc Microbiol Clin (Engl Ed). 2023. https://doi.org/10.1016/j.eimce.2022.11.021. (Epub Ahead of Print).
Jang YR, Oh YJ, Kim JY. Regdanvimab for patients with mild-to-moderate COVID-19: a retrospective cohort study and subgroup analysis of patients with the Delta variant. Int J Infect Dis. 2023;130:94–100.
Razonable RR, O’Horo JC, Challener DW, et al. Curbing the Delta surge: clinical outcomes after treatment with bamlanivimab-etesevimab, casirivimab-imdevimab, or sotrovimab for mild to moderate coronavirus disease 2019. Mayo Clin Proc. 2022;97:1641–8.
Kip KE, McCreary EK, Collins K, et al. Evolving real-world effectiveness of monoclonal antibodies for treatment of COVID-19: a cohort study. Ann Intern Med. 2023;176:496–504.
Planas D, Saunders N, Maes P, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602:671–5.
Iketani S, Liu L, Guo Y, et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature. 2022;604:553–6.
VanBlargan LA, Errico JM, Halfmann PJ, et al. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med. 2022;28:490–5.
Acknowledgements
We thank all patients and investigators involved in the study.
Medical Writing Assistance
Medical writing support, including the development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing, was provided by Duncan Campbell PhD, CMPP (Aspire Scientific Limited, Bollington, UK). Funding for medical writing support for this article was provided by Celltrion Inc. (Incheon, Republic of Korea).
Funding
This study and the journal’s Rapid Service fee was funded by Celltrion Inc. (Incheon, Republic of Korea).
Author information
Authors and Affiliations
Contributions
Sunghyun Kim, Keumyoung Ahn, Nahyun Jung, and Yeonmi Lee designed the study and, together with Yoobin Jung and Chankyoung Hwang, were responsible for data analysis and/or interpretation. Ji Yeon Lee, Seon Hee Bu, EunHyang Song, Seongcheol Cho, Sungbong Yu, Jungok Kim, Sungmin Kym, Kwang Won Seo, Ki Tae Kwon, Jin Yong Kim, and Sang Won Park acquired the data. All authors were responsible for manuscript preparation, review and revision, approved the final version for submission, and are accountable for the accuracy and integrity of the data presented.
Corresponding author
Ethics declarations
Conflict of Interest
Ji Yeon Lee has been an investigator on COVID-19 clinical trials sponsored by Daewoong Pharmaceuticals, Hyundai Bioscience, Shin Poong Pharmaceutical, and Ildong Pharmaceutical and on an investigator-initiated trial with Celltrion Inc. Seongcheol Cho has been an investigator on a COVID-19 clinical trial sponsored by Shin Poong Pharmaceutical. Jungok Kim and Sungmin Kym have been investigators on COVID-19 trials sponsored by Daewoong Pharmaceuticals, Hyundai Bioscience, Shin Poon Pharmaceutical, Ildong Pharmaceutical, ImmuneMed, Bioleaders, Dong-Wha, Pfizer, Gilead, and Celltrion Inc. Ki Tae Kwon has been an investigator on COVID-19 clinical trials sponsored by GC Holdings, Pfizer, Hyundai Bioscience, APRG Inc., Shin Poong Pharmaceutical, and Ildong Pharmaceutical and on an investigator-initiated trial with Celltrion Inc. Jin Yong Kim has been an investigator on COVID-19 clinical trials sponsored by Daewoong Pharmaceuticals, Enzychem Lifesciences, GC Pharma, Bukwang Pharmaceutical and Pfizer outside the scope of the submitted work, and on an investigator-initiated trial with Celltrion Inc. Sunghyun Kim, Keumyoung Ahn, Nahyun Jung, Yeonmi Lee, Yoobin Jung, and Chankyoung Hwang are employees of Celltrion Inc. Seon Hee Bu, EunHyang Song, Sungbong Yu, and Kwang Won Seo declare no interests related to this article. Sang Won Park has been an investigator on a COVID-19 clinical trial sponsored by Celltrion Inc. Seon Hee Bu was affiliated with Seoul Metropolitan City Bukbu Hospital at the time of the study, and is now affiliated with Kyung Hee University Hospital.
Ethical Approval
The study was conducted according to the ethical principles of the Declaration of Helsinki and in compliance with the International Council for Harmonisation Good Clinical Practice and applicable regulatory requirements. The study protocol was approved by the MFDS of the Republic of Korea. Per South Korean laws, additional approval from individual institutional review boards is not required for surveillance studies. All patients provided informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lee, J.Y., Bu, S.H., Song, E. et al. Safety and Effectiveness of Regdanvimab for COVID-19 Treatment: A Phase 4 Post-marketing Surveillance Study Conducted in South Korea. Infect Dis Ther 12, 2417–2435 (2023). https://doi.org/10.1007/s40121-023-00859-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00859-1