
Abstract
Cardiac fibrosis is closely associated with multiple heart diseases, which are a prominent health issue in the global world. Neurohormones and cytokines play indispensable roles in cardiac fibrosis. Many signaling pathways participate in cardiac fibrosis as well. Cardiac fibrosis is due to impaired degradation of collagen and impaired fibroblast activation, and collagen accumulation results in increasing heart stiffness and inharmonious activity, leading to structure alterations and finally cardiac function decline. Herbal plants have been applied in traditional medicines for thousands of years. Because of their naturality, they have attracted much attention for use in resisting cardiac fibrosis in recent years. This review sheds light on several extracts from herbal plants, which are promising therapeutics for reversing cardiac fibrosis.
Similar content being viewed by others
Cardiac fibrosis is the most common consequence after myocardial infarction and other cardiovascular conditions. |
Cardiac fibrosis is triggered by numerous risk factors and cytokines. |
Multiple signaling pathways are involved in cardiac fibrosis. |
Herbal plant extracts are promising therapeutics in treating cardiac fibrosis. |
Introduction
Cardiovascular diseases and sudden cardiac death are major health concerns globally, leading to significant hospitalization and about 17 million deaths annually [1]. Mounting evidence indicates that cardiac fibrosis occurs in multiple common cardiac diseases, such as myocardial infarction (MI), heart failure (HF), atrial fibrillation (AF), and diabetic cardiomyopathy. Most of these diseases are relevant to cardiac remodeling, including electrical remodeling and structure remodeling (fibrosis and hypertrophy). Collagen production and fibrogenesis are enhanced by neurohumor regulation after MI [2]. Commonly, HF is mostly caused by MI and approximately one billion cardiomyocytes are killed as a result of ischemia [3]. Triggered by inflammation, oxidative stress, hormone environment, and neuroregulation, fibrosis precipitates the development of HF and promotes sudden death because it facilitates cardiac inharmonious electrical and mechanical activities by increased cardiac hypertrophy and stiffness [4]. Arrhythmia is also closely related to cardiac fibrosis. AF is one of the most common arrhythmias in clinical practice. It is estimated that about 85% of patients with AF present atrial fibrosis [5]. Besides drugs, cardiac fibrosis can also be induced by hemodynamic alterations, ion imbalance, re-entry, and tachyarrhythmia [6]. The area around periphery of sleeves is most prominently affected by fibrosis because it is distant from the coronary blood supply [7].
This review is based on previously conducted studies and does not contain any new studies with human participants or animals performed by the author.
Development of Cardiac Fibrosis
Myocardium fibrosis is characterized by fibroblast proliferation, activation, and accumulation of extracellular matrix (ECM) [8]. Cardiac fibroblasts, which are motile cells with the shape of a flat spindle, derive from resident fibroblasts, bone marrow, endothelial cells, and epithelial cells, accounting for most cell populations in the heart. Cardiac fibroblasts participate in ECM production and maintain ECM homeostasis (including producing interstitial collagen I and III, less collagen IV, V, VI, laminin and elastin, glycoproteins, growth factors, and cytokines) in response to cytokines and mechanical stimulation [9]. In addition, fibroblasts provide an ideal mechanical scaffold between cardiomyocytes and other types of cells in the heart. Plenty of stretch-activated ion channels permeable to Na+, K+, and Ca2+ are expressed on cardiac fibroblasts, constituting a critical part of cardiac electrophysiology [10, 11]. Cardiac fibroblasts are elevated significantly during injuries [12]. With chronic stimulation of various cytokines, cardiac fibroblasts are likely to utilize a hypersecretion mode by transforming into myofibroblasts to function in tissue repair [13, 14]. Myofibroblasts normally do not occur in healthy myocardium unless injury or stress occurs. They possess combined features of fibroblasts and smooth muscle cells, and are typically characterized by abilities to migrate and contract due to the expression of α-smooth muscle actin (α-SMA) [15]. The persistent existence of myofibroblasts leads to excessive ECM production [13].
Homeostasis of collagen in myocardium is strictly controlled under normal conditions. Stimulated by injuries, cardiac fibroblasts and myofibroblasts become insensitive to regulatory mechanisms and collagen production is dramatically enhanced, even though these cells may undergo apoptosis [16]. Replacement fibrosis and reactive fibrosis are two major types of fibrosis occurring after MI. Replacement fibrosis in the myocardium is accompanied by a gradual loss of cardiomyocytes. It is critical to prevent cardiac rupture and increases in mechanical stress after MI. Scar formation is a good example [17, 18]. In contrast, reactive fibrosis is an adaptation without loss of cardiomyocytes. It originates from areas around vessels and spreads through the whole myocardium, preserving the pressure generated by the heart, changing chamber compliance, and compromising cardiac output by increasing ventricular stiffness, but it can progressively develop into replacement fibrosis [15, 17]. Overproduction of fibrosis impairs mechanic-electric coupling in myocardium, increasing susceptibility to arrhythmia. Besides, fibrosis causes myocardium stiffness and promotes the development of HF [19].
Common Causes of Cardiac Fibrosis
Ischemia
Evidence indicates that ischemia and infarction are direct injuries causing fibrosis [20]. At the early stage of cardiomyocyte death, neutrophils infiltrate into the infarcted area, triggering the activation of neurohormonal and intracellular signaling pathways. In the meantime, renin–angiotensin–aldosterone system (RAAS) and sympathetic nervous system are stimulated to promote endothelin-1 (ET-1) release, which further stimulates cardiac fibrosis and myocardial hypertrophy. Activation of matrix metalloproteinases (MMPs) promotes the degradation of collagens in ECM. In the late remodeling stage, transforming growth factor beta (TGFβ1) is increased and released from macrophages. It stimulates the transformation of fibroblasts into myofibroblasts and enhances type I and III collagen production, leading to fibrosis synthesis [2]. Formation of fibrosis and scar at infarcted and interstitial areas further prevents cardiac repair and increases susceptibility to arrhythmia.
Inflammation and Oxidative Stress
Acute death of cardiomyocytes often triggers inflammation and oxidative stress immediately. Inflammation clears ECM and dead cells around the infarcted area in preparation for the healing process. Simultaneously, cytokines like ET-1, interleukin-1 (IL-1), interleukin-6 (IL-6), platelet-derived growth factor (PDGF), and tumor necrosis factor alpha (TNFα) are secreted by macrophages and mononuclear cells. Besides, a large amount of ECM is produced by myofibroblasts to participate in the healing process and maintain the integrity of the chamber. After healing, reparative cells will undergo apoptosis and scars will be finally formed with cross-linked collagens [21]. Damaged ECM activates innate immune cells, identifying injury signals through transmembrane receptors, such as Toll-like receptors, stimulating proinflammatory cascades. The complement system is activated during myocardial infarction as well, which further triggers inflammation. Oxidative stress triggers the synthesis of free radicals resulting in cellular dysfunction, cell apoptosis, necrosis, and tissue damage. These free radicals are released from damaged antioxidant defenses and impaired mitochondrial metabolism. They are enhanced because of ischemia, tachycardiomyopathy, and pressure and volume overload. Infarcted myocardium promotes reactive oxygen species (ROS) production, stimulating recruitment of inflammatory cells and activating inflammation in the injured myocardium. ROS generated in oxidative stress might regulate intracellular signaling pathways with high specificity. Moreover, free radicals promote activation of leukocyte integrins and adhesion molecules, facilitating the expression of multiple chemokines and cytokines [21]. Metabolic oxidative products can be determined in body fluids, such as serum and urine. Accumulating evidence indicates that levels of metabolic products associated with the oxidative stress are positively correlated with severity of cardiac fibrosis in MI, HF, and AF [22,23,24]. Reduction of inflammatory factors and/or inhibition of oxidative stress can significantly reduce cardiac fibrosis in different heart diseases [25, 26]. Despite several large clinical trials showing that inhibition of complement and P-selectin did not benefit acute coronary syndromes, inhibition of inflammation and oxidative stress are still promising therapeutic strategies for cardiac fibrosis [27, 28].
Other Common Causes of Cardiac Fibrosis
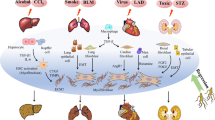
Infection, many cardiac conditions (arrhythmia, hypertension, dilated/restricted/hypertrophic cardiomyopathy, MI, HF, etc.), systemic autoimmune disease (sarcoidosis, amyloidosis, etc.), and aging are also the most common causes of cardiac fibrosis [29]. Moreover, genetic propensity plays a significant role in the development of cardiac fibrosis as well; for example, Japanese patients with sarcoidosis who have polymorphisms along with HLA-DQB1 (in particular DQB1*0601) are reported to have increased susceptibility for cardiac sarcoidosis, which is characterized by patchy cardiac fibrosis, myocardial granuloma, and lymphocytic infiltration [30] (Fig. 1).

The etiologies, risk factors, cytokines, and signaling pathways involved in cardiac fibrosis, and the functions herbal medicines exert in reversing cardiac fibrosis (this figure was made using www.biorender.com)
Major Chemokines Associated with Cardiac Fibrosis
Angiotensin II (Ang II)
Ang II is a predominant factor in the RAAS, taking part in many physiological and pathological processes [31]. A receptor for Ang II named Ang II type 1 receptor (AT1R) dominatingly coupled to Gq/11 protein, is responsible for promoting collagen production, hypertrophy, and cell proliferation and migration in fibroblasts, endothelial cells, and smooth muscle cells [32,33,34,35,36]. Triggered by different stimuli, AT1R may present different conformations. Mechanical stress can lead to the activation of AT1R directly, which stimulates cardiac fibroblast proliferation, ECM production, and fibrogenesis. This process can be suppressed by the Ang II receptor blocker candesartan [37]. Moreover, Ang II enhances TGFβ1 expression and activates TGFβ signaling to increase collagen production in cardiomyocytes and fibroblasts [31]. There is also a crosstalk between Ang II and TGFβ signaling to mediate collagen homeostasis. Studies show that cardiac fibrosis induced by angiotensin II could be ameliorated by improving vagal activity, AT1R blockers, and statins [38,39,40].
TGFβ
TGFβ has been extensively studied as a key regulator for fibrogenesis via the activation of fibroblasts. It mediates the fibrotic process in heart, lung, liver, and renal diseases [41,42,43,44]. Stimulated by cardiac injuries and tissue repair, TGFβ can be significantly upregulated and TGFβ signaling is activated to promote phosphorylation of Smad2/3, binding Smad4, translocating to the nucleus, and eventually promoting expression of fibrotic genes. TGFβ prominently increases the accumulation of ECM in the myocardium by inhibiting MMP expression [45, 46]. Downregulation of TGFβ by antibodies could suppress ECM expression and deposition [47, 48]. After cardiac infarction, TGFβ suppresses the inflammation in cardiac healing processes, regulating fibroblast phenotypes, and stimulating ECM deposition at the infarcted zone by increasing collagen synthesis and inhibiting the degradation of the matrix via the induction of protease inhibitors [45]. Moreover, TGFβ and ET-1 collaboratively induce the differentiation of myofibroblasts to stimulate cardiac fibrosis [49].
Connective Tissue Growth Factor (CTGF)
CTGF is a key regulator in cardiac fibrosis in response to cytokines and growth factors generated from injuries. It can be expressed in cardiomyocytes and cardiac fibroblasts [50]. Extensive studies demonstrate that CTGF is elevated notably in MI and HF in animals and humans [51,52,53,54]. CTGF is increased in other fibrosis-related disorders as well, such as idiopathic pulmonary fibrosis, chronic hepatitis, cirrhosis, diabetic nephropathy, and focal segmental glomerulosclerosis [55,56,57]. CTGF promotes fibroblast proliferation and ECM production [58]. It is positively correlated with the formation of cardiac fibrosis. CTGF is a downstream mediator of TGFβ, and TGFβ is a driving force for CTGF expression as well. CTGF synthesis is closely relevant to RhoA/ROCK, Ras/MEK/ERK, and ROS-related signaling pathways [59,60,61,62]. Blocking pro-fibrogenic pathways involving CTGF by antagonists is effective in inhibiting fibrosis in animal models [63]. CTGF seems to be a potential therapeutic intervention for cardiac fibrosis [51].
ET-1
ET-1 is an outstanding profibrotic peptide synthesized in endothelial cells and epithelial cells. Inflammatory cells and fibroblasts are also involved in ET-1 secretion. ET-1 has multiple biological functions. It regulates inflammation, cardiac contractility, water and sodium reabsorption, and vascular constriction [64, 65]. In recent studies, ET-1 has received much more attention because of its contribution to tissue fibrosis, especially in cardiac fibrosis. ET-1 binds to its receptors named endothelin-A (ETA) and endothelin-B (ETB) in cardiac cells, fibroblasts, endothelial cells, and smooth muscle cells to perform normal functions [65]. ET-1 can promote cardiac fibrosis via endothelial-to-mesenchymal transition (EMT) [66]. Besides, ET-1 promotes EMT by inducing TGFβ expression in cardiac development and fibrosis [67]. TGFβ upregulates ET-1 via the JNK signaling pathway, which accelerates the synthesis of ECM [13]. Blocking ET-1 signaling with endothelin receptor blockers like bosentan can inhibit cardiac fibrosis in animals with cardiac hypertrophy [68]. Clinical investigations into using ET-1 receptor blockade in treating heart failure are still ongoing.
PDGF
PDGF is a growth factor family comprising PDGF-AA, AB, BB, CC, and DD. PDGF has two receptors, α and β [13, 69]. During the healing process, enhanced expression of PDGF attracts macrophages, neutrophils, and fibroblasts to migrate to the wounded area [70]. PDGF induces differentiation of myofibroblasts and elevates the expression of collagens together with Ang II, TGFβ, and ET-1 [71]. After cardiac infarction, PDGF signaling is activated in perivasculature and mononuclear-like cells to facilitate cardiac repair [72]. Moreover, myocardial infarction can promote the expression of TGFβ, collagen I, and TIMP1 in a PDGF-dependent manner [73].
Osteopontin (OPN)
OPN is an acidic glycoprotein with an arginine–glycine–aspartic acid (RGD) sequence that can bind and interact with integrins and the CD44 receptor. OPN is expressed with a basal level in the adult heart and its expression will be increased during cardiac remodeling and heart failure. Current evidence indicates that OPN is closely associated with inflammation and fibrogenesis, which are critical in regulating cardiac fibrosis and wound healing after cardiac injury. Moreover, OPN expression is significantly increased during dilated and hypertrophic cardiomyopathy, MI, and HF; decreased OPN is closely related to improved heart function and reduced apoptosis in cardiomyocytes [74].
Periostin
Periostin is an adhesive matricellular glycoprotein secreted by activated cardiac fibroblasts via the stimulation of cytokines, mechanical stress, and the TGFβ signaling pathway. It is involved in cell proliferation, inflammation response, and tumorigenesis. It is also a crucial matricellular factor which participates in cardiac mesenchymal tissue development and cell–matrix crosstalk in myocardium during fibrogenesis. It contains a fasciclin domain that is highly produced by activated fibroblasts and released into the ECM after cardiac damage. Periostin can be expressed in epicardium, smooth muscle cells in vasculatures, and valvular interstitial cells, which can be used as targets for treating cardiac fibrosis. Moreover, it controls ECM assembly and plays indispensable roles in hearts from neonatal mice [75].
Secreted Protein Acidic and Rich in Cysteine (SPARC)
SPARC is a glycoprotein that has a high affinity calcium-binding domain and a strong affinity for collagen. It is involved in the activation of fibroblasts, collagen synthesis, and fibrogenesis, promoting growth factor signaling and facilitating angiogenesis. SPARC can induce TGFβ signaling and stimulate ADAMTS1, which can enhance collagen assembly and stabilization in cardiac fibrosis [76].
Thrombospondin (TSP)-1
Thrombospondins (TSPs) are major ECM glycoproteins that include five members. All TSP members have useful but complicated multidomains which allow interactions with numerous cytokines, chemokines, receptors, growth factors, and other ECM proteins. Among TSPs, TSP1 is the most well-known and investigated member. TSP1 exerts antiangiogenic activity and is able to activate TGFβ, which is a key regulator in profibrotic and anti-inflammatory processes. TSP1 regulates cardiac fibrogenesis through multiple ways, such as influencing collagen production, affecting activity of MMPs, activating TGFβ signaling, promoting myofibroblast differentiation, regulating cardiomyocyte apoptosis, and controlling myocardial contraction. TSP-1-deficient animal models have been evaluated for many cardiovascular conditions like MI, cardiac hypertrophy, and HF. TSPs are promising therapeutic targets for treating cardiac fibrosis [77].
Tenascin C (TNC)
TNC is a large extracellular matrix glycoprotein and transiently expressed in the heart at several important stages during embryonic development. It is weakly present in adult healthy hearts, but can be re-expressed, upregulated, and deposited in myocardium in a spatiotemporally constrained manner post cardiac injury (e.g., MI, HF). TNC participates in inflammation and fibrogenesis and can activate integrin signaling in macrophages to promote fibrogenesis. Although TNC knockout mice can present a grossly normal phenotype, multiple disease mouse models with TNC knockout have proved that TNC is a key regulator in controlling inflammation, tissue repair, and cardiac regeneration. TNC has both detrimental and beneficial effects in injured hearts. However, it seems that TNC exhibits more harmful effects because of its proinflammatory and profibrotic functions. Nevertheless, TNC is a promising diagnostic and prognostic biomarker for inflammatory heart diseases, and can be a potential therapeutic target for cardiac fibrosis [78].
Signaling Pathways Involved in Cardiac Fibrosis
TGFβ Signaling
TGFβ signaling is a critical mediator in the regulation of cell growth, proliferation, differentiation, cell injury, inflammation, immune function, and tissue repair [79]. It is critical in fibrogenesis, including cardiac fibrosis. The canonical TGFβ signaling pathway involves transcriptional activators of the Smad family [80]. Mammalian Smads are categorized into three groups, receptor-activated Smads (including R-Smads, Smad1, 2, 3, 5, and 8), inhibitory Smads (I-Smads, Smad 6 and 7), and common mediator Smads (Co-Smad, Smad4) [81]. When R-Smads and Smad2 and Smad3 are phosphorylated, the complexes formed by R-Samds, Co-Smad, and Smad4 will translocate to the nucleus, which further activates gene transcription [82]. Studies illustrate that TGFβ-Smad2/3 signaling could activate fibroblasts and promote ECM production in cardiac remodeling [13, 83]. Activation of TGFβ receptors mediates the activation of MAPK signaling, leading to high expression of p38, JNK1/2, and ERK1/2 signaling [84]. TGFβ can regulate the phenotypes and functions of fibroblasts. Stimulated by TGFβ, fibroblasts can be activated and transformed into myofibroblasts, characterized by synthesis of contractile proteins like α-SMA [85]. During the healing process and pathological conditions, myofibroblasts are activated from resident fibroblasts, pericytes, endothelial cells, and progenitor cells derived from bone marrow. The derivation of epithelial cells into myofibroblasts requires stimulation of TGFβ/Smad signaling [86]. In vivo experiments show that the TGFβ-Smad2/3 signaling pathway participates in pressure overload-induced cardiac fibrosis. Deletion of TGFβ receptor Tgfbr1/2 and Smad3 could notably reduce fibrosis induced by pressure overload. Loss of Smad2/3 decreases expression of fibrosis-related genes and attenuates ECM deposition [87]. The balance between ECM synthesis and accumulation is regulated by TGFβ. It elevates collagen I by activation of the Smad3-dependent pathway. TGFβ induces the generation of protease inhibitors to inhibit the activity of MMPs. Activation of TGFβ signaling leads to tissue fibrosis in many animals and human beings [83, 88]. Moreover, animal and human failing hearts present high expression of TGFβ [89, 90]. Inhibition of TGFβ/Smad signaling and EMT by drugs like pioglitazone or TGFβ receptor-specific antagonists can significantly alleviate cardiac fibrosis [91, 92]. Smad-dependent pathway (TGFβ/Smads, TGFβ/Sirtuins, TGFβ/BMP, TGFβ/miRNAs, TGFβ/MAPK) and Smad-independent pathway (TGFβ/PI3K/Akt, TGFβ/Rho/ROCK, TGFβ/Wnt/β-catenin) signaling is also indispensable in cardiac fibrosis [79].
Transcription Factor Nuclear Factor (NF)-κB Signaling
NF-κB signaling mediates cell growth, stress, and inflammation [93]. The effect of NF-κB on cell survival may be either pro- or anti-apoptotic according to different cell types and stimuli [94]. NF-κB is a key signal leading to induction of cell apoptosis in response to endoplasmic reticulum (ER) stress [95]. Mounting evidence indicates that NF-κB activity is enhanced in many cardiological diseases, and NF-κB signaling is strongly associated with cardiac fibrosis [96,97,98]. Clinical studies prove that NF-κB is activated in failing human hearts [99, 100]. NF-κB signaling inducing cardiac fibrosis and hypertrophy involves multiple mechanisms. It regulates microRNA expression to modulate fibrosis-associated gene expression [97]. NF-κB signaling facilitates cardiac fibrosis by ER stress as well [101]. Additionally, NF-κB signaling has crosstalk with inflammation signaling to induce cardiac fibrosis [102]. Inflammatory factor IL-6 is upregulated by arginine vasopressin in rat cardiac fibroblasts through regulation of the GRK2/NF-κB pathway [103]. Activation of NF-κB can elevate inflammatory factor interleukin-17 (IL-17)-induced MMP-1 in primary human cardiac fibroblasts to enhance cardiac fibrosis [104]. NF-κB signaling can strengthen cardiac fibrosis by interacting with oxidative stress. Inhibition of NF-κB signaling and ROS generation can decrease cell necrosis and apoptosis in myocardial ischemia/reperfusion [105]. Inhibition of NF-κB signaling by applying transgenic animals or pharmacological NF-κB antagonists was shown to be cardioprotective [106,107,108,109]. Activation of NF-κB can lead to adaptive cardiac hypertrophy, and inhibition of NF-κB can ameliorate cardiac hypertrophy induced by chronic pressure overload [110]. Inhibition of NF-κB can also attenuate Ang II-induced hypertrophy accompanied by altered expression of IL-6 receptor protein gp130 [111]. Suppression of the TLR4/NF-κB signaling pathway can also ameliorate cardiac fibrosis in mice with myocardial hypertrophy [112]. Thus, NF-κB signaling is important in regulating cardiac remodeling.
mTOR Signaling
The mammalian target of rapamycin, mTOR, plays a critical role in regulating mRNA translation and determining cell and organ size [113]. mTOR has two functional complexes: mTORC1 is sensitive to rapamycin and mTORC2 is insensitive to rapamycin. Studies indicate that inhibition of mTOR can activate expression of proinflammatory cytokines through NF-κB in immune cells [114]. Accumulating evidence indicates that mTOR activity and its associated signaling are activated in cardiac fibrosis and hypertrophy in response to Ang II, β-adrenergic stimulation, and insulin growth factor 1 [115,116,117]. Cardiac fibrosis induced by NADPH oxidase 4 is due to the activation of Akt/mTOR and NF-κB [118]. Cardiac fibrosis induced by leptin is also correlated with increased oxidation and the activation of the mTOR signaling pathway in cardiac myofibroblasts [119]. Blocking mTOR with rapamycin can decrease heart weight in models with hypertrophy, and mitigate the size increase of cardiomyocytes induced by Ang II and hydrogen peroxide [120]. Cardiac fibrosis induced by TGFβ1 can be inhibited by mediating the AMPKα/mTOR signaling pathway [121]. Suppression of mTOR mitigates cardiac fibrosis and hypertrophy induced by chronic pressure overload [113]. However, some studies point out that mTOR ameliorates adverse outcomes induced by pressure overload, and it plays a cardioprotective role by suppressing the release of cytokines and inhibiting NF-κB activity [122]. Moreover, when cardiac function deteriorates, mTORC1 is inactivated and HF may develop [123]. Thus, mTOR signal might play a complicated role in regulating cardiac fibrosis in a time- and space-dependent manner.
Mitogen-Activated Protein Kinase (MAPK) Signaling
The MAPK signaling cascade contains extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal protein kinase (JNK), and p38 mitogen-activated protein kinase (p38 MAPK). p38 MAPK contains four isoforms, p38α, p38β, p38γ, and p38δ. P38α is an important ingredient expressed in healthy hearts whereas p38β expression is much lower [124]. Extensive evidence demonstrates that the MAPK signaling cascade is involved in cardiac fibrosis [125,126,127]. p38α MAPK contributes to cardiac hypertrophy and p38 MAPK signaling is activated in cardiac fibrosis induced by MI [128]. Impaired cardiac function and ECM remodeling are also associated with the activation of the β2-AR-Nox-ROS-p38 MAPK axis [129]. ROS and p38 MAPK exert critical roles in cardiac fibrosis; ROS can trigger cardiac fibrosis by activating MMPs [130]. As part of MAPK signaling, the ERK1/2 signaling pathway is activated by ROS and EN-1 in cardiac fibrosis in dilated cardiomyopathy, and antioxidant agents could inhibit ROS production and ERK1/2 signaling to prevent cardiac fibrosis [131, 132]. Studies also believe that cardiac fibrosis and hypertrophy induced by pressure overload can be associated with the activation of JNK, ERK, and p38 MAPK. JNK activation is related to the reduction of gap junction, ERK activation is correlated with the growth of cardiomyocytes, and p38 MAPK activation is associated with enhanced cardiac fibrosis [133]. Thus, regulation of the MAPK signaling pathway is a promising method to treat cardiac fibrosis.
Na/K-ATPase Signaling
Na/K-ATPase, a canonical ion transporter on cell membranes mediating Na+ and K+ transport, functions as a signal transducer to prolong the activation of Na/K-ATPase signaling. There is about 700 pmol/g wet weight of Na/K-ATPase on cardiomyocytes in normal myocardium from the human left ventricle [134]. Interestingly, about 40% of Na/K-ATPase is lost in human dilated cardiomyopathy [135]. Studies prove that Na/K-ATPase is involved in cardiac fibrosis [136, 137]. Mutant rodents expressing a ouabain-sensitive form of Na/K-ATPase α1 subunit exposed to transverse aortic coarctation (TAC) were subject to earlier and severe cardiac fibrosis and hypertrophy compared to wild types. Mice treated with an ovine anti-digoxin antibody digibind show inhibition of cardiac fibrosis and hypertrophy [138]. Moreover, Na/K-ATPase inhibited by digitalis triggers myofibroblast differentiation via enhanced expression of COX-2 and stimulation of PKA [139]. These studies imply that Na/K-ATPase plays a critical role during cardiac fibrosis.
Wnt/β-Catenin Signaling
Wnt/β-catenin signaling is activated during numerous cellular responses, such as cell differentiation and proliferation, inflammation, ischemic injury, tissue repair, and scar formation. Numerous studies have shown that Wnt/β-catenin signaling participates in cardiac fibrosis. It is reported that the Wnt signaling can promote fibroblast activation and proliferation [140]. Wnt signaling participates in heart development and is quiescent in normal adult cardiomyocytes. However, it becomes reactivated in many cardiovascular conditions, such as cardiac hypertrophy, or cardiac fibrogenesis [141].
Receptors/Ion Channels Involved in Cardiac Fibrosis
Ryanodine Receptor Type 2 (RyR-2)
RyR-2, the most important Ca2+ release channel in the sarcoplasmic reticulum in cardiomyocytes, is involved in cardiomyocyte contraction and cardiac hypertrophy. Mechanical stretch can enhance the expression of TGFβ1 and induce fibrosis in cultured cardiomyocytes from neonatal rats. Knockdown of RyR-2 can significantly reduce TGFβ1 expression in cardiomyocytes and suppress collagen gene expression in cardiac fibroblasts with mechanical stretch, suggesting that RyR-2 promotes cardiac fibrosis through regulating TGFβ1 under mechanical stretch [142]. Another study indicated that RyR-2± mice exhibited less compensated cardiac fibrosis, hypertrophy, and contractility under pressure overload compared with wild-type mice, suggesting that RyR-2 contributes to cardiac fibrosis and hypertrophy through regulation of Ca2+ release from the sarcoplasmic reticulum [143].
Transient Receptor Potential Cation Channel 3 (TRPC3)
TRPC proteins, which are non-selective Ca2+ permeable channels, are involved in maladaptive cardiac remodeling. TRPC3 plays a positive role in the regulation of ROS in the heart. It can modulate fibroblast activities and functions in rats with AF [144]. A membrane-bound ROS enzyme named NADPH oxidase 2 (Nox2) promotes fibrosis by regulating TGFβ and Ang II-mediated Akt and Wnt signaling pathways in cardiomyocytes and cardiac fibroblasts. TRPC3 reduces fibrosis by stabilizing Nox2. TRPC3 interacts with Nox2 physically at C-terminal sites of TRPC3 and inhibits Nox2 from proteasome-dependent degradation. Nox2 stabilizes TRPC3 proteins to intensify TRPC3 channel activity. TRPC3 enhances Nox2-induced ROS production in cardiomyocytes from neonatal rats [145, 146]. Moreover, peptides at TRPC3 C-terminal abolish TRPC3-mediated ROS synthesis [146, 147]. Studies indicate that inhibition of TRPC3 pharmacologically can significantly attenuate cardiac reactive fibrosis and left ventricular diastolic dysfunction in mice with dilated cardiomyopathy [148]. Moreover, blockage of TRPC3 and TRPC6 together with genetic deletion and selective small molecules in mice and rats can prominently suppress pathological cardiac fibrosis and hypertrophy induced by sustained pressure overload [149]. These results suggest TRPC3 might be a potential therapeutic target for fibrosis reversal.
Sphingosine 1-Phosphate Receptor (S1PR)
S1P is a bioactive lipid and lysophospholipid mediator exerting regulation of cell proliferation, differentiation, and migration. S1P plays a cardioprotective role in many animal models and exerts pro- and antifibrotic roles during injuries depending on different cell types [150]. S1P promotes the transformation of myofibroblasts by activation of the TGFβ signaling pathway. TGFβ promotes sphingosine kinase 1(Sphk1) expression, and inhibition of Sphk1 may decrease S1P in the blood. Silent Sphk1 notably decreases collagen production induced by TGFβ [151, 152]. Neutralization of S1P in the extracellular space with specific antibodies inhibits collagen production induced by TGFβ [152]. Besides, overexpression of Sphk1 promotes fibrosis and degeneration of cardiomyocytes [153]. The binding between S1P and its G-protein receptor sphingosine 1-phosphate receptors (S1PRs) activates signaling involved in cardiac fibrosis [154]. S1PRs have five subtypes, of which S1PR1 is the most abundant in the myocardium. S1PR on cardiomyocytes is involved in hypertrophy and cardiac protection. Activation of S1PR can inhibit cAMP production and reduce adrenergic receptor-induced contractility [155]. Activation of S1PR on fibroblasts can mediate migration and proliferation which are necessary for fibrosis and cardiac remodeling. Activation of cardiac S1PR also affects heart rate and cardiac contractility, and promotes hypertrophy, protecting the heart from ischemia. S1PR knockout mice display embryonic lethality and exhibit progressive heart failure [156]. However, excessive exposure to S1PR agonists may result in a vascular leak and fibrosis [157].
Other ion channels are also involved in cardiac fibrosis, such as sarcoendoplasmic reticulum calcium ATPase (SERCA) in cardiomyocytes; Ca2+ release-activated Ca2+ (CRAC) channels; potassium channels; sodium channels, and voltage-gated Ca2+ channels (VGCCs) in myocardial fibroblasts [158]. Thus, modifying and targeting ion channels in cardiomyocytes and cardiac fibroblasts are potential ways to regulate cardiac fibrosis after cardiac injury.
Noncoding RNAs Involved in Cardiac Fibrosis
MicroRNAs
MicroRNAs are a class of small noncoding RNAs with a length of about 22–25 nt. They participate in the vast majority of biological and pathological processes involved in cell proliferation, cell apoptosis, organ development, and disease progression. Recent studies demonstrate that microRNAs contribute to the progression of cardiac hypertrophy, cardiac fibrosis, and angiogenesis under cardiomyocyte injuries in many cardiac diseases. MicroRNAs can act as both positive and negative regulators for cardiac fibrosis [159]. A recent study shows that miR-130a is significantly increased in mice infused with angiotensin II and human failing hearts. Upregulation of miR-130a in cardiac fibroblasts enhances profibrotic response, and downregulation of miR-130a can reverse this phenotype [160]. miR-133 is a negative regulator of fibrosis by downregulation of CTGF [161]. miR-433 is shown to be a trigger of cardiac fibrosis through inhibition of antizyme inhibitor 1 (AZIN1) and JNK1 in cardiac fibroblasts [162]. In addition, miR-1, miR-133a, miR-199, and miR-21 increase the expression of TGFβ/Smad3 by downregulation of AZIN1, whereas miR-15, miR-199, miR-21, miR-29, and miR-208 can decrease the expression of JNK1, leading to increase of pSmad3 expression. These microRNAs are critical in regulating fibrosis synthesis and degradation in the myocardium [163].
Long Noncoding RNAs (lncRNAs)
LncRNAs are another class of noncoding RNAs with lengths over 200 nt and seldom encode proteins. Mounting evidence states that lncRNAs are involved in multiple biological and pathological conditions at transcriptional, post-transcriptional, and translational levels [164, 165]. Recent studies indicate that lncRNAs can modulate cardiac fibrosis as well. LncRNA Wisp2 super-enhancer-associated RNA (Wisper) is enriched in cardiac fibroblasts. Expression of Wisper is positively correlated with cardiac fibrosis-related markers collagen type 1 alpha 1 chain (COL1A1) and collagen type III alpha 1 chain (COL3A1) in both MI models in murine and human heart with aortic stenosis. Besides, Wisper is important in regulating cardiac fibroblast proliferation and migration. Downregulation of Wisper attenuates fibrosis induced by MI [1]. IL-17 is involved in the pathological formation of cardiac fibrosis. A recent study demonstrates that lncRNA AK081284 is upregulated in cardiac fibroblasts treated with IL-17, and increased AK081284 enhances collagen and TGFβ production [166]. Another study clarifies that miR-455 is negatively correlated with the expression of collagen I and III, and miR-455 could downregulate the expression of CTGF. Moreover, mitigating expression of lncRNA H19 can notably upregulate miR-455, leading to attenuated expression of CTGF [167]. Another lncRNA named growth arrest-specific 5 (GAS5) can regulate cardiac fibrosis as well. GAS5 is downregulated and miR-21 is increased in cardiac fibrosis tissue. Elevating GAS5 expression suppresses cardiac fibroblast proliferation by mitigating the expression of miR-21 through regulation of the PTEN-MMP-2 signaling pathway [168]. As lncRNAs are also involved in cardiac development and fibrogenesis, more underlying mechanisms should be further determined, an they are potential therapeutic targets involved in cardiac fibrosis.
Circular RNAs
Circular RNAs are a group of noncoding RNAs important in cellular biology. However, their functions in cardiac fibrosis and cardiac regeneration are largely unknown. In recent years, circular RNAs have been reported to regulate cardiac repair. A study demonstrated that mitochondria-derived ROS production can be regulated by a mitochondria-localized circular RNA named circSamd4, which are expressed in fetal and neonatal cardiomyocytes. Moreover, the transcription factor Nrf2 can regulate the expression of circSamd4 to control oxidative stress in mitochondria. Upregulation of circSamd4 can induce proliferation and prevent apoptosis in cardiomyocytes, and reduce fibrogenesis after MI [169].
Mitochondria Are Involved in Cardiac Fibrosis
Mitochondria are one of the most critical cell organs in cardiomyocytes. They account for one-third of the cell volume and participate in ATP production and energy balance to maintain the normal contraction and dilation of the heart. Different cardiac injuries can result in mitochondrial membrane damage, ion channel opening, mitochondrial membrane potential loss, mitochondrial permeability transition pore (mPTP) opening, water and sodium entering mitochondria leading to mitochondrial swelling and breakage, and activation of apoptosis. Moreover, mitochondrial DNA can be damaged and plenty of ROS can be produced, which can negatively control the TCA cycle and ATP production. Moreover, multiple signaling pathways are activated during this process to promote cardiac fibrosis.
Promising Herbal Plants in Inhibition of Cardiac Fibrosis
As fibrosis occurs in numerous heart diseases, therapeutic strategies targeting cardiac fibrosis are promising treatments for cardiac diseases. In recent decades, increasing attention is paid to plant components because of their safety and easy accessibility. Accumulating evidence indicates that multiple traditional herbs restrain or reverse fibrosis via anti-inflammation, anti-oxidative stress, and relevant signaling. These findings indicate that herbal plants are promising in treating cardiac fibrosis.
Andrographolide
Andrographolide, a major botanical compound from a medical herb named Andrographis paniculata, has been used in Chinese traditional medicine for decades. Accumulating studies illustrate that andrographolide has multiple biological functions, e.g., it can protect against inflammation [170], oxidative stress [171], and hyperglycemia [172]. Andrographolide can also alleviate liver fibrosis by activation of the Nrf2-associated antioxidant pathway, and it inhibits fibrogenesis and inflammation in non-alcoholic steatohepatitis [173,174,175]. Andrographolide treatment can ameliorate pulmonary fibrosis by decreasing the level of oxidative stress via reducing malondialdehyde (MDA) and increasing the glutathione/oxidized glutathione ratio, and decreasing the production of collagen I and collagen III [176, 177]. Andrographolide also attenuates diabetic nephropathy by reducing oxidative stress and inflammation mediated by the Akt/NF-κB pathway [178]. Wu et al. reported that administration of andrographolide for 7 weeks orally (25 mg/kg/day) significantly improves cardiac function and attenuates cardiac fibrosis in mice with cardiac hypertrophy induced by aortic banding. Andrographolide mitigates transcription of hypertrophy-related genes (ANP, BNP, and β-MHC), and fibrosis-associated genes (collagen I, collagen III, TGFβ, and CTGF). Cardiomyocytes treated with andrographolide exhibit a blunt response to angiotensin II. Andrographolide prevents activation of cardiac fibroblasts and cardiac hypertrophy in mice through inhibiting the MAPK signaling pathway [179].
Arctiin
Arctiin is an active ingredient from Arctium lappa L. in traditional Chinese medicine. Evidence shows that arctiin is administered to fight against viral infection [180] and cancers [181]. Arctiin can inhibit inflammation by suppressing COX-2 expression via the NF-κB signaling pathway [182]. Arctiin can reduce serum glucose and hemoglobin levels in rats with diabetic mellitus, decreasing the incidence of diabetic retinopathy [183]. It reduces endoplasmic reticulum stress-induced EMT of podocytes [184]. It is also effective in attenuating glomerulosclerosis and albuminuria by regulation of nephrin and podocin expression [185]. Arctiin aglucone arctigenin inhibits renal interstitial fibrosis in rats with obstructive nephropathy by decreasing collagen deposition, epithelial atrophy, and tubular dilatation via downregulating inflammatory cytokines TNFα, interferon-γ (IFNγ), and interleukin-1β (IL-1β) and upregulating anti-oxidation manganese superoxide dismutase, implying that arctiin has potential in treating renal fibrosis [186]. Recently, Li et al. discovered that actiin can prevent pressure-overload and phenylephrine-induced cardiac hypertrophy in mice by inhibition MAPKs and AKT signaling [187]. These studies indicate that arctiin might play an anti-fibrosis role in heart diseases.
Arjunolic Acid
Arjunolic acid is one of the elementary bioactive ingredients of arjuna extracts. Purified arjunolic acid has multiple biological functions, such as anti-oxidative stress, anticoagulant, anti-apoptosis, and anti-inflammation [188,189,190]. Arjunolic acid restrains intracellular ROS-dependent JNK-p38 and p53-mediated cardiac apoptosis induced by doxorubicin [191]. Arjunolic acid can reduce cardiac toxicity induced by sodium nitrite by balancing cell apoptosis [191]. In addition, arjunolic acid inhibits collagen expression and elevates cardiac function in cardiac hypertrophy, because it can bind and stabilize its ligand binding domain named peroxisome proliferator-activated receptor alpha (PPARα). Moreover, it promotes PPARα expression and represses TGFβ signaling specifically by suppressing TGFβ activated kinase 1 (TAK1) phosphorylation in cardiac hypertrophy. PPARα N-terminal transactivation domain (AF-1) interacts with TAK1 directly, thereby masking TAK1 kinase domain. The level of arjunolic acid-induced PPARα-bound TAK1 presents an inverse correlation with TAK1 phosphorylation and subsequently reduces activation of p38 MAPK and NF-κBp65, ameliorating excess collagen synthesis in cardiac hypertrophy. This study implies that arjunolic acid is a PPARα agonist which could inactivate non-canonical TGFβ signaling in cardiac fibrosis [192].
Astragaloside IV
Astragaloside IV is a bioactive ingredient in Astragalus membranaceus Bunge (Fabaceae). Mounting evidence indicates that astragalus resists cell apoptosis, oxidative stress, and viral infection. Astragaloside is identified to prevent hepatic fibrosis by suppressing the PAR2 signaling pathway in diabetic rats and inhibiting the Notch signaling pathway in rats with cholestatic liver fibrosis induced by common bile duct ligation [193, 194]. Astragaloside IV effectively restrains pulmonary fibrosis in rats induced by bleomycin via mitigating ECM deposition [195]. Moreover, astragaloside IV can prevent glucose-mediated podocyte apoptosis and protects against renal fibrosis by inhibition of TGFβ1-induced fibrosis in mouse renal fibroblasts through suppression of MAPK and NF-κB signaling pathways [196, 197]. Astragaloside IV alleviates myocardial ischemia reperfusion-induced cardiac injury by activating HIF-1α signaling and energy regulation [198, 199]. Another study proves that astragaloside IV could target calcineurin, angiotensin-converting enzyme, and c-JNK to block calcium influx to exert a protective role in cardiac diseases [200]. Lu et al. found that astragaloside IV could decrease the expression of PAR1, PAR4, NF-κB, and inhibit TGFβ/p-AKT/p-GSK-3β signaling and cell apoptosis in cardiac fibroblasts. Moreover, astragaloside IV can reduce cardiac fibrosis, and improve cardiac functions in rats with diabetic cardiomyopathy [201]. In addition, cardiac fibrosis induced by isoprenaline can be inhibited by astragaloside IV via inhibition of ROS expression, phosphorylation of profibrotic family members of MAPKs, extracellular signal-regulated kinase, p38 MAPK, and JNK; cardiac fibrosis could be inhibited by astragaloside IV by suppression of ROS-induced MAPK activation [202]. Studies also demonstrate that astragaloside IV can prevent isoproterenol-induced cardiac hypertrophy by modulating NF-κB/PGC-1α signaling [203]. Astragaloside IV can attenuate cardiac fibrosis in coxsackievirus B3-induced cardiomyopathy by inhibiting the TGFβ1 signaling pathway [204].
Baicalin (BA)
BA, a major active flavonoid ingredient in skullcap, is reported to have antioxidant, anti-inflammation, and anti-fibrosis functions. Studies show that baicalin could attenuate liver fibrosis in nonalcoholic fatty liver disease (NAFLD) mice by inhibiting the expression of inflammation-related factors like TNFα, IL-1β, and MCP-1, suppressing macrophage influx and nuclear factor-κB, and inhibition of α-SMA, TGFβ1, and Col1A1 [205]. BA has also been shown to protect against heart and vascular injuries. BA attenuates atherosclerosis by lipid regulation and inhibition of dendritic cells in ApoE−/− mice [206, 207]. BA also exerts a cardiac protective role in ER stress-induced cardiomyocyte apoptosis, cardiac infarction, and hypoxic pulmonary hypertension [208,209,210]. In chronic pressure overload-induced cardiac hypertrophy mice models, BA can induce the expression of PPARα and mitigate cardiac fibrosis and cell apoptosis [211]. In cardiac fibroblasts treated with angiotensin II, BA inhibits cell proliferation and collagen production by reducing the expression of fibronectin and CTGF. Moreover, BA inhibits cardiac fibrosis by regulating AMPK/TGFβ/Smads signaling [212].
Corydalis hendersonii Hemsl. (CH)
CH mainly grows in northern temperate regions of Tibet with an altitude of 4200–4500 m [213]. It is a well-described folk medicine with effects of clearing heat and detoxifying. It has been applied for the treatment of high-altitude polycythemia for centuries in Chinese traditional medicine [214]. It is also used for therapeutic treatments of hepatitis, hypertension, gastritis, edema, and infectious diseases [213]. Bai et al. showed that CH treatment in mice with acute myocardial infarction (AMI) has a dose-dependent cardioprotective effect. It decreases left ventricular end-diastolic diameter (LVEDs), improves EF compared to those without CH treatment; CH alleviates the increase of LDH and CK-MB in serum; reduces plasma inflammation factors like Ang II, TNFα, IL-6, and IL-1β and expression of cardiac MMP-2 and MMP-9. Moreover, CH decreases p-p65, p-IκBα, p-JAK2, p-STAT3, MMP-2, and MMP-9 in myocardium from mice with AMI. CH also reduces inflammatory cell infiltration around infarcted areas and inhibits platelet aggregation. Thus, CH exerts as a cardiac protector against MI by inhibition of myocardial fibrosis, inflammation, and platelet aggregation via NF-κB and JAK2-STAT3 signaling [213].
Kaempferol
Kaempferol is a remarkable component of Kaempferia exerting anti-inflammatory and anti-oxidative stress effects in many diseases. Kaempferol is abundant in apples, beans, citrus, strawberries, and tea [215]. Kaempferol is very important because it regulates cell apoptosis, cell cycle, and inflammation in cancer cells [216,217,218]. Kaempferol shows many benefits in treating atherosclerosis, hyperlipidemia, and coronary heart diseases owing to its anti-inflammatory and anti-oxidative stress properties [219]. It reduces fibrosis formation induced by angiotensin II as it inhibits Ang II- or TGFβ-induced EMT and suppresses the proliferation and activation of cardiac fibroblasts, which can alleviate cardiac fibrosis production [220]. Kaempferol mitigates cardiac injuries induced by hyperglycemia through the alleviation of inflammation and oxidative stress. It protects myocardium of diabetic cardiomyopathy by suppressing NF-κB nucleus translocation and exciting a nuclear factor named erythroid 2 p45-related factor-2 (Nrf2). Kaempferol also prevents cardiac fibrosis and apoptosis in streptozotocin (STZ)-induced diabetic mice [221]. Kaempferol can reduce cardiac fibrosis and hypertrophy, and improve cardiac functions in mice treated with aorta banding by modulation of oxidative stress and the ASK1/MAPK signaling pathway [219]. Another plant containing kaempferol named Boerhavia diffusa also exerts a cardioprotective role in rats with Ang II-induced cardiac fibrosis and hypertrophy. It reduces ANP and BNP expression and cardiac mass index induced by Ang II and upregulates endogenous antioxidant enzymes with elevated translocation of Nrf2 from cytoplasm to the nucleus. Moreover, it attenuates cardiac fibrosis significantly by decreasing the lipid and protein oxidation induced by Ang II [222]. These results demonstrate that kaempferol has potential for treating cardiac fibrosis.
Matrine
Matrine, an extract from Chinese traditional medicine named Kushen (Sophora alopecuroides L.), exerts anti-inflammatory and anti-oxidative stress effects in many diseases [223, 224]. It exhibits an anti-fibrosis role by inhibiting the TGFβ/Smad signaling pathway in liver and renal fibrosis [225, 226]. Zhang et al. discovered that matrine is a negative regulator of the TGFβ signaling pathway. Administration of matrine improves left ventricle function, heart compliance, and reduces cardiac fibrosis via inhibition of the TGFβ1/Smad signaling pathway in diabetic cardiomyopathy rat models. High glucose increases collagen production by activation of the TGF-1/R-Smad signaling pathway and suppression of I-Smad signaling in cultured cardiac fibroblasts. However, treatment with matrine at non-cytotoxic concentrations of glucose without affecting I-Smad in cardiac fibroblasts, matrine blocks TGFβ1/R-Smad signaling transduction and represses collagen production and deposition [227]. Liu et al. also found that giving 200 mg/kg/day matrine for 10 consecutive days to rats with diabetic cardiomyopathy could attenuate cardiac fibrosis and mitigate the damage of cardiac function via inhibiting ATF6 signaling, which causes accumulation of intracellular calcium and activation of ECM expression triggered by NFAT. Moreover, cardiac fibroblasts treated with matrine and high glucose could significantly decrease the activity of calcineurin and reduce the expression of fibronectin and collagen I compared with cardiac fibroblasts treated with high glucose alone [228]. All these studies suggest that matrine plays a significant cardioprotective role in cardiac fibrosis.
Myricitrin
Myricitrin is a major flavone component in the root bark of Myrica esculenta, Myrica cerifera, Ampelopsis grossedentata, and some other plants. Some analysis indicates that myricitrin inhibits apoptosis of retinal pericytes induced by high glucose [229]. Myricitrin regulates the production of NADPH oxidase-dependent ROS, which can inhibit endotoxin-induced inflammation through inhibiting JAK/STAT1 and NOX2/p47phox signaling pathways [230]. Moreover, myricitrin can protect against hypoxia/reoxygenation-induced injury in cardiomyocytes [231]. Myricitrin mitigates high glucose-mediated apoptosis of H9C2 myocardiocytes by activation of the Akt-Nrf2 signaling pathway [232]. Another study elucidated that myricitrin prevents against cardiotoxicity induced by doxorubicin via inhibiting oxidative stress and mitochondrial apoptosis through the ERK/P53 pathway [233]. Besides, pretreatment with myricitrin prominently decreases advanced glycation end product (AGE)-induced TGFβ1 and collagen I expression, mitigates ROS increase, and inhibits cell apoptosis and fibrosis in H9C2 cells through Nrf2 activation and NF-κB inhibition. Oral administration of myricitrin to STZ-induced diabetic mice with a concentration of 300 mg/kg/day for 8 weeks can significantly improve heart function and myocardial fiber arrangement. Furthermore, fibrosis and collagen accumulation are significantly reduced in myocardium. Myricitrin can significantly increase anti-oxidation enzymes Nrf2, HO-1, and NQO-1, and decreases inflammation related IL-6 and TNFα genes. Apoptosis of cardiomyocytes is attenuated by myricitrin via activation of Akt and inhibition of ERK signaling. These findings suggest that myricitrin protects myocardium through inhibiting apoptosis of cardiomyocytes and reducing fibrosis synthesis by inhibition of inflammation, oxidative stress, and apoptosis [234].
Nigella sativa
Nigella sativa is a protective traditional medicine applied in many Asian countries. It is reported N. sativa helps resist inflammation, cancer, and fights against immune disorders and parasitic diseases [235]. N. sativa is reported to attenuate pulmonary fibrosis induced by bleomycin in rats [236], and it demonstrated an anti-fibrosis effect in liver, oral, and renal fibrosis in different animal models and clinical practice [237,238,239]. N. sativa (2.5, 5, and 10 ml/kg) administered to rats for 12 consecutive weeks while receiving 80 mg/kg sodium nitrite orally prominently reduced sodium nitrite-induced increases of serum urea and creatinine, and significantly reduced the expression of fibrosis markers like MCP-1 and TGFβ1. N. sativa can ameliorate nephrotoxicity induced by sodium nitrite via inhibition of oxidative stress, restoration of fibrosis and inflammation, amelioration of cytochrome c oxidase, and attenuation of apoptosis [237]. Norouzi et al. found that N. sativa reduces heart IL-6 and TNFα expression, ameliorates expression of the oxidative stress marker MDA and collagen production in myocardium from rat inflammation models induced by lipopolysaccharide in a concentration-dependent manner. After treatment with N. sativa, the total thiol, superoxide dismutase (SOD), and catalase are increased. Cardiac fibrosis is also decreased by N. sativa in a concentration-dependent manner. All these experiments suggest that N. sativa mitigates cardiac fibrosis by affecting oxidative/antioxidative balance, and increasing antioxidative enzymes [240].
Roselle
Roselle, also known as Hibiscus sabdariffa L., is often consumed as juice or hot tea because of its nutraceutical benefits. It is an antioxidant and could protect insulin-resistant rats against hyperglycemia and hyperlipidemia [241]. A study indicates that rosemary extract can inhibit pulmonary fibrosis induced by bleomycin [242]. Roselle extract can also protect against cardiac oxidative damage in diabetic rats. Si et al. find that roselle could improve heart functions in obese rats with MI, as it increases left ventricular diastolic pressure (LVDP), LVdP/dtmax, LVdP/dtmin, coronary flow, and rate pressure product (RPP). Besides, roselle decreases collagen production, ANP, BNP expression, fibrosis production, and inhibits oxidative stress by reducing Nox2 expression, enhancing SOD enzyme activity and increasing GSH concentration [243]. Moreover, roselle attenuates cardiac fibrosis in rats with MI induced by isoproterenol.
Rosemary (Rosmarinus officinalis L.)
Rosemary is a medical herb with antioxidant and anti-inflammatory effects. One study reported that intraperitoneal administration of 75 mg/kg of rosemary leaf extract for 4 weeks could significantly attenuate pulmonary fibrosis in rats by inhibiting oxidative stress induced by bleomycin [242]. Rosemary extract also relieves liver cirrhosis mediated by thioacetamide in male rats [244]. Murino Rafacho et al. discovered that dietary supplementation of rosemary could significantly reduce cardiac fibrosis and improve cardiac function after myocardial infarction by decreasing collagen production, LDH activity, oxidative stress, and increasing ATP synthase activity, 3-hydroxyacyl coenzyme A dehydrogenase activity, and citrate synthase activity [245]. Another study showed that rosmarinic acid from rosemary can significantly reduce cardiac fibrosis induced by MI through regulation of the AT1R/p38 MAPK signaling pathway [128]. These results suggest that rosemary might be a protective agent for reducing cardiac fibrosis induced by cardiac injuries.
Scutellarin
Scutellarin is a flavonoid from a Chinese herb named Erigeron breviscapus. This traditional Chinese medicine has been applied in cardiovascular diseases for decades [246]. Scutellarin also has a protective role in cerebrovascular diseases [247, 248]. Pan et al. discovered that scutellarin (3 mg/kg, 10 mg/kg, 30 mg/kg) can significantly improve cardiac function and reduce fibrosis production in left anterior descending artery (LAD)-ligation rats. Moreover, scutellarin can significantly decrease the expression of pro-fibrosis cytokine and inflammation-associated factors TGFβ1 and fibrosis-related glycoprotein fibronectin. Scutellarin remarkably attenuates elevated phosphorylation of p38-MAPK and ERK1/2 in infarcted myocardium and cardiac fibroblasts induced by Ang II [127]. Other studies also show scutellarin has a potential cardiac protective role in cardiac ischemia, as it attenuates the increase of intracellular free calcium during hypoxia in neonatal cardiomyocytes [249,250,251]. Moreover, scutellarin could inhibit cardiac fibrosis by suppressing EMT in rats with cardiac fibrosis induced by isoprenaline, and it also reduces the synthesis of collagen I and collagen III and increases microvascular density [252]. These results suggest that scutellarin is a potential drug which could be further investigated in cardiac fibrosis.
Ulmus wallichiana Planchon
Ulmus wallichiana Planchon which belongs to the Ulmaceae family is a traditional Indian herbal medicine used as an astringent, emollient, expectorant, demulcent, and diuretic. It contains quercetin analogue flavonoids. Ethanolic extracts (EE) and butanoic fractions (BF) from U. wallichiana are useful for anabolic effects on osteoporotic bone by enhancing osteoblast differentiation in ovariectomized rats [253]. It also can regulate osteoblast differentiation by regulating cytokeratin 14 via mTOR/Akt signaling. Besides, EE and BF also lower blood glucose in diabetic rats [254]. Syed et al. demonstrated that rats with cardiac hypertrophy induced by isoprenaline (ISO) present significantly reduced blood pressure and heart rate after treatment of EE and BF from U. wallichiana (orally, 500 and 50 mg/kg/day, respectively). In addition, activities of circulating renin, Ang II, ACE are notably decreased and NO, cGMP levels are markedly increased by EE and BF. Levels of ANP, BNP, TNFα, IL-6, MMP-9, β1-AR, and TGFβ1 are decreased and NOS3, ACE2, and Mas are increased by these two extracts, respectively, suggesting that U. wallichiana can protect against ISO-induced cardiac hypertrophy [255].
Zingerone
Zingerone, an active chemical compound from Zingiber officinale, is used in spice oils because of its spicy aroma in the food. Research discovered that it exerts a protective role against fructose-induced NAFLD. Muniandy Narayanan and Jesudoss discovered that intragastric intubation of NAFLD rats with 100 mg/kg/day of zingerone could markedly reduce microvesicular steatosis and sinusoidal fibrosis, and inhibit the infiltration of inflammatory cells compared with the group without zingerone treatment, indicating that zingerone act as an anti-fibrosis factor in NAFLD [256]. Another study illustrates that zingerone inhibits cardiac fibrosis. It improves reduced catalase activity, reduces expression of angiotensin receptor 1, and inhibits production of oxidative stress factor 8-isoprostane and uric acid. Moreover, zingerone remarkedly reduces TGFβ1 expression and inhibits fibrosis in STZ-induced diabetic rats [257].
Discussion
Cardiac fibrosis contributes to cardiac remodeling in multiple heart diseases and is characterized by elevated cardiac fibroblast activity and excessive ECM synthesis and accumulation. Current studies indicate that angiotensin-converting enzyme inhibitors (ACEIs), Ang II receptor II blockers (ARBs), and beta-blockers can attenuate increased cardiac fibrosis induced by different injuries [258, 259]. However, these drugs have some limitations. Some beta-blockers, such as metoprolol, induce cardiac fibrosis by regulation of a G-protein-independent signaling pathway [260]. It is also known that chronic inhibition of Ang II can lead to an escape phenomenon. Some studies demonstrate that chronic treatment with ACEI can enhance cardiac fibrosis in rats subjected to early ovarian failure [261].
Natural herbal plants and their active biocomponents have a huge potential as anti-fibrosis treatments in cardiologic diseases. They exert anti-inflammation, anti-oxidative stress, anti-proliferation, and anti-migration effects on cardiac fibroblasts and myofibroblasts. Although many of them are still being investigated in the laboratory and few have entered clinical practice, many of them have a great potential for treating cardiologic diseases on the basis of in vivo and in vitro experiments. Herbal plants can be natural, safe, and easily accessible. Numerous experiments demonstrated that herbal plants exhibit a range of benefits for diseases, and their anti-inflammatory, anti-oxidative stress, and anti-fibrosis synthesis properties imply a great significance in therapeutics.
Despite advantages, the challenges of these natural products should also be considered before their application in clinical trials and clinical practice. Natural products have multiple targets, which may lead to off-target activity, and their effects on humans could be either beneficial or detrimental. Secondly, efficient extraction and purification of an effective component from herbal plants is a long and difficult process. It is hard to extract effective biocomponents with 100% purity, which causes imprecision in relevant studies. The identification of effective components is even more challenging. Obtaining a specific amount of plant extract will often require a large amount of raw herbal plants, which can sometimes be time consuming and expensive. It is more challenging to make an extract effective in clinical practice with little dose but high effectiveness. Furthermore, different methods for extracting effective components have prominent impacts on the production of bioactive components, which need to be carefully selected before extraction. More intriguingly, sometimes an active component with lower and higher doses could lead to totally opposite results and this also requires deep investigation. Another big concern is that the current herbal plant research about cardiac fibrosis is still far from enough. The underlying mechanisms of effective extracts from herbal plants, such as their bioactivity, metabolism, specificity, drug interactions, delivered methods, and side effects, etc., are still largely unknown and waiting to be further explored. All in all, extracts from natural herbal plants are promising therapeutics for cardiac fibrosis.
References
Micheletti R, Plaisance I, Abraham BJ, et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med. 2017. https://doi.org/10.1126/scitranslmed.aai9118.
Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–8.
Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–56.
Gulati A, Jabbour A, Ismail TF, et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013;309:896–908.
Janse MJ. Why does atrial fibrillation occur? Eur Heart J. 1997;18(Suppl C):C12–8.
Nguyen MN, Kiriazis H, Gao XM, Du XJ. Cardiac fibrosis and arrhythmogenesis. Compr Physiol. 2017;7:1009–49.
Yang F, Tiano J, Mittal S, Turakhia M, Jacobowitz I, Greenberg Y. Towards a mechanistic understanding and treatment of a progressive disease: atrial fibrillation. J Atr Fibrillation. 2017;10:1627.
Weber KT. Fibrosis and hypertensive heart disease. Curr Opin Cardiol. 2000;15:264–72.
Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200:423–8.
Li GR, Sun HY, Chen JB, Zhou Y, Tse HF, Lau CP. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE. 2009;4: e7307.
Hu H, Sachs F. Stretch-activated ion channels in the heart. J Mol Cell Cardiol. 1997;29:1511–23.
Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15.
Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–80.
Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac fibroblasts. J Mol Cell Cardiol. 2014;70:2–8.
Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016;365:563–81.
Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21.
Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–7.
van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–7.
Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. 1997;20:397–413.
Ptaszek LM, Portillo Lara R, Shirzaei Sani E, et al. Gelatin methacryloyl bioadhesive improves survival and reduces scar burden in a mouse model of myocardial infarction. J Am Heart Assoc. 2020;9:e014199.
Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–65.
Toyama K, Yamabe H, Uemura T, et al. Analysis of oxidative stress expressed by urinary level of 8-hydroxy-2′-deoxyguanosine and biopyrrin in atrial fibrillation: effect of sinus rhythm restoration. Int J Cardiol. 2013;168:80–5.
Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–35.
Neri M, Fineschi V, Di Paolo M, et al. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr Vasc Pharmacol. 2015;13:26–36.
Ye J, Liu L, Ji Q, et al. Anti-interleukin-22-neutralizing antibody attenuates angiotensin II-induced cardiac hypertrophy in mice. Mediat Inflamm. 2017;2017:5635929.
Zheng D, Dong S, Li T, et al. Exogenous hydrogen sulfide attenuates cardiac fibrosis through reactive oxygen species signal pathways in experimental diabetes mellitus models. Cell Physiol Biochem. 2015;36:917–29.
Tardif JC, Tanguay JF, Wright SR, et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J Am Coll Cardiol. 2013;61:2048–55.
Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014;63:1593–603.
Yu XJ, Zou LH, Jin JH, et al. Long noncoding RNAs and novel inflammatory genes determined by RNA sequencing in human lymphocytes are up-regulated in permanent atrial fibrillation. Am J Transl Res. 2017;9:2314–26.
Naruse TK, Matsuzawa Y, Ota M, et al. HLA-DQB1*0601 is primarily associated with the susceptibility to cardiac sarcoidosis. Tissue Antigens. 2000;56:52–7.
Gao X, He X, Luo B, Peng L, Lin J, Zuo Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur J Pharmacol. 2009;606:115–20.
Sutsch G, Bertel O, Rickenbacher P, et al. Regulation of aldosterone secretion in patients with chronic congestive heart failure by endothelins. Am J Cardiol. 2000;85:973–6.
Yao D, Sun NL. Hyperhomocysteinemia accelerates collagen accumulation in the adventitia of balloon-injured rat carotid arteries via angiotensin II type 1 receptor. Int J Mol Sci. 2014;15:19487–98.
Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88.
Kassab S, Garadah T, Abu-Hijleh M, et al. The angiotensin type 1 receptor antagonist valsartan attenuates pathological ventricular hypertrophy induced by hyperhomocysteinemia in rats. J Renin Angiotensin Aldosterone Syst. 2006;7:206–11.
Sarkar C, Ganju RK, Pompili VJ, Chakroborty D. Enhanced peripheral dopamine impairs post-ischemic healing by suppressing angiotensin receptor type 1 expression in endothelial cells and inhibiting angiogenesis. Angiogenesis. 2017;20:97–107.
Zou Y, Akazawa H, Qin Y, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506.
Liu JJ, Huang N, Lu Y, et al. Improving vagal activity ameliorates cardiac fibrosis induced by angiotensin II: in vivo and in vitro. Sci Rep. 2015;5:17108.
Wang J, Duan L, Gao Y, et al. Angiotensin II receptor blocker valsartan ameliorates cardiac fibrosis partly by inhibiting miR-21 expression in diabetic nephropathy mice. Mol Cell Endocrinol. 2017. https://doi.org/10.1016/j.mce.2017.12.005.
Kudo S, Satoh K, Nogi M, et al. SmgGDS as a crucial mediator of the inhibitory effects of statins on cardiac hypertrophy and fibrosis: novel mechanism of the pleiotropic effects of statins. Hypertension. 2016;67:878–89.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–38.
Oruqaj G, Karnati S, Vijayan V, et al. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-beta signaling. Proc Natl Acad Sci USA. 2015;112:E2048–57.
Xu F, Liu C, Zhou D, Zhang L. TGF-beta/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64:157–67.
Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82.
Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95.
Leask A. Targeting the TGFbeta, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal. 2008;20:1409–14.
Shah M, Foreman DM, Ferguson MW. Neutralising antibody to TGF-beta 1,2 reduces cutaneous scarring in adult rodents. J Cell Sci. 1994;107(Pt 5):1137–57.
Cordeiro MF, Mead A, Ali RR, et al. Novel antisense oligonucleotides targeting TGF-beta inhibit in vivo scarring and improve surgical outcome. Gene Ther. 2003;10:59–71.
Shi-wen X, Kennedy L, Renzoni EA, et al. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007;56:4189–94.
Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–19.
Dean RG, Balding LC, Candido R, et al. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53:1245–56.
Behnes M, Brueckmann M, Lang S, et al. Connective tissue growth factor (CTGF/CCN2): diagnostic and prognostic value in acute heart failure. Clin Res Cardiol. 2014;103:107–16.
de Sousa C, Lopes SM, Feijen A, et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn. 2004;231:542–50.
Koitabashi N, Arai M, Niwano K, et al. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur J Heart Fail. 2008;10:373–9.
Kono M, Nakamura Y, Suda T, et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin Chim Acta. 2011;412:2211–5.
Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol Res. 2003;26:1–9.
Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2)–a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol. 2010;114:e83-92.
Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell. 1993;4:637–45.
Ahmed MS, Oie E, Vinge LE, et al. Connective tissue growth factor–a novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J Mol Cell Cardiol. 2004;36:393–404.
Iwanciw D, Rehm M, Porst M, Goppelt-Struebe M. Induction of connective tissue growth factor by angiotensin II: integration of signaling pathways. Arterioscler Thromb Vasc Biol. 2003;23:1782–7.
Black SA Jr, Palamakumbura AH, Stan M, Trackman PC. Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase. J Biol Chem. 2007;282:15416–29.
Ruperez M, Lorenzo O, Blanco-Colio LM, Esteban V, Egido J, Ruiz-Ortega M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation. 2003;108:1499–505.
Brigstock DR. Strategies for blocking the fibrogenic actions of connective tissue growth factor (CCN2): from pharmacological inhibition in vitro to targeted siRNA therapy in vivo. J Cell Commun Signal. 2009;3:5–18.
Loke WM, Proudfoot JM, Hodgson JM, et al. Specific dietary polyphenols attenuate atherosclerosis in apolipoprotein E-knockout mice by alleviating inflammation and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2010;30:749–57.
Vignon-Zellweger N, Heiden S, Miyauchi T, Emoto N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci. 2012;91:490–500.
Widyantoro B, Emoto N, Nakayama K, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–18.
Yoshimatsu Y, Watabe T. Roles of TGF-beta signals in endothelial-mesenchymal transition during cardiac fibrosis. Int J Inflamm. 2011;2011: 724080.
Roldan Ramons S, Pieles GE, Sun M, Slorach C, Hui W, Friedberg MK. Early versus late cardiac remodelling during right ventricular pressure load and impact of preventive versus rescue therapy with endothelin-1 receptor blockers. J Appl Physiol. 2018;124:1349–62.
Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–73.
Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–316.
Jinnin M, Ihn H, Mimura Y, Asano Y, Yamane K, Tamaki K. Regulation of fibrogenic/fibrolytic genes by platelet-derived growth factor C, a novel growth factor, in human dermal fibroblasts. J Cell Physiol. 2005;202:510–7.
Zymek P, Bujak M, Chatila K, et al. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–23.
Liu C, Zhao W, Meng W, et al. Platelet-derived growth factor blockade on cardiac remodeling following infarction. Mol Cell Biochem. 2014;397:295–304.
Abdelaziz Mohamed I, Gadeau AP, Hasan A, Abdulrahman N, Mraiche F. Osteopontin: a promising therapeutic target in cardiac fibrosis. Cells. 2019;8:1558.
Landry NM, Cohen S, Dixon IMC. Periostin in cardiovascular disease and development: a tale of two distinct roles. Basic Res Cardiol. 2018;113:1.
Maruyama K, Imanaka-Yoshida K. The pathogenesis of cardiac fibrosis: a review of recent progress. Int J Mol Sci. 2022;23:2617.
Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Thrombospondins: a role in cardiovascular disease. Int J Mol Sci. 2017;18:1540.
Imanaka-Yoshida K, Tawara I, Yoshida T. Tenascin-C in cardiac disease: a sophisticated controller of inflammation, repair, and fibrosis. Am J Physiol Cell Physiol. 2020;319:C781–96.
Ma Y, Zou H, Zhu XX, et al. Transforming growth factor beta: a potential biomarker and therapeutic target of ventricular remodeling. Oncotarget. 2017;8:53780–90.
Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700.
ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73.
Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93.
Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–6.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63.
Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72.
Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Investig. 2017;127:3770–83.
Bottinger EP. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309–20.
Rosenkranz S, Flesch M, Amann K, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol. 2002;283:H1253–62.
Li RK, Li G, Mickle DA, Weisel RD, et al. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation. 1997;96:874–81.
Wei WY, Zhang N, Li LL, et al. Pioglitazone alleviates cardiac fibrosis and inhibits endothelial to mesenchymal transition induced by pressure overload. Cell Physiol Biochem. 2018;45:26–36.
Sakata Y, Chancey AL, Divakaran VG, Sekiguchi K, Sivasubramanian N, Mann DL. Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res Cardiol. 2008;103:60–8.
Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–48.
Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death–a new approach to cancer therapy. J Clin Investig. 2005;115:2625–32.
Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Investig. 2005;115:2656–64.
Gaspar-Pereira S, Fullard N, Townsend PA, et al. The NF-kappaB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am J Pathol. 2012;180:929–39.
Wei C, Kim IK, Kumar S, et al. NF-kappaB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol. 2013;228:1433–42.
Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem. 2005;280:19230–42.
Frantz S, Fraccarollo D, Wagner H, et al. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–56.
Saito T, Giaid A. Cyclooxygenase-2 and nuclear factor-kappaB in myocardium of end stage human heart failure. Congest Heart Fail. 1999;5:222–7.
Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–38.
Maier HJ, Schips TG, Wietelmann A, et al. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci USA. 2012;109:11794–9.
Xu F, Sun S, Wang X, Ni E, Zhao L, Zhu W. GRK2 mediates arginine vasopressin-induced interleukin-6 production via nuclear factor-kappaB signaling neonatal rat cardiac fibroblast. Mol Pharmacol. 2017;92:278–84.
Cortez DM, Feldman MD, Mummidi S, et al. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol. 2007;293:H3356–65.
Li D, Wang X, Huang Q, Li S, Zhou Y, Li Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-kappaB pathway in vivo and in vitro. Redox Biol. 2018;15:62–73.
Gupta S, Young D, Sen S. Inhibition of NF-kappaB induces regression of cardiac hypertrophy, independent of blood pressure control, in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H20–9.
Kawano S, Kubota T, Monden Y, et al. Blockade of NF-kappaB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res. 2005;67:689–98.
Kawano S, Kubota T, Monden Y, et al. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–44.
Timmers L, van Keulen JK, Hoefer IE, et al. Targeted deletion of nuclear factor kappaB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ Res. 2009;104:699–706.
Zelarayan L, Renger A, Noack C, et al. NF-kappaB activation is required for adaptive cardiac hypertrophy. Cardiovasc Res. 2009;84:416–24.
Esposito G, Rapacciuolo A, Naga Prasad SV, et al. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92.
Ma D, Zhang J, Zhang Y, et al. Inhibition of myocardial hypertrophy by magnesium isoglycyrrhizinate through the TLR4/NF-kappaB signaling pathway in mice. Int Immunopharmacol. 2018;55:237–44.
Gao XM, Wong G, Wang B, et al. Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:1663–70.
Weichhart T, Costantino G, Poglitsch M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–77.
Sadoshima J, Izumo S. Rapamycin selectively inhibits angiotensin II-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6 kinase in angiotensin II-induced cardiac hypertrophy. Circ Res. 1995;77:1040–52.
Simm A, Schluter K, Diez C, Piper HM, Hoppe J. Activation of p70(S6) kinase by beta-adrenoceptor agonists on adult cardiomyocytes. J Mol Cell Cardiol. 1998;30:2059–67.
Lavandero S, Foncea R, Perez V, Sapag-Hagar M. Effect of inhibitors of signal transduction on IGF-1-induced protein synthesis associated with hypertrophy in cultured neonatal rat ventricular myocytes. FEBS Lett. 1998;422:193–6.
Zhao QD, Viswanadhapalli S, Williams P, et al. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation. 2015;131:643–55.
Martinez-Martinez E, Jurado-Lopez R, Valero-Munoz M, et al. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: potential role in obesity. J Hypertens. 2014;32:1104–14 (discussion 1114).
Proud CG. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc Res. 2004;63:403–13.
Xiao Y, Chang W, Wu QQ, et al. Aucubin protects against TGFbeta1-induced cardiac fibroblasts activation by mediating the AMPKalpha/mTOR signaling pathway. Planta Med. 2018;84:91–9.
Song X, Kusakari Y, Xiao CY, et al. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–66.
Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–64.
Tian J, Zhao Y, Liu Y, Liu Y, Chen K, Lyu S. Roles and mechanisms of herbal medicine for diabetic cardiomyopathy: current status and perspective. Oxid Med Cell Longev. 2017;2017:8214541.
Zhang S, Weinheimer C, Courtois M, et al. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Investig. 2003;111:833–41.
Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4.
Pan Z, Zhao W, Zhang X, et al. Scutellarin alleviates interstitial fibrosis and cardiac dysfunction of infarct rats by inhibiting TGFbeta1 expression and activation of p38-MAPK and ERK1/2. Br J Pharmacol. 2011;162:688–700.
Liu Q, Tian J, Xu Y, Li C, Meng X, Fu F. Protective effect of RA on myocardial infarction-induced cardiac fibrosis via AT1R/p38 MAPK pathway signaling and modulation of the ACE2/ACE ratio. J Agric Food Chem. 2016;64:6716–22.
Di Lisa F, Kaludercic N, Paolocci N. Beta(2)-adrenoceptors, NADPH oxidase, ROS and p38 MAPK: another “radical” road to heart failure? Br J Pharmacol. 2011;162:1009–11.
Essick EE, Ouchi N, Wilson RM, et al. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am J Physiol Heart Circ Physiol. 2011;301:H984–93.
Ko SY, Lin IH, Shieh TM, et al. Cell hypertrophy and MEK/ERK phosphorylation are regulated by glyceraldehyde-derived AGEs in cardiomyocyte H9c2 cells. Cell Biochem Biophys. 2013;66:537–44.
Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol. 2001;37:676–85.
Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600.
Schmidt TA, Allen PD, Colucci WS, Marsh JD, Kjeldsen K. No adaptation to digitalization as evaluated by digitalis receptor (Na, K-ATPase) quantification in explanted hearts from donors without heart disease and from digitalized recipients with end-stage heart failure. Am J Cardiol. 1993;71:110–4.
Norgaard A, Bagger JP, Bjerregaard P, Baandrup U, Kjeldsen K, Thomsen PE. Relation of left ventricular function and Na,K-pump concentration in suspected idiopathic dilated cardiomyopathy. Am J Cardiol. 1988;61:1312–5.
Drummond CA, Hill MC, Shi H, et al. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol Genomics. 2016;48:220–9.
Kjeldsen K, Norgaard A, Gheorghiade M. Myocardial Na,K-ATPase: the molecular basis for the hemodynamic effect of digoxin therapy in congestive heart failure. Cardiovasc Res. 2002;55:710–3.
Wansapura AN, Lasko VM, Lingrel JB, Lorenz JN. Mice expressing ouabain-sensitive alpha1-Na,K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2011;300:H347–55.
La J, Reed EB, Koltsova S, et al. Regulation of myofibroblast differentiation by cardiac glycosides. Am J Physiol Lung Cell Mol Physiol. 2016;310:L815–23.
Pistocchi A, Fazio G, Cereda A, et al. Cornelia de lange syndrome: NIPBL haploinsufficiency downregulates canonical Wnt pathway in zebrafish embryos and patients fibroblasts. Cell Death Dis. 2013;4:e866.
Tao H, Yang JJ, Shi KH, Li J. Wnt signaling pathway in cardiac fibrosis: new insights and directions. Metabolism. 2016;65:30–40.
Ding Z, Yuan J, Liang Y, et al. Ryanodine receptor type 2 plays a role in the development of cardiac fibrosis under mechanical stretch through TGFbeta-1. Int Heart J. 2017;58:957–61.
Zou Y, Liang Y, Gong H, et al. Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertension. 2011;58:1099–110.
Harada M, Luo X, Qi XY, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–64.
Kitajima N, Watanabe K, Morimoto S, et al. TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem Biophys Res Commun. 2011;409:108–13.
Kitajima N, Numaga-Tomita T, Watanabe M, et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci Rep. 2016;6:37001.
Numaga-Tomita T, Oda S, Shimauchi T, Nishimura A, Mangmool S, Nishida M. TRPC3 channels in cardiac fibrosis. Front Cardiovasc Med. 2017;4:56.
Numaga-Tomita T, Kitajima N, Kuroda T, et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci Rep. 2016;6:39383.
Seo K, Rainer PP, Shalkey Hahn V, et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci USA. 2014;111:1551–6.
Vestri A, Pierucci F, Frati A, Monaco L, Meacci E. Sphingosine 1-phosphate receptors: do they have a therapeutic potential in cardiac fibrosis? Front Pharmacol. 2017;8:296.
Yamanaka M, Shegogue D, Pei H, et al. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J Biol Chem. 2004;279:53994–4001.
Gellings Lowe N, Swaney JS, Moreno KM, Sabbadini RA. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc Res. 2009;82:303–12.
Takuwa N, Ohkura S, Takashima S, et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc Res. 2010;85:484–93.
Takuwa Y, Ikeda H, Okamoto Y, Takuwa N, Yoshioka K. Sphingosine-1-phosphate as a mediator involved in development of fibrotic diseases. Biochem Biophys Acta. 2013;1831:185–92.
Means CK, Brown JH. Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc Res. 2009;82:193–200.
Liu Y, Wada R, Yamashita T, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Investig. 2000;106:951–61.
Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol. 2010;43:662–73.
Xing C, Bao L, Li W, Fan H. Progress on role of ion channels of cardiac fibroblasts in fibrosis. Front Physiol. 2023;14:1138306.
Chen Z, Li Y, Dian K, Rao L. Modulating microRNAs as novel therapeutic targets in cardiac fibrosis. Theranostics. 2017;7:2287–8.
Li L, Bounds KR, Chatterjee P, Gupta S. MicroRNA-130a, a potential antifibrotic target in cardiac fibrosis. J Am Heart Assoc. 2017;6:e006763.
Duisters RF, Tijsen AJ, Schroen B, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–8 (6p following 178).
Oh JG, Hajjar RJ, Park WJ. Cardiac fibrosis and miR-433. Ann Transl Med. 2016;4:511.
Ooi JY, Bernardo BC, McMullen JR. Therapeutic potential of targeting microRNAs to regulate cardiac fibrosis: miR-433 a new fibrotic player. Ann Transl Med. 2016;4:548.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751–62.
Zhang Y, Zhang YY, Li TT, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol. 2018;115:64–72.
Huang ZW, Tian LH, Yang B, Guo RM. Long noncoding RNA H19 acts as a competing endogenous RNA to mediate CTGF expression by sponging miR-455 in cardiac fibrosis. DNA Cell Biol. 2017;36:759–66.
Tao H, Zhang JG, Qin RH, et al. LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology. 2017;386:11–8.
Zheng H, Huang S, Wei G, et al. CircRNA Samd4 induces cardiac repair after myocardial infarction by blocking mitochondria-derived ROS output. Mol Ther. 2022;30:3477–98.
Ren J, Liu Z, Wang Q, et al. Andrographolide ameliorates abdominal aortic aneurysm progression by inhibiting inflammatory cell infiltration through downregulation of cytokine and integrin expression. J Pharmacol Exp Ther. 2016;356:137–47.
Chen HW, Huang CS, Li CC, et al. Bioavailability of andrographolide and protection against carbon tetrachloride-induced oxidative damage in rats. Toxicol Appl Pharmacol. 2014;280:1–9.
Li Y, Yan H, Zhang Z, et al. Andrographolide derivative AL-1 improves insulin resistance through down-regulation of NF-kappaB signalling pathway. Br J Pharmacol. 2015;172:3151–8.
Yan H, Huang Z, Bai Q, et al. Natural product andrographolide alleviated APAP-induced liver fibrosis by activating Nrf2 antioxidant pathway. Toxicology. 2018;396–397:1–12.
Cabrera D, Wree A, Povero D, et al. Andrographolide ameliorates inflammation and fibrogenesis and attenuates inflammasome activation in experimental non-alcoholic steatohepatitis. Sci Rep. 2017;7:3491.
Lee TY, Chang HH, Wen CK, Huang TH, Chang YS. Modulation of thioacetamide-induced hepatic inflammations, angiogenesis and fibrosis by andrographolide in mice. J Ethnopharmacol. 2014;158(Pt A):423–30.
Yin JN, Li YN, Gao Y, Li SB, Li JD. Andrographolide plays an important role in bleomycin-induced pulmonary fibrosis treatment. Int J Clin Exp Med. 2015;8:12374–81.
Zhu HL, Huang CL, Wang WJ, Zhan XQ, Fan XM. [Effects of andrographolide on the concentration of cytokines in BALF and the expressions of type I and III collagen mRNA in lung tissue in bleomycin-induced rat pulmonary fibrosis]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin J Cell Mol Immunol. 2011;27:725–9.
Ji X, Li C, Ou Y, et al. Andrographolide ameliorates diabetic nephropathy by attenuating hyperglycemia-mediated renal oxidative stress and inflammation via Akt/NF-kappaB pathway. Mol Cell Endocrinol. 2016;437:268–79.
Wu QQ, Ni J, Zhang N, Liao HH, Tang QZ, Deng W. Andrographolide protects against aortic banding-induced experimental cardiac hypertrophy by inhibiting MAPKs signaling. Front Pharmacol. 2017;8:808.
Hayashi K, Narutaki K, Nagaoka Y, Hayashi T, Uesato S. Therapeutic effect of arctiin and arctigenin in immunocompetent and immunocompromised mice infected with influenza A virus. Biol Pharm Bull. 2010;33:1199–205.
Hirose M, Yamaguchi T, Lin C, et al. Effects of arctiin on PhIP-induced mammary, colon and pancreatic carcinogenesis in female Sprague-Dawley rats and MeIQx-induced hepatocarcinogenesis in male F344 rats. Cancer Lett. 2000;155:79–88.
Lee S, Shin S, Kim H, et al. Anti-inflammatory function of arctiin by inhibiting COX-2 expression via NF-kappaB pathways. J Inflamm. 2011;8:16.
Lu LC, Zhou W, Li ZH, et al. Effects of arctiin on streptozotocin-induced diabetic retinopathy in Sprague–Dawley rats. Planta Med. 2012;78:1317–23.
Zhang J, Guo TT, Yang L, et al. [Effect of arctiin on mouse podocyte epithelial-mesenchymal transition induced by advanced oxidation protein products]. Nan Fang Yi Ke Da Xue Xue Bao = J South Med Univ. 2012;32:379–82.
Ma ST, Liu DL, Deng JJ, Niu R, Liu RB. Effect of arctiin on glomerular filtration barrier damage in STZ-induced diabetic nephropathy rats. Phytother Res. 2013;27:1474–80.
Li A, Zhang X, Shu M, et al. Arctigenin suppresses renal interstitial fibrosis in a rat model of obstructive nephropathy. Phytomedicine. 2017;30:28–41.
Li J, Yuan YP, Xu SC, et al. Arctiin protects against cardiac hypertrophy through inhibiting MAPKs and AKT signaling pathways. J Pharmacol Sci. 2017;135:97–104.
Maulik SK, Talwar KK. Therapeutic potential of Terminalia arjuna in cardiovascular disorders. Am J Cardiovasc Drugs. 2012;12:157–63.
Amalraj A, Gopi S. Medicinal properties of Terminalia arjuna (Roxb) Wight & Arn: a review. J Tradit Complement Med. 2017;7:65–78.
Ghosh J, Sil PC. Arjunolic acid: a new multifunctional therapeutic promise of alternative medicine. Biochimie. 2013;95:1098–109.
Al-Gayyar MM, Al Youssef A, Sherif IO, Shams ME, Abbas A. Protective effects of arjunolic acid against cardiac toxicity induced by oral sodium nitrite: effects on cytokine balance and apoptosis. Life Sci. 2014;111:18–26.
Bansal T, Chatterjee E, Singh J, et al. Arjunolic acid, a peroxisome proliferator-activated receptor alpha agonist, regresses cardiac fibrosis by inhibiting non-canonical TGF-beta signaling. J Biol Chem. 2017;292:16440–62.
Wang Z, Li Q, Xiang M, et al. Astragaloside alleviates hepatic fibrosis function via PAR2 signaling pathway in diabetic rats. Cell Physiol Biochem. 2017;41:1156–66.
Yongping M, Zhang X, Xuewei L, et al. Astragaloside prevents BDL-induced liver fibrosis through inhibition of notch signaling activation. J Ethnopharmacol. 2015;169:200–9.
Li LC, Xu L, Hu Y, et al. Astragaloside IV improves bleomycin-induced pulmonary fibrosis in rats by attenuating extracellular matrix deposition. Front Pharmacol. 2017;8:513.
Gui D, Guo Y, Wang F, et al. Astragaloside IV, a novel antioxidant, prevents glucose-induced podocyte apoptosis in vitro and in vivo. PLoS ONE. 2012;7:e39824.
Che X, Wang Q, Xie Y, et al. Astragaloside IV suppresses transforming growth factor-beta1 induced fibrosis of cultured mouse renal fibroblasts via inhibition of the MAPK and NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2015;464:1260–6.
Si J, Wang N, Wang H, et al. HIF-1alpha signaling activation by post-ischemia treatment with astragaloside IV attenuates myocardial ischemia-reperfusion injury. PLoS ONE. 2014;9:e107832.
Tu L, Pan CS, Wei XH, et al. Astragaloside IV protects heart from ischemia and reperfusion injury via energy regulation mechanisms. Microcirculation. 2013;20:736–47.
Zhao J, Yang P, Li F, et al. Therapeutic effects of astragaloside IV on myocardial injuries: multi-target identification and network analysis. PLoS ONE. 2012;7:e44938.
Lu J, Wang QY, Zhou Y, et al. Astragaloside against cardiac fibrosis by inhibiting TRPM7 channel. Phytomedicine. 2017;30:10–7.
Dai H, Jia G, Lu M, Liang C, Wang Y, Wang H. Astragaloside IV inhibits isoprenaline-induced cardiac fibrosis by targeting the reactive oxygen species/mitogen-activated protein kinase signaling axis. Mol Med Rep. 2017;15:1765–70.
Zhang S, Tang F, Yang Y, et al. Astragaloside IV protects against isoproterenol-induced cardiac hypertrophy by regulating NF-kappaB/PGC-1alpha signaling mediated energy biosynthesis. PLoS ONE. 2015;10:e0118759.
Chen P, Xie Y, Shen E, et al. Astragaloside IV attenuates myocardial fibrosis by inhibiting TGF-beta1 signaling in coxsackievirus B3-induced cardiomyopathy. Eur J Pharmacol. 2011;658:168–74.
Zhang J, Zhang H, Deng X, et al. Baicalin attenuates non-alcoholic steatohepatitis by suppressing key regulators of lipid metabolism, inflammation and fibrosis in mice. Life Sci. 2018;192:46–54.
Liao P, Liu L, Wang B, Li W, Fang X, Guan S. Baicalin and geniposide attenuate atherosclerosis involving lipids regulation and immunoregulation in ApoE−/− mice. Eur J Pharmacol. 2014;740:488–95.
Liu L, Liao P, Wang B, Fang X, Li W, Guan S. Oral administration of baicalin and geniposide induces regression of atherosclerosis via inhibiting dendritic cells in ApoE-knockout mice. Int Immunopharmacol. 2014;20:197–204.
Liu X, Gu J, Fan Y, Shi H, Jiang M. Baicalin attenuates acute myocardial infarction of rats via mediating the mitogen-activated protein kinase pathway. Biol Pharm Bull. 2013;36:988–94.
Shen M, Wang L, Yang G, et al. Baicalin protects the cardiomyocytes from ER stress-induced apoptosis: inhibition of CHOP through induction of endothelial nitric oxide synthase. PLoS ONE. 2014;9:e88389.
Liu P, Yan S, Chen M, et al. Effects of baicalin on collagen Iota and collagen IotaIotaIota expression in pulmonary arteries of rats with hypoxic pulmonary hypertension. Int J Mol Med. 2015;35:901–8.
Zhang Y, Liao P, Zhu M, et al. Baicalin attenuates cardiac dysfunction and myocardial remodeling in a chronic pressure-overload mice model. Cell Physiol Biochem. 2017;41:849–64.
Xiao Y, Ye J, Zhou Y, et al. Baicalin inhibits pressure overload-induced cardiac fibrosis through regulating AMPK/TGF-beta/Smads signaling pathway. Arch Biochem Biophys. 2018;640:37–46.
Bai R, Yin X, Feng X, et al. Corydalis hendersonii Hemsl protects against myocardial injury by attenuating inflammation and fibrosis via NF-kappaB and JAK2-STAT3 signaling pathways. J Ethnopharmacol. 2017;207:174–83.
Tao L, Shen S, Fu S, et al. Traditional Chinese medication Qiliqiangxin attenuates cardiac remodeling after acute myocardial infarction in mice. Sci Rep. 2015;5:8374.
Somerset SM, Johannot L. Dietary flavonoid sources in Australian adults. Nutr Cancer. 2008;60:442–9.
Kim BW, Lee ER, Min HM, et al. Sustained ERK activation is involved in the kaempferol-induced apoptosis of breast cancer cells and is more evident under 3-D culture condition. Cancer Biol Ther. 2008;7:1080–9.
Luo H, Daddysman MK, Rankin GO, Jiang BH, Chen YC. Kaempferol enhances cisplatin’s effect on ovarian cancer cells through promoting apoptosis caused by down regulation of cMyc. Cancer Cell Int. 2010;10:16.
Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–40.
Feng H, Cao J, Zhang G, Wang Y. Kaempferol attenuates cardiac hypertrophy via regulation of ASK1/MAPK signaling pathway and oxidative stress. Planta Med. 2017;83:837–45.
Liu Y, Gao L, Guo S, et al. Kaempferol alleviates angiotensin II-induced cardiac dysfunction and interstitial fibrosis in mice. Cell Physiol Biochem. 2017;43:2253–63.
Chen X, Qian J, Wang L, et al. Kaempferol attenuates hyperglycemia-induced cardiac injuries by inhibiting inflammatory responses and oxidative stress. Endocrine. 2018;60:83–94.
Prathapan A, Varghese MV, Abhilash S, et al. Polyphenol rich ethanolic extract from Boerhavia diffusa L. mitigates angiotensin II induced cardiac hypertrophy and fibrosis in rats. Biomed Pharmacother Biomed Pharmacother. 2017;87:427–36.
Liu ZW, Wang JK, Qiu C, et al. Matrine pretreatment improves cardiac function in rats with diabetic cardiomyopathy via suppressing ROS/TLR-4 signaling pathway. Acta Pharmacol Sin. 2015;36:323–33.
Zhang L, Zhang H, Zhu Z, et al. Matrine regulates immune functions to inhibit the proliferation of leukemic cells. Int J Clin Exp Med. 2015;8:5591–600.
Gao HY, Li GY, Lou MM, Li XY, Wei XY, Wang JH. Hepatoprotective effect of Matrine salvianolic acid B salt on carbon tetrachloride-induced hepatic fibrosis. J Inflamm. 2012;9:16.
Fu P, Feng M, Zhang Z. Experimental study on effect of matrine in alleviating renal tubulointerstitial fibrosis in unilateral ureteral obstruction model. Zhongguo Zhong Xi Yi Jie He Za Zhi = Chin J Integr Tradit West Med. 2006;26:140–3.
Zhang Y, Cui L, Guan G, et al. Matrine suppresses cardiac fibrosis by inhibiting the TGFbeta/Smad pathway in experimental diabetic cardiomyopathy. Mol Med Rep. 2018;17:1775–81.
Liu Z, Zhang Y, Tang Z, et al. Matrine attenuates cardiac fibrosis by affecting ATF6 signaling pathway in diabetic cardiomyopathy. Eur J Pharmacol. 2017;804:21–30.
Pyun BJ, Kim YS, Lee IS, Kim JS. Homonoia riparia and its major component, myricitrin, inhibit high glucose-induced apoptosis of human retinal pericytes. Integr Med Res. 2017;6:300–9.
Qi S, Feng Z, Li Q, Qi Z, Zhang Y. Myricitrin modulates NADPH oxidase-dependent ROS production to inhibit endotoxin-mediated inflammation by blocking the JAK/STAT1 and NOX2/p47(phox) pathways. Oxid Med Cell Longev. 2017;2017:9738745.
Wang M, Sun GB, Du YY, et al. Myricitrin protects cardiomyocytes from hypoxia/reoxygenation injury: involvement of heat shock protein 90. Front Pharmacol. 2017;8:353.
Zhang B, Chen Y, Shen Q, et al. Myricitrin attenuates high glucose-induced apoptosis through activating Akt-Nrf2 signaling in H9c2 cardiomyocytes. Molecules. 2016;21:880.
Sun J, Sun G, Cui X, Meng X, Qin M, Sun X. Myricitrin protects against doxorubicin-induced cardiotoxicity by counteracting oxidative stress and inhibiting mitochondrial apoptosis via ERK/P53 pathway. Evid-Based Complement Altern Med eCAM. 2016;2016:6093783.
Zhang B, Shen Q, Chen Y, et al. Myricitrin alleviates oxidative stress-induced inflammation and apoptosis and protects mice against diabetic cardiomyopathy. Sci Rep. 2017;7:44239.
Majdalawieh AF, Fayyad MW. Immunomodulatory and anti-inflammatory action of Nigella sativa and thymoquinone: a comprehensive review. Int Immunopharmacol. 2015;28:295–304.
Abidi A, Robbe A, Kourda N, Ben Khamsa S, Legrand A. Nigella sativa, a traditional Tunisian herbal medicine, attenuates bleomycin-induced pulmonary fibrosis in a rat model. Biomed Pharmacother. 2017;90:626–37.
Al-Gayyar MM, Hassan HM, Alyoussef A, Abbas A, Darweish MM, El-Hawwary AA. Nigella sativa oil attenuates chronic nephrotoxicity induced by oral sodium nitrite: effects on tissue fibrosis and apoptosis. Redox Rep. 2016;21:50–60.
Turkdogan MK, Agaoglu Z, Yener Z, Sekeroglu R, Akkan HA, Avci ME. The role of antioxidant vitamins (C and E), selenium and Nigella sativa in the prevention of liver fibrosis and cirrhosis in rabbits: new hopes. Dtsch Tierarztl Wochenschr. 2001;108:71–3.
Pipalia PR, Annigeri RG, Mehta R. Clinicobiochemical evaluation of turmeric with black pepper and Nigella sativa in management of oral submucous fibrosis-a double-blind, randomized preliminary study. Oral Surg Oral Med Oral Pathol Oral Radiol. 2016;122:705–12.
Norouzi F, Abareshi A, Asgharzadeh F, et al. The effect of Nigella sativa on inflammation-induced myocardial fibrosis in male rats. Res Pharma Sci. 2017;12:74–81.
Peng CH, Chyau CC, Chan KC, Chan TH, Wang CJ, Huang CN. Hibiscus sabdariffa polyphenolic extract inhibits hyperglycemia, hyperlipidemia, and glycation-oxidative stress while improving insulin resistance. J Agric Food Chem. 2011;59:9901–9.
Bahri S, Ben Ali R, Gasmi K, et al. Prophylactic and curative effect of rosemary leaves extract in a bleomycin model of pulmonary fibrosis. Pharm Biol. 2017;55:462–71.
Si LY, Ali SAM, Latip J, Fauzi NM, Budin SB, Zainalabidin S. Roselle is cardioprotective in diet-induced obesity rat model with myocardial infarction. Life Sci. 2017;191:157–65.
Al-Attar AM, Shawush NA. Influence of olive and rosemary leaves extracts on chemically induced liver cirrhosis in male rats. Saudi J Biol Sci. 2015;22:157–63.
Murino Rafacho BP, Portugal-Dos-Santos P, Goncalves AF, et al. Rosemary supplementation (Rosmarinus oficinallis L) attenuates cardiac remodeling after myocardial infarction in rats. PLoS ONE. 2017;12:e0177521.
Chen X, Shi X, Zhang X, et al. Scutellarin attenuates hypertension-induced expression of brain Toll-like receptor 4/nuclear factor kappa B. Mediat Inflamm. 2013;2013:432623.
Wang S, Wang H, Guo H, Kang L, Gao X, Hu L. Neuroprotection of scutellarin is mediated by inhibition of microglial inflammatory activation. Neuroscience. 2011;185:150–60.
Liu H, Yang X, Tang R, Liu J, Xu H. Effect of scutellarin on nitric oxide production in early stages of neuron damage induced by hydrogen peroxide. Pharmacol Res. 2005;51:205–10.
Jia JH, Chen KP, Chen SX, Liu KZ, Fan TL, Chen YC. Breviscapine, a traditional Chinese medicine, alleviates myocardial ischaemia reperfusion injury in diabetic rats. Acta Cardiol. 2008;63:757–62.
Li XL, Li YQ, Yan WM, et al. A study of the cardioprotective effect of breviscapine during hypoxia of cardiomyocytes. Planta Med. 2004;70:1039–44.
Wang M, Zhang WB, Zhu JH, Fu GS, Zhou BQ. Breviscapine ameliorates cardiac dysfunction and regulates the myocardial Ca(2+)-cycling proteins in streptozotocin-induced diabetic rats. Acta Diabetol. 2010;47(Suppl 1):209–18.
Zhou H, Chen X, Chen L, et al. Anti-fibrosis effect of scutellarin via inhibition of endothelial-mesenchymal transition on isoprenaline-induced myocardial fibrosis in rats. Molecules. 2014;19:15611–23.
Sharan K, Siddiqui JA, Swarnkar G, et al. Extract and fraction from Ulmus wallichiana Planchon promote peak bone achievement and have a nonestrogenic osteoprotective effect. Menopause. 2010;17:393–402.
Rawat P, Kumar M, Rahuja N, Lal Srivastava DS, Srivastava AK, Maurya R. Synthesis and antihyperglycemic activity of phenolic C-glycosides. Bioorg Med Chem Lett. 2011;21:228–33.
Syed AA, Lahiri S, Mohan D, et al. Cardioprotective effect of Ulmus wallichiana planchon in beta-adrenergic agonist induced cardiac hypertrophy. Front Pharmacol. 2016;7:510.
Muniandy Narayanan J, Jesudoss VA. Hepatoprotective potential of zingerone against nonalcoholic fatty liver disease in rats fed with fructose-enriched diet. Gen Physiol Biophys. 2016;35:185–94.
El-Bassossy HM, Al-Thubiani WS, Elberry AA, et al. Zingerone alleviates the delayed ventricular repolarization and AV conduction in diabetes: effect on cardiac fibrosis and inflammation. PLoS ONE. 2017;12:e0189074.
Weber KT, Sun Y, Gerling IC, Guntaka RV. Regression of established cardiac fibrosis in hypertensive heart disease. Am J Hypertens. 2017;30:1049–52.
von Lueder TG, Wang BH, Kompa AR, et al. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. 2015;8:71–8.
Nakaya M, Chikura S, Watari K, et al. Induction of cardiac fibrosis by beta-blocker in G protein-independent and G protein-coupled receptor kinase 5/beta-arrestin2-dependent Signaling pathways. J Biol Chem. 2012;287:35669–77.
Dutra SGV, Felix ACS, Gastaldi AC, De Paula FT, Vieira S, De Souza HCD. Chronic treatment with angiotensin-converting enzyme inhibitor increases cardiac fibrosis in young rats submitted to early ovarian failure. Auton Neurosci. 2017;206:28–34.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Author Contributions
Xuejing Yu contributed to the conception, literature searching and writing.
Disclosures
Xuejing Yu has nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Yu, X. Promising Therapeutic Treatments for Cardiac Fibrosis: Herbal Plants and Their Extracts. Cardiol Ther 12, 415–443 (2023). https://doi.org/10.1007/s40119-023-00319-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-023-00319-4