
Abstract
This paper describes the hydrothermal synthesis, full characterization, and architectural diversity of three intriguingly bioactive cobalt–organic frameworks, namely, 3D [Co(HL −)2(BPY)] n ·4nH2O (1), 2D [Co(HL −)2(BPE)] n (2), and 2D [Co(HL −)2(DPP)] n (3) coordination polymers, synthesized through a mixed ligand strategy using H 2 L (1-H-indazole-3-carboxylic acid) as a main structural block and the flexible bipyridine and its derivatives (BPY = 4,4′-bipydine, BPE = 1,2-bis(4-pyridyl)ethane, DPP = 1,3-bis(4-pyridyl)propane) as auxiliary ligand sources. Complexes 1–3 were isolated as air stable and slightly soluble crystalline solids and characterized using elemental analysis, FT-IR, electrochemical technique, thermogravimetric analysis, powder X-ray diffractometer, and single-crystal X-ray crystallography. The bipyridine derivatives played key roles in defining the structural space group and dimensionality feature of the obtained networks. The abundant H-bonding and π–π stacking interactions in complexes 1–3 gave rise to their intricate metal–organic structures of 3D (1), 2D (2), and 2D (3). In addition, the solutions of complexes 1–3 showed profound antifungal activities against the selected strain of Colletotrichum musae compared with the controlled group using benomyl as a traditional agrochemical fungicide.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
1-H-indazole-3-carboxylic acid (H 2 L) is a common moiety in the pharmaceutical industry and exhibits profound anticancer and antioxidant activities and other intriguingly therapeutic properties. To date, much effort has been devoted to design and synthesize a new structure of indazole carboxylic acid derivatives through molecular docking and chemical modification [1–4]. However, only a few researchers focused on the property and application of its coordination complexes. The latest studies in H 2 L coordination polymers were reported by Gryca et al. [5] and Hawes et al. [6]. Therefore, we intended to synthesize three new cobalt coordination polymers with the combination of H 2 L and bipyridine compounds. The three cobalt–organic frameworks were characterized and described, namely, 3D [Co(HL −)2(BPY)] n ·4nH2O (1), 2D [Co(HL −)2(BPE)] n (2), and 2D [Co(HL −)2(DPP)] n (3) coordination polymers. Complexes 1–3 were synthesized using the main structural block of 1-H-indazole-3-carboxylic acid (H 2 L) assembled by the flexible ligands of 4,4′-bipyridine (BPY), 1,2-bis(4-pyridyl)ethane (BPE), and 1,3-bis(4-pyridyl)propane (DPP), respectively. In addition, the antifungal activity of complexes 1–3 was evaluated against the fruit pathogen of Colletotrichum musae using the mycelium growth rate method. The experimental results indicated that Co compounds dissolved in N,N-dimethylformamide (DMF) under two concentrations (100 and 10 μg/L) inhibited fungal mycelium growth with varying degrees of effectiveness compared with the control. Complex 1 exhibited the most growth inhibition against C. musae [7–9].
Experimental
Materials and methods
All reagents were purchased from commercial sources and used without further purification. H 2 L was synthesized according to the literature [10], and its crystal structure is illustrated in Fig. 1. Elemental analysis for C, H, and N was conducted using a Perkin-Elmer 2400II elemental analyzer. Infrared spectra were recorded on an FT-IR spectrometer 6700 instrument with samples dispersed in powdered KBr in the range of 4000–400 cm−1 (abbreviation: vs, very strong; s, strong; m, medium; and w, weak). Cyclic voltammetry (CV) curves were measured using an advanced electrochemical workstation of Princeton Applied Research PARSTAT 2273. A conventional three-electrode system was used. A glassy carbon electrode, Ag–AgCl, and Pt wire were used as working, reference, and auxiliary electrodes, respectively. Powder X-ray diffraction (PXRD) patterns were obtained on Rigaku MiniFlex 600 diffractometer instrument at the Faculty of Materials, University of Guangxi. The apparatus parameters were set at 20 kV and 20 mA with Cu Kα (λ = 1.5406 Å) radiation. Thermogravimetric analysis (TGA) was recorded on a Perkin-Elmer SAT-6000 instrument where the samples were heated on the surface of ceramic crucibles under constant N2 flow (10 mL/min) at a rate of 5 °C/min.

Single-crystal structural diagram of 1-H-indazole-3-carboxylic acid (H 2 L). Irrelevant H atoms were omitted for clarity (cyan dashed line shows H bonds)
X-ray crystallography
X-ray crystallographic data for complexes 1–3 were collected on a Bruker SMART CCD area detector at room temperature using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures of complexes 1–3 were addressed using direct methods and refined by the full-matrix least squares on F 2 using the SHELXL-97 program [11–14]. All non-H atoms were refined with anisotropic displacement parameters. The H atom was inserted at a geometrically calculated position and included in the refinement using the riding model approximation, Uiso (H) = 1.2 × Ueq (C). Moreover, the H atom of water molecule was assigned with isotropic thermal parameter setting at 1.5 times the associated O atoms. Crystallographic data and structure determination statistics are listed in Table 1, and the selected bond lengths and angles for complexes 1–3 are represented in Table 2. Furthermore, the PXRD patterns of complexes 1–3 were obtained using a Rigaku MiniFlex instrument (Cu-Kα radiation, scanning rate of 1°/min, 293.15 K).
Synthesis and analytical data [Co(HL−)2(BPY)] n ·4nH2O (1)
H 2 L (2 mmol, 324.30 mg), BPY (1 mmol, 156.18 mg), and CoCl2·6H2O (1 mmol, 237.93 mg) were combined in a solution containing 2 mL of DMF, 3 mL of MeOH, and 5 mL of H2O. The obtained mixture was stirred for 60 min in air at room temperature. An aqueous 1 M NaOH solution (100 μL) was then added. The mixture was further stirred at room temperature for 30 min and then transferred to a 25 mL Teflon reactor, which was sealed, heated at 125 °C for 3 days, and cooled at a ramp rate of 10 °C/6 h. Orange polyhedral-shaped crystals of complex 1 were obtained, along with a 53 % yield (based on Co salt). Analytical calculations for C26H26CoN6O8 (mol wt. 609.46): C, 51.19; N, 13.78; H, 4.27. Found: C, 52.05; N, 13.69; H, 4.25. FTIR (KBr pellet, cm−1): 3513–3466 m, 1632 s, 1605 s, 1323 m, 1243 w, 1100 m, 823 s, 723 s, and 615 vs.
Synthesis and analytical data [Co(HL−)2(BPE)] n (2)
The mixture of CoCl2·6H2O (1 mmol, 237.93 mg), H 2 L (2 mmol, 324.30 mg), and BPE (1 mmol, 184.24 mg) were combined in a solution with 3 mL of DMF and 10 mL of MeOH. The obtained mixture was stirred for 30 min in air, moved to a 25 mL Teflon reactor, heated to 130 °C for 2 days, and cooled at 5 °C h−1 at room temperature. Pink rhombus-shaped crystals were collected and dried in air to obtain a 60 % yield for complex 2 (based on Co salt). Analytical calculations for C28H22CoN6O4 (mol wt. 565.45): C, 59.42; N, 14.86; H, 3.89. Found: C, 59.35; N, 14.80; H, 4.02. FTIR (KBr pellet, cm−1): 2953 m, 1626 vs, 1589 s, 1320 s, 1230 m, 1100 s, 742 m, 734 m, and 620 vs.
Synthesis and analytical data [Co(HL−)2(DPP)] n (3)
Complex 3 was synthesized following the method described for complex 2, except the auxiliary ligand of BPE was replaced by DPP molecule. Purple block crystals were obtained and dried in air to obtain a 56 % yield for complex 3 (based on Co salt). Analytical calculations for C29H24CoN6O4 (mol wt. 579.47): C, 60.05; N, 14.50; H, 4.14. Found: C, 60.01; N, 14.35; H, 4.05. FTIR (KBr pellet, cm−1): 2947 w, 1624 vs, 1574 s, 1322 s, 1220 m, 1070 m, 751 s, and 620 s.
Antifungal activity studies
The biological activities of complexes 1–3 and organic ligand molecules were evaluated using the mycelium growth rate method with potato dextrose agar [15, 16]. The strain Colletotrichum musarum was used as a test organism obtained from the Guangxi Academy of Agricultural Sciences in Nanning, Guangxi. The general procedure of the experiment is that the agar plates involving the blank and controlled groups were separately pretreated with DMF or test compounds dissolved in DMF. The plates were then inoculated by spreading 3 µL of a solution containing 1 × 105 spores/mL onto the agar surface and incubated in a growth chamber at 25 ± 1 °C. Inhibition of fungal growth was monitored several days after the treatment. Sensitivity of fungal species to each test compound was determined by comparing the size of the inhibitory zones (Table 3) drawn on Eq. (1), where R i and R C represent the spore percentages germinating within the treated and controlled samples, respectively [17, 18]. In addition, fungicide standard benomyl was used as a control at 2 mM in 4 μL of DMF.
Results and discussion
Crystal structure of complex [Co(HL−)2(BPY)] n ·4nH2O (1)
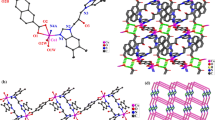
Single-crystal XRD revealed that complex 1 was a 3D coordination polymer that crystallized in the tetragonal space group I4/a. The symmetric unit of complex 1 contained one Co(II) cation, two deprotonated HL − ligand motifs, one BPY auxiliary ligand, and four lattice water molecules. As shown in Fig. 2a, the crystallographic inversion center of Co(II) showed an octahedral coordination, and it was coordinated by Co(1)–N(2) and Co(1)–O(1) distances of 2.140(4) and 2.056(3) Å from the bidentate ligand moieties of H 2 L, Co(1)–N(1) distance of 2.171(3) Å from the auxiliary ligand of Bpy molecule, and two lattice water (O3w, O4w) molecules per symmetric unit. The angles of O1–Co1–N1 and N2–Co1–N1 were 91.12° and 87.81°, respectively, whereas the aromatic ring of BPY molecule was bent and twisted, forming a dihedral angle of 29.9°; all bond lengths and angles were within normal ranges [6]. In complex 1, BPY adopted an N-coordination mode, bridged the neighboring Co(II) cation, and formed overlapping linear chains with cationic charge. Furthermore, each two symmetrically distributed molecules of deprotonated H 2 L linked the center atom of Co(II) ion through the N2 and O2 heteroatom, constructed the 1D linear chain, and neutralized the charge of the linear chain. Mutually paralleled 1D chain motifs were reinforced through the π–π stacking interactions generated from two adjacent indazole blocks (Fig. 2b). The metal–organic framework of complex 1 was further extended into an intricate 3D supramolecular network through helical H-bonding interactions (O3–H3A···O2) between the crystallization water molecule (O3) and non-chelating carboxylate groups of deprotonated H 2 L blocks (Fig. 2c).

a Coordination environment of the center Co(II) atom in complex 1 with the thermal ellipsoids drawn at 30 % probability. H atoms were omitted for clarity, and the following symmetry code was used: A = 1 − x, −y + 2, −z + 2; b π–π stacking interactions between two adjacent aromatic rings (red dashed lines); and c 3D supramolecular framework formed by helical H-bond interactions (green dashed lines), which were separately enlarged
Crystal structure of complex [Co(HL−)2(BPE)] n (2)
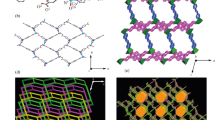
Complex 2 exhibited a crystallized structure in the monoclinic C2/c space group and a 2D metal–organic framework. The asymmetric unit included a Co(II) cation, a doubly deprotonated molecules of HL −, and one BPE flexible ligand, as depicted in Fig. 3a. The six-coordinated Co atom {CoN4O2} adopted a distorted octahedral geometry (τ 6 = 0.21) [19, 20] filled with N1 and O1 atoms from two H 2 L ligands (Co1–N1 2.174(3), Co1–O1 2.063(2) Å) and N3 atoms from two different BPE moieties (Co1–N3 2.143(3) Å). The bond angles between the center Co atom and the coordinated O and N atoms ranged from 76.77(9)° to 100.15(0)°. In complex 2, each of the two symmetrically positioned HL − anions displayed a bidentate mode, linking the Co(II) ion to generate a neutral monomer [Co(HL −)2]. The flexible ligand of BPE positioned in the equatorial axis linked the two neighboring monomers through the pyridyl N atom to generate extended 1D zigzag chain motifs (Fig. 3b). Furthermore, two mutually parallel linear chains were pillared through intermolecular H-bond interactions (N2–H2···O2) and gave rise to a 2D H-bonded layer in the linear motifs (Fig. 3c). The metal–organic network of complex 2 was reinforced by a series of π-π stacking interactions between two adjacent indazole blocks (Fig. 3d).

a Coordination environment of the Co(II) atom in complex 2 with thermal ellipsoids drawn at 30 % probability. H atoms were omitted for clarity, and a symmetry code was used to label the equivalent atoms: A = −x + 1, y, −z + 3/2; b 1D zigzag chain (H 2 L and BPE color code as cyan and red, respectively); c 2D network formed by H-bond interactions (illustrated in light yellow dashed lines); and d π–π stacking interactions (red cylinder)
Crystal structure of complex [Co(HL−)2(DPP)] n (3)
Complex 3 revealed a crystallized structure in the triclinic P-1 space group and a 2D coordination network. The asymmetric unit contained one metal cation Co(II) atom, two anion HL − moieties, and one DPP ligand molecule. As depicted in Fig. 4, the structural composition and coordination mode {CoN4O2} of complex 3 were similar to those of complex 2, except that the higher flexibility ligand of DPP occurred in the BPE molecule in complex 2. Although complexes 2 and 3 were constructed from the same main building block and auxiliary ligand, the length modification and flexibility of the backbone chain –(CH2)– (n = 2, 3) exerted a slight effect on the polymeric structure embodied in the molecular distance involving H-bond and π–π stacking interactions. The distance of H bonds (N1–H1···O1 1.804(2) Å) and interchain π–π interactions (Cg1···Cg1A 3.646(2) Å, Cg1, and Cg1A are the centroid of the two adjacent aromatic rings of indazole blocks) in complex 3 was shorter than that of H bonds (N2–H2···O2 1.901(6) Å) and π–π stacking interactions (Cg2···Cg2A 3.805(8) Å, Cg2, and Cg2A defined similarly as Cg1) in complex 2 [21]. In addition, the elongated aliphatic (n = 3) chain along with the auxiliary ligand of DPP that acts as a linker increased the cavity size between the metal–organic networks and diminished the symmetry of complex 3 in the triclinic system compared with complex 2 in the monoclinic system.

Coordination environment of the Co(II) atom in complex 3 with thermal ellipsoids drawn at 30 % probability. H atoms were omitted for clarity and a symmetry code was used to label the equivalent atoms: A = x + 1, y + 1, z
IR spectrum
The IR spectra of complexes 1–3 showed the expected bands, revealing N-containing carboxylate ligand; bipyridine ligand molecules involving BPY, BPE, and DPP; and lattice water. As shown in Supplementary Figs. S1–3, the strong board band in the range of 3550–3462 cm−1 was assigned to the O–H stretching vibration of lattice water [22]. The characteristic bands at 1605 cm−1 for 1, 1589 cm−1 for 2, and 1574 cm−1 for 3 may be attributed to the ν C=N stretching vibration of the indazole rings [23]. The bands at 2953 cm−1 for 2 and 2937 cm−1 for 3 could identify the existence of an aliphatic chain in the –(CH) n – (n = 2 or 3) group that resulted from the flexibility ligand molecules BPE and DPP. In addition, the strong absorption peak separately observed at 1632 and 1323 cm−1 for 1, 1626 and 1320 cm−1 for 2, and 1624 and 1322 cm−1 for 3 revealed the asymmetric and symmetric vibrations of the carboxylic groups. The calculated frequency difference (Δ = 302–309 cm−1) between the symmetric and asymmetric COO− stretching in complexes 1–3 was in good agreement with the μ1–η1:η0 coordination mode of the carboxylate groups in the μ-H 2 L moiety [24].
Electrochemical behavior
Complexes 1–3 and H 2 L were illustrated using CV with a Pt working electrode at room temperature in 0.1 M LiClO4–MeCN solution [25, 26]. Two single-electron irreversible reduction waves were detected under N2 ambience for complex 1. The former at potential values (Table 4) of −0.45 to −0.49 V versus the Ag/AgCl range was assigned to the process of Co(III) → Co(II) reduction (wave Ired), whereas the latter in −0.84 to −0.92 V versus the Ag/AgCl range was attributed to the Co(II) → Co(I) process (IIred). Furthermore, a repeated trial was conducted to clarify the influence of the reaction condition of detection under N2 atmosphere or in air on electrochemical behavior. Interestingly, the former cationic wave Ired vanished while being detected in air without bubbling N2. The curve of complexes 2 and 3 showed similar electrochemical behavior in air when detected under ambient conditions. No anodic waves were observed for every compound upon scan reversal. The electrochemical property of ligand molecule of H 2 L was also unveiled. A cathodic wave that occurred at the potential of −1.03 V was easily found. Furthermore, no obvious anodic waves have been monitored for H 2 L upon scan reversal. An integrated CV curve is shown in Fig. 5.

CV curves of complexes 1–3 and H 2 L were obtained while being dissolved in 0.1 M LiClO4/MeCN solution, respectively, under a scan rate of 100 mV s−1. The blue and black curves indicate detection under N2 ambience, whereas the red curve indicates the collecting date in air
The reduction potential (Ired) of complex 1 reflected that Co(III) compound could exist in acetonitrile solution. Nevertheless, the former cathodic wave easily vanished while being detected without any further inert gas protection through bubbling N. This wave repeatedly appeared when detected under N2 atmosphere. This phenomenon manifested that complex 1 was an oxidant-hypersensitive substance that easily reacts with oxidant components, such as O [27]. Furthermore, no cathodic waves (Ired) in complexes 2 and 3 were observed, except the reduction wave (IIred). This finding showed that the auxiliary ligands BPE and DPP could stabilize complexes 2 and 3. In addition, the structural difference between complex 1 and complexes 2 and 3, in which abundant lattice water molecules permeated in the complex 1 cavity, may have resulted in these different electrochemical properties.
Thermal properties and PXRD analysis
TGA was conducted to complexes 1–3 to probe the thermal stabilities of coordination polymers 1–3 (Fig. 6). In the case of complex 1, the weight losses were observed to be as follows: ~11.05 % (calcd., 11.81 %), 48–150 °C, attributable to the loss of lattice water molecules. Decomposition of ligand molecules of H 2 L and BPE was achieved beyond 500 °C with 78.51 % (calcd., 78.53 %) mass loss. Finally, the residue was 10.21 % (calcd., 9.66 %), which was defined as CoO. Complexes 2 and 3 showed a similar variation trend involving a two-step weight loss process on account of the similar structural composition and coordination mode. A weight loss of ~55.87 % (calcd., 56.71 %) for complex 2 and ~55.04 % (calcd., 55.62 %) for complex 3 under 316–362 °C accounted for the loss of organic compound H 2 L. Furthermore, weight loss of ~31.02 (calcd., 32.58 %) and ~32.84 (calcd., 34.21 %) was initially observed at 375 and 390 °C, corresponding to the decomposition of auxiliary ligand molecules BPE and DPP, respectively.

TG curves of complex 1 (blue), complex 2 (red), and complex 3 (black)
The simulated PXRD patterns derived from the data of complexes 1–3 matched well with the experimental PXRD curve collected at room temperature (Fig. 7), indicating that the structural integrity and phase purity in the synthesized materials were in good agreement with the as-grown crystals.

PXRD diffraction patterns for complexes 1–3
Antifungal activity studies
The ligand molecules and their cobalt chelates were individually tested in vitro using the mycelium growth rate method under two different concentrations (10 and 100 μM) to evaluate antifungal activity against C. musarum [16]. The inhibition rates varied (Fig. 8). Compound 5 in vitro containing 10 or 100 μM 3.21 or 78.33 % fungal suspension, respectively, showed strong antifungal activities. Compound 6 containing 10 or 100 μM 57.46 or 62.07 % fungal suspension, respectively, and compound 7 containing 10 or 100 μM 47.27 or 51.16 % fungal suspension, respectively, demonstrated similar results. Compounds 1–4 showed comparatively weak activities against C. musarum growth at the highest concentration of 100 μM with 46.10, 32.70, 29.30, and 24.81 %, respectively. The experimental statistics showed that complexes 1–3 were more activated than their parent ligands against the same microorganism under the identical experimental conditions. The increasing antifungal activities of metal complexes were mainly attributed to the interaction between the metal ion and cell membrane. A reasonable illustration can be consulted in Tweedy’s chelation theory [28, 29]. Chelations partly decrease the polarity of the metal core in consequence of partial sharing of its positive charge with N- or O-containing ligand molecules. Furthermore, the possible π-electron can be delocalized over the whole chelating ring. Such chelation effect can enhance the lipophilic character of the central metal atom, which subsequently promotes its permeation through the lipid layers of the fungus cell membrane. In addition, the outstandingly biological activities of Co complexes 1–3 may result from the formation of H bonds through the non-chelating carboxyl group and the heteroatom of indazole blocks with the active component of cell membrane. The general tendency of growth inhibition against C. musarum was observed in the following reactivity order: [Co(HL −)2(BPY)] n ·4nH2O > [Co(HL −)2-(BPE)] n > [Co(HL −)2(DPP)] n > H 2 L > BPY > BPE > DPP. The results showed that the antifungal effectiveness of Co complexes 1–3 could reach the same level of conventional fungicide as benomyl in inhibiting the growth of banana anthracnose. Complexes 1–3 may potentially act as a good source of ‘green pesticide’ in preventing a great number of economic losses in the main plantation of Guangxi, China.

Growth inhibition of Colletotrichum aimed at compounds 1–8 under two concentrations of 100 and 10 μg/L with fungal suspension. Numbers 1–8 represent DMF + H2L, DMF + BPY, DMF + BPE, DMF + DPP, DMF + complex 1, DMF + complex 2, DMF + complex 3, and benomyl (agrochemical fungicides), respectively
Conclusion
In summary, three new structures of Co complexes assembled using the organic compound of indazole carboxylic acid and flexibility ligand of bipyridine and its derivatives have been successfully synthesized through the hydrothermal method. The coordination mode and supramolecular framework of complexes 1–3 demonstrated that the structural diversity mainly resulted from the auxiliary ligand and molecular interactions involving H bonds and π–π stacking. In addition, the study of the electrochemical behavior of complex 1 has a potential application in organic reduction reaction. Similar studies in our laboratory are currently underway using these characteristics of metal–organic compounds in the preparation of a novel organic catalyst. Moreover, research on antifungal activities indicated that the biological activity of Co(II) complexes can extend its application in preventing the growth of C. musarum, which causes serious fruit diseases in bananas and mangoes.
Supplementary materials
CCDC Nos. 1032742, 1032743, 1032744, and 1032745 contain the supplementary crystallographic data for 1, 2, 3, and H 2 L. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk. In addition, the FT-IR spectroscopy patterns of complexes 1–3 were distributed as the supplementary materials labeled as Supplementary Figs. S1–3.
References
J. Bermudez et al., 5-Hydroxytryptamine (5-HT3) receptor antagonists. 1. Indazole and indolizine-3-carboxylic acid derivatives. J. Med. Chem. 33(7), 1924–1929 (1990)
K.P. Harish, K.N. Mohana, L. Mallesha, Synthesis of indazole substituted-1,3,4-thiadiazoles and their anticonvulsant activity. Drug Invent. Today 5(2), 92–99 (2013)
J. Sun et al., Synthesis, biological evaluation and molecular docking studies of pyrazole derivatives coupling with a thiourea moiety as novel CDKs inhibitors. Eur. J. Med. Chem. 68, 1–9 (2013)
R. Yefidoff-Freedman et al., 3-Substituted indazoles as configurationally locked 4EGI-1 mimetics and inhibitors of the eIF4E/eIF4G interaction. ChemBioChem 15(4), 595–611 (2014)
I. Gryca et al., New p-tolylimido rhenium(v) complexes with carboxylate-based ligands: synthesis, structures and their catalytic potential in oxidations with peroxides. Dalton Trans. 43(15), 5759 (2014)
C.S. Hawes, P.E. Kruger et al., Preparation of open and closed forms of the lvt network with Cu(RSC. Advances 4(30), 15770 (2014)
Z.H. Chohan, C.T. Supuran, Organometallic compounds with biologically active molecules: in vitro antibacterial and antifungal activity of some 1, 1′-(dicarbohydrazono)ferrocenes and their cobalt(II), copper(II), nickel(II) and zinc(II) complexes. Appl. Organomet. Chem. 19(12), 1207–1214 (2005)
S. Srinivasan, J. Annaraj, P. Athappan, Spectral and redox studies on mixed ligand complexes of cobalt(III) phenanthroline/bipyridyl and benzoylhydrazones, their DNA binding and antimicrobial activity. J. Inorg. Biochem. 99(3), 876–882 (2005)
S.U. Rehman et al., In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J. Enzyme Inhib. Med. Chem. 20(4), 333–340 (2005)
T. Yoshida et al., Practical synthesis of 1H-indazole-3-carboxylic acid and its derivatives. Heterocycles 43(12), 2701–2712 (1996)
Bruker. SMART. Bruker AXS Inc., Madison, Wisconsin, USA. 2001
S. Bruker, SAINT (Bruker AXS Inc., Madison, 2002)
G.M. Sheldrick, SADABS (University of Göttingen, Germany, 1996)
G.M. Sheldrick, Acta Cryst. A64, 112–122 (2008)
S.H. Khan, J. Aked, N. Magan, Control of the anthracnose pathogen of banana (Colletotrichum musae) using antioxidants alone and in combination with thiabendazole or imazalil. Plant Pathol. 50(5), 601–608 (2001)
N. Suwannarach et al., Biofumigation with the endophytic fungus Nodulisporium spp. CMU-UPE34 to control postharvest decay of citrus fruit. Crop Protect. 45, 63–70 (2013)
A.P. Martínez-Camacho et al., Chitosan composite films: thermal, structural, mechanical and antifungal properties. Carbohydr. Polym. 82(2), 305–315 (2010)
M.M. Radwan et al., Antifungal compounds from turmeric and nutmeg with activity against plant pathogens. Fitoterapia 99, 341–346 (2014)
O. Fabelo et al., 1,2,4,5-Benzenetetracarboxylate- and 2,2′-bipyrimidine-containing cobalt(II) coordination polymers: preparation, crystal structure, and magnetic properties. Inorg. Chem. 47(9), 3568–3576 (2008)
O. Fabelo et al., (4,4) Rectangular lattices of cobalt(II) with 1,2,4,5-benzenetetracarboxylic acid: influence of the packing in the crystal structure. Cryst. Growth Des. 8(11), 3984–3992 (2008)
S.M. Sony, M.N. Ponnuswamy, Nature of pi-interactions inn nitrogen-containing heterocyclic systems: a structural database analysis. Cryst. Growth Des. 6(3), 736–742 (2006)
K. Ohno et al., The effect of cooperative hydrogen bonding on the OH stretching-band shift for water clusters studied by matrix-isolation infrared spectroscopy and density functional theory. Phys. Chem. Chem. Phys. 7(16), 3005–3014 (2005)
M. Boca et al., Selective imidazolidine ring opening during complex formation of iron(III), copper(II), and zinc(II) with a multidentate ligand obtained from 2-pyridinecarboxaldehyde N-oxide and triethylenetetramine. Inorg. Chem. 39(15), 3205–3212 (2000)
S.W. Jaros et al., Aliphatic dicarboxylate directed assembly of silver(I) 1,3,5-triaza-7-phosphaadamantane coordination networks: topological versatility and antimicrobial activity. Cryst. Growth Des. 14(11), 5408–5417 (2014)
F. Jaouen and A.Morozan, Metal organic frameworks for electrochemical applications. Energy Environ. Sci. 5, 9269–9290 (2012)
S.R. Ahrenholtz, C.C. Epley, A.J. Morris, Solvothermal preparation of an electrocatalytic metalloporphyrin MOF thin film and its redox hopping charge-transfer mechanism. J. Am. Chem. Soc. 136(6), 2464–2472 (2014)
J.A. Miranda, C.J. Wade, R.D. Little, Indirect electroreductive cyclization and electrohydrocyclization using catalytic reduced nickel(II) salen. J. Org. Chem. 70(20), 8017–8026 (2005)
H.K.P. Porto, et al., Synthesis of a ruthenium(II) tryptophan-associated complex and biological evaluation against Ehrlich murine breast carcinoma. Transit. Metal Chem. 40,1–10 (2015)
B.G. Tweedy, Phytopathology 55, 910 (1964)
Acknowledgments
We gratefully acknowledge the support from Prof. Xian-Hong Yin of the School of Chemistry and Chemical Engineering, Guangxi University for Nationalities, Nanning, China, for providing single-crystal structural refinement. Furthermore, the authors wish to thank the Natural Science Foundation of Guangxi (Grant No. 2014GXNSFAA118047) for the financial aid.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Xiao, H., Li, XB., Qin, GF. et al. Directed assembly of cobalt(II) 1-H-indazole-3-carboxylic acid coordination networks by bipyridine and its derivatives: structural versatility, electrochemical properties, and antifungal activity. J IRAN CHEM SOC 13, 793–802 (2016). https://doi.org/10.1007/s13738-015-0792-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-015-0792-8